Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(16):4523-4531. doi:10.7150/ijms.121550 This issue Cite
Research Paper
Mapping the Somatic Mutation Landscape of Familial NF2-Related Schwannomatosis using Whole-Exome Sequencing
Fushu Luo1#, Liangqi Jiang1#, Yimin Pan1, Haoyu Li1,2,3, Jun Tan1,2,3 ![]() , Changwu Wu1,2,3
, Changwu Wu1,2,3 ![]() , Qing Liu1,2,3
, Qing Liu1,2,3 ![]()
1. Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha, Hunan, China.
2. National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, Hunan, China.
3. Clinical Research Center for Skull Base Surgery and Neuro-oncology in Hunan Province, Changsha, Hunan, China.
# Fushu Luo and Liangqi Jiang contribute equally, co-first authors.
Received 2025-7-12; Accepted 2025-9-23; Published 2025-10-27
Abstract

Background: Neurofibromatosis type 2 (NF2), currently more accurately named NF2-related schwannomatosis (NF2-SWN), is classified as a multiple tumor syndrome, caused by impaired expression of the merlin protein. Approximately 50% of affected individuals inherit a germline mutation from their parents, while reports on the somatic mutation landscape of other genes are infrequent.
Aim: To further explore the somatic mutations of NF2-SWN and provide a theoretical basis for the treatment of NF2-SWN.
Design: A retrospective study was conducted to follow up on NF2-SWN patients who underwent surgical treatment in the Department of Neurosurgery of Xiangya Hospital. Whole-exome sequencing (WES) was performed on patients with a clear family history.
Methods: This study compiled clinical data from 29 patients diagnosed with NF2-SWN, conducted WES on 7 patients with well-documented genetic histories, and subsequently analyzed their genetic mutations.
Results: Whole-exome sequencing identified frequent somatic mutations in genes such as TTN, FLG, CR2, and FSIP2. Missense mutations and C>T transitions were the most common alteration types.
Conclusion: TTN, CR2, FLG, and FSIP2 demonstrated elevated mutation frequencies in these familial NF2-SWN patients, indicating that these mutations may contribute to the development and progression of familial NF2-SWN.
Keywords: Neurofibromatosis type 2, Whole-exome sequencing, Somatic mutation landscape, Genetic history
Introduction
Neurofibromatosis type 2 (NF2), now more accurately referred to as NF2-related schwannomatosis (NF2-SWN)[1], is a rare autosomal dominant genetic disorder with an estimated incidence of 1 in 25,000 to 40,000 live births, affecting both genders equally[2-4]. It is characterized by the development of multiple neoplasms of the central and peripheral nervous systems, most notably bilateral vestibular schwannomas, which are present in nearly all affected individuals[5]. The phenotypic manifestation of NF2-SWN exhibits variability in terms of severity, age of onset, and tumor presentation, encompassing intracranial and/or spinal schwannomas, intracranial meningiomas, and ependymomas[2, 3]. Moreover, the specific type or locus of mutations within the NF2 gene may affect the phenotypic diversity of tumor manifestations[6].
The disorder arises from mutations in the NF2 tumor suppressor gene located on chromosome 22q12, which encodes the protein merlin (MIM 607379), a critical regulator of cell proliferation and apoptosis through the Hippo tumor suppressor pathway[3, 4, 7]. Merlin plays a critical role in the formation of cell junctions, activation of anti-mitogenic signaling pathways, and inhibition of oncogenic gene expression, all of which are encoded by the NF2 gene[7, 8]. More than 50% of individuals diagnosed with NF2-SWN have no family history of the disease, suggesting that they likely have de novo mutations in the NF2 gene, which highlights the genetic heterogeneity of this disorder[9, 10]. Nonetheless, certain cases exhibit a distinct familial predisposition, and it remains uncertain whether the pattern of NF2 mutations in these patients aligns with that of non-inherited NF2-SWN. Knudson's two-hit hypothesis offers an explanation for the onset of NF2-SWN[4]. Beyond mutations in the NF2 gene, alterations in other genes may also contribute to the initiation of NF2-SWN[11, 12]. The disease is relentlessly progressive, with patients often developing multiple tumors over their lifetime, necessitating a multidisciplinary approach to management[4].
Diagnosis of NF2-SWN is primarily based on clinical criteria, including the presence of bilateral vestibular schwannomas or a combination of other characteristic tumors and a family history of the disorder[4]. Advances in genetic testing have allowed for the identification of NF2 mutations in both germline and somatic tissues, providing a more definitive diagnosis and enabling early intervention[13, 14]. Genetic studies have also revealed that the clinical phenotype of NF2-SWN can vary significantly, even within families, due to factors such as mosaicism and the timing of biallelic inactivation of the NF2 gene[13]. This variability underscores the importance of personalized medicine in the management of NF2-SWN, as treatment strategies must be tailored to the individual tumor burden and clinical symptoms of each patient[4, 5].
In this study, we conducted a retrospective analysis of the clinical and molecular pathological features of 29 patients diagnosed with NF2-SWN treated at the Department of Neurosurgery, Xiangya Hospital, China. Moreover, we reported novel related gene mutations in the exon sequencing results of patients with 7 cases of familial NF2-related schwannomatosis.
Methods
Patients
A total of twenty-nine NF2-related schwannomatosis patients diagnosed and treated surgically in the Department of Neurosurgery at Xiangya Hospital, Central South University between June 2013 and June 2023 were included in this study. All diagnoses were confirmed based on the updated diagnostic criteria for NF2-SWN (Table 1) Clinical data, imaging findings, pathological results, and other treatment-related information were retrieved from the hospital information system. The cohort consisted of 15 male and 14 female patients. A comprehensive family history review identified 7 individuals with a potential familial predisposition to NF2-SWN. Blood samples were collected from these patients for whole-exome sequencing (WES). This research was conducted in accordance with the principles outlined in the Declaration of Helsinki. The study was approved by the Medical Ethics Committee of Xiangya Hospital, Central South University (Approval number: 202407141), and written informed consent was obtained from all participants.
Diagnosis criteria for NF2-related schwannomatosis, formerly known as NF2.
| Diagnostic criteria for NF2-related schwannomatosis | ||||
|---|---|---|---|---|
| A diagnosis of NF2-related schwannomatosis (previously termed neurofibromatosis 2, NF2) can be made when an individual has one of the following: | ||||
| 1. Bilateral vestibular schwannomas (VS) | ||||
| 2. An identical NF2 pathogenic variant in at least 2 anatomically distinct NF2-related tumors (schwannoma, meningioma, and/or ependymoma). (Note: if the variant allele fraction (VAF) in unaffected tissues such as blood is clearly <50%, the diagnosis is mosaic NF2-related schwannomatosis) | ||||
| 3. Either 2 major or 1 major and 2 minor criteria as described in the following: | ||||
| Major criteria: • Unilateral VS • First-degree relative other than sibling with NF2-related schwannomatosis • 2 or more meningiomas (Note: single meningioma qualifies as minor criteria) • NF2 pathogenic variant in an unaffected tissue such as blood (Note: if the VAF is clearly if the VAF is clearly <50%, the diagnosis is mosaic NF2-related schwannomatosis) | ||||
| Minor criteria: Can count >1 of a type (e.g., 2 distinct schwannomas would count as 2 minor criteria) • Ependymoma, meningioma (Note: multiple meningiomas qualify as a major criteria), schwannoma (Note: if the major criterion is unilateral VS, at least 1 schwannoma must be dermal in location) Can count only once (e.g., bilateral cortical cataracts count as a single minor criterion) • Juvenile subcapsular or cortical cataract, retinal hamartoma, epiretinal membrane in a person aged <40 years, meningioma Pattern of genetic changes in unaffected and tumor tissue in NF2-related schwannomatosis | ||||
| Gene locus | Unaffected Tissue | Tumor 1 | Tumor 2 | Comment |
| NF2 | ||||
| Allele 1 | PV1 | PV1 | PV1 | Shared NF2 pathogenic variant |
| Allele 2 | WT | LOH or NF2 PV2 | LOH or NF2 PV3 | Tumor-specific partial loss of 22q in trans position or a different NF2 somatic second PV in every anatomically unrelated tumor |
DNA extraction and library construction
Genomic DNA was extracted from intracranial tumor tissue using Agilent SureSelect Human All Exon V6 kit. DNA quantification and integrity were determined by the Nanodrop spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE) and the 1% agarose electrophoresis, respectively. Human genomic DNA samples were captured using Agilent SureSelect Human All Exon v6 library (Agilent Technologies, USA) following the manufacturer's protocol. Briefly, the genomic DNA was sheared into short fragments using the kit's enzyme. The sheared deoxyribonucleic acid (DNA) was purified and treated with reagents supplied with the kit according to the protocol. Adapters from Agilent were ligated onto the polished ends and the libraries were amplified by polymerase chain reaction (PCR). The amplified libraries were hybridized with the custom probes. The DNA fragments bound with the probes were washed and eluted with the buffer provided in the kit. Then these libraries were sequenced on the Illumina sequencing platform (NovaSeq 6000, Illumina, Inc., San Diego, CA) and 150 bp paired-end reads were generated. The WES and analysis were conducted by OE Biotech Co., Ltd. (Shanghai, China).
Whole-exome sequencing analysis
The raw data was compiled in fastq format. To obtain high-quality sequences suitable for subsequent analysis, the raw reads were preprocessed using fastq (Version: 0.20.0). Initially, adapter sequences were removed, and bases with an average base quality score below 20 were filtered out. Additionally, reads containing ambiguous bases or shorter than 75 bp were also filtered. The clean reads were then aligned to the human reference genome (GRCh37) using BWA (Version: 0.7.17). SAMtools (Version: 1.9) was utilized to convert the mapped reads to the appropriate format. Using paired blood sample sequencing or the 1000 Genomes Project data as a reference[15], somatic single nucleotide polymorphisms (SNPs) and insertions/deletions (INDELs) were identified. When the data from the 1000 Genomes Project was used as a reference, variants with a population frequency greater than 0.1% in the database were filtered out to minimize the inclusion of common germline polymorphisms. Similar to previous studies[16, 17], the R package “maftools” was used to generate a summary plot of single nucleotide variation (SNV), while the R package “ComplexHeatmap” was utilized to visualize the overall somatic alterations in an OncoPrint plot[18].
Results
The clinical characteristics of NF2-SWN
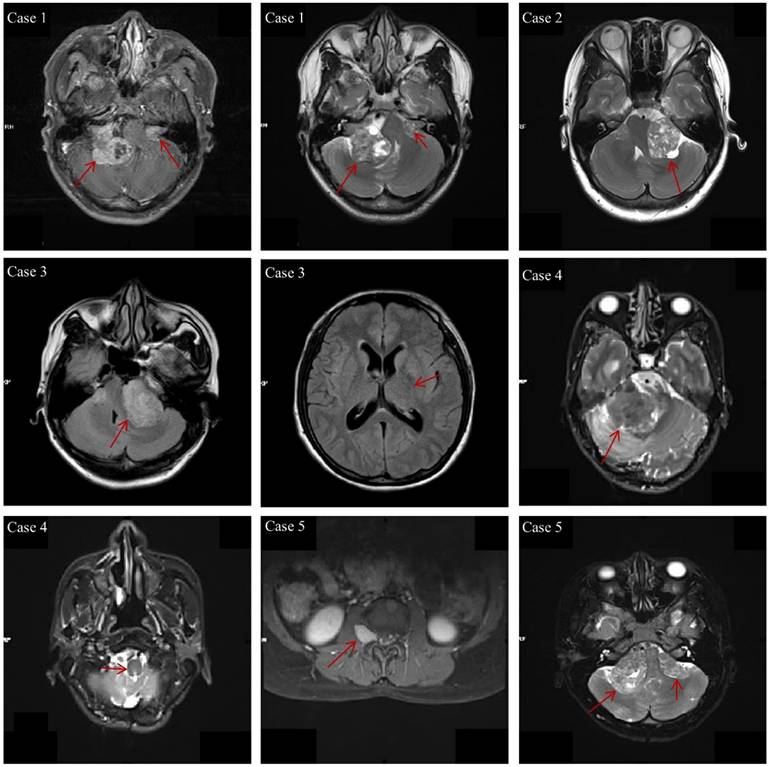
The clinical characteristics of the 29 NF2-SWN patients are listed in Table 2. The age of onset for almost all NF2-SWN patients was less than 40 years (93.1%), and most patients presented with tinnitus and hearing loss when visiting the hospital (72.4%). The majority of tumors were located in the cerebellar horn region (subtentorial, 96.7%), which is a primary cause of hearing loss among these patients. Furthermore, most patients exhibited multiple intracranial tumors (93.1%), with pathological results indicating that schwannomas were the most common tumor type (79.3%). Notably, patients with multiple tumors predominantly presented with a combination of schwannomas and meningiomas, which supports the second diagnostic criterion for NF2-SWN. Figure 1 demonstrates the typical radiological findings of intracranial tumors in NF2-SWN patients, including those with schwannomas combined with meningiomas.
The typical radiological findings of intracranial tumors in NF2-SWN patients. Case 1: Bilateral vestibular schwannomas; Case 2: Unilateral vestibular tumor, first-level relatives suffer from NF2-SWN; Case 3: Multiple meningiomas and unilateral vestibular schwannoma; Case 4: Heterozygous schwannomas (schwannoma and perineurium); Case 5: Vestibular schwannoma with intraspinal and subcutaneous masses.
Clinical characteristics of all patients in this study
| Characteristics | Number (cases) | Proportion (%) |
|---|---|---|
| Total | 29 | 100 |
| Gender | ||
| Male | 15 | 51.7 |
| Female | 14 | 48.3 |
| Age of onset | ||
| <40 | 27 | 93.1 |
| ≥ 40 | 2 | 6.9 |
| Initial symptoms | ||
| Tinnitus, hearing loss | 21 | 72.4 |
| Dizziness, headache | 7 | 24.1 |
| Limb weakness, walking instability | 9 | 31 |
| Loss of consciousness | 1 | 3.4 |
| Family history | ||
| Familial | 7 | 24.1 |
| Sporadic | 22 | 75.9 |
| Tumor number | ||
| Single | 2 | 6.9 |
| Multiple | 27 | 93.1 |
| Pathology | ||
| Schwannoma | 23 | 79.3 |
| Meningioma | 8 | 27.6 |
| Perineurioma | 1 | 3.4 |
| Tumor location | ||
| Supratentorial | 18 | 60 |
| Subtentorial | 29 | 96.7 |
| Intraspinal | 5 | 16.7 |
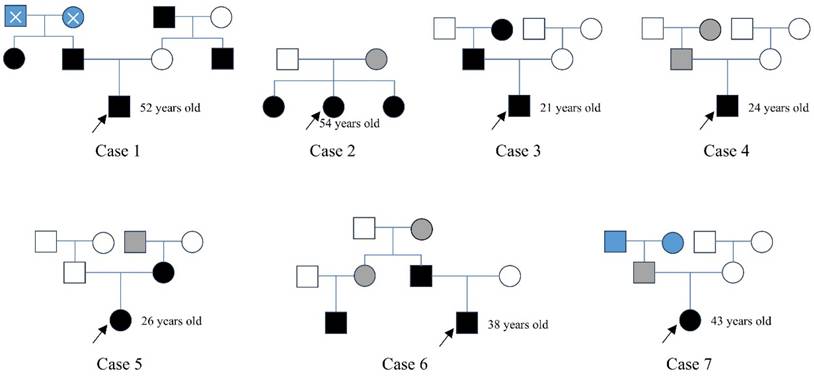
For NF2-SWN patients with a suspected family history, we conducted follow-up investigations into their family backgrounds and constructed family pedigrees (Figure 2). In Case 1, the patient's father, aunt, uncle, and maternal grandfather were all diagnosed with NF2-SWN; however, his paternal grandparents had passed away, and their NF2-SWN status could not be confirmed. In Case 2, a 54-year-old female patient and her two sisters were diagnosed with NF2-SWN, while their mother exhibited hearing loss symptoms but lacked definitive imaging findings, thus being classified as a suspected NF2-SWN case. In Case 3, both the patient's father and grandmother were confirmed NF2-SWN patients, clearly indicating a familial inheritance pattern. In Case 4, a 24-year-old male patient reported that his father and grandmother exhibited hearing loss symptoms, but no further clinical evidence supported an NF2-SWN diagnosis, so they were also categorized as suspected cases. In Case 5, both the patient and her mother were diagnosed with NF2-SWN, and it was reported that her maternal grandfather had suffered from acoustic neuroma (no medical records available; marked as a suspected case). In Case 6, the patient's father and cousin were confirmed to have NF2-SWN, and he reported that his aunt and grandmother had experienced hearing loss, although no relevant diagnostic reports were available. In Case 7, the female patient reported unilateral hearing loss in her father, while the health status of her grandparents remained unknown. Analysis of the constructed pedigrees did not identify any specific genetic transmission patterns, such as maternal or paternal inheritance.
The family maps of patients with inherited NF2-SWN. In the pedigrees, black symbols represent individuals diagnosed with NF2-SWN, white symbols indicate unaffected individuals, grey symbols denote suspected cases, and blue symbols indicate individuals with unknown disease status. A cross-symbol marks deceased individuals. Squares represent males, and circles represent females. A black arrow highlights the proband, i.e., the individual who sought medical care at our hospital.
The whole-exome sequencing analysis of familial NF2-SWN
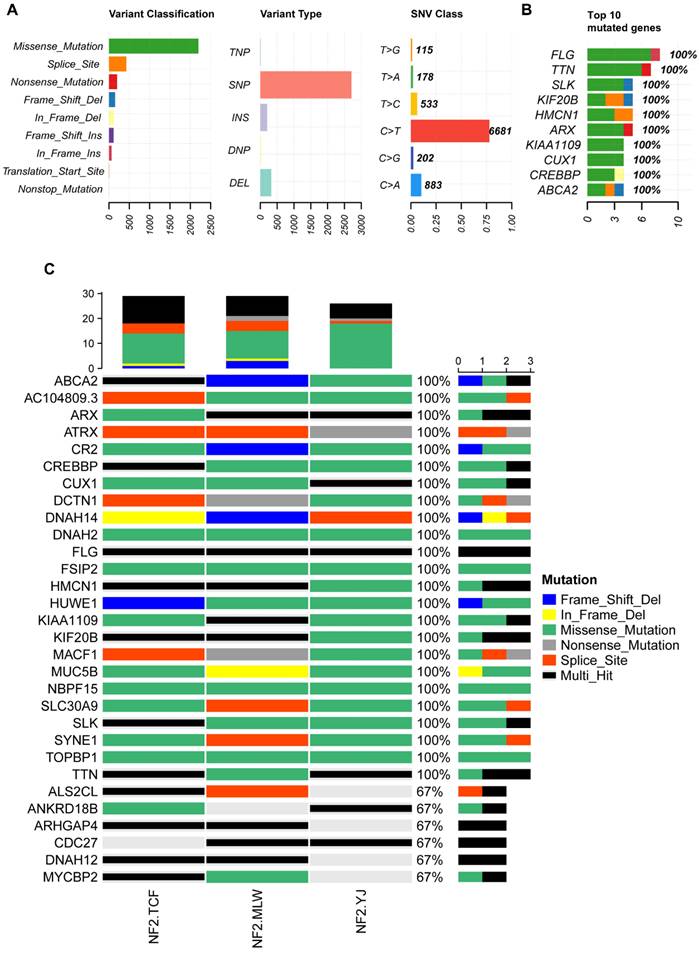
Familial NF2-SWN is relatively rare, and we aim to comprehensively understand the somatic mutation landscape of familial NF2-SWN in order to identify potential therapeutic targets. We collected tumor samples from 7 cases of familial NF2-SWN, among which 3 cases had matched blood samples. Through WES of the NF2-SWN tumors from the 3 cases with blood samples as controls, we identified a total of 2648 genes that were mutated in at least one sample (Supplementary Table S1). Figure 3 presents a landscape of the somatic mutations in the samples with blood controls. The results indicated that missense mutations represented the predominant mutation type, with SNPs comprising over 80% of all mutations. Among these, the C>T substitution was identified as the most frequent SNV type, accounting for more than 75% (Figure 3A). Figure 3B illustrated the top 10 genes with the highest mutation counts, among which FLG and TTN exhibit the most frequent mutations, with 8 and 7 mutations respectively (Supplementary Table S1). Figure 3C displayed the top 30 genes with the highest mutation frequencies, 24 of which—including ATRX—were found to be mutated in all samples.
The somatic mutation landscape of three NF2-SWN patients with paired blood samples. A, Landscape and counts of mutation types. B, The top 10 genes with the highest mutation counts. C, The top 30 frequently mutated genes in NF2-SWN patients with paired blood samples.
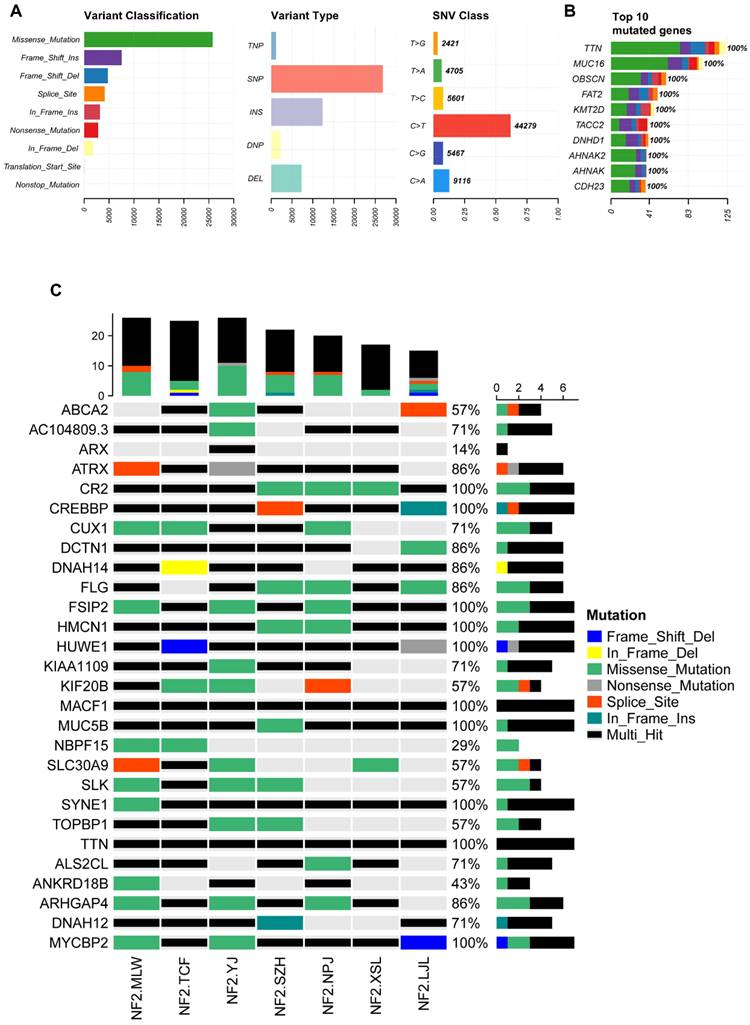
Due to limitations in sample collection, we were unable to obtain paired blood samples for all NF2-SWN tumors. Therefore, we analyzed seven NF2-SWN samples using data from the 1000 Genomes Project as a control[19]. As illustrated in Figure 4A, within the framework of the 1000 Genomes Project, gene mutations in familial NF2-SWN remained predominantly missense mutations and were primarily composed of SNPs, with C>T being the most common type of SNVs (>50%). This finding aligned with the results presented in Figure 3. Figure 4B displays the top 10 genes with the highest mutation counts in this background, among which TTN and MUC16 have the highest number of mutations. A total of 273 genes were mutated across all 7 samples (Supplementary Table S2). Considering that using paired blood samples as controls is more accurate, we validated the top 30 genes with the highest mutation frequency identified previously in the background of the 1000 Genomes Project. Most of these genes, such as ATRX, CR2, FLG, and FSIP2, still exhibited high mutation frequencies (Figure 4C), suggesting that mutations in these genes may play a role in the development and progression of familial NF2-SWN.
The somatic mutation landscape of all seven NF2-SWN patients. A, Landscape and counts of mutation types. B, The top 10 genes with the highest mutation counts. C, Mutation profiles of 30 previously identified genes in the background of the 1000 Genomes Project.
Discussion
NF2-SWN presents a significant challenge in neurosurgery due to its low prevalence and poor prognosis. Currently, surgical resection is the primary treatment modality; however, the management of symptoms and postoperative complications necessitates the development of novel therapeutic approaches[20-22]. Despite early detection and surgical intervention, the prognosis for NF2-SWN is often poor, characterized by recurrent neurological tumors and hearing loss[21, 23]. Whole-exome sequencing demonstrates high sensitivity in identifying common, rare and low-frequency mutations, and effectively detecting most disease-associated mutations within exonic regions while requiring sequencing of only approximately 1% of the genome[24, 25]. Whole-exome sequencing is employed in the study of NF2-SWN to identify mutations within the population, thereby offering novel insights into the clinical treatment and prognosis of NF2-SWN. This research seeks to identify specific mutated genes in patients with a familial genetic history of NF2-SWN by utilizing WES, with the goal of elucidating the genetic or developmental mechanisms underlying the disease. Such findings have the potential to inform and innovate clinical treatment strategies for NF2-SWN.
Previous research has investigated the pathogenesis of NF2-SWN with a focus on gene expression. It is now widely recognized that the pathogenesis is attributed to the dysfunction of Merlin, a protein encoded by the tumor suppressor gene NF2. Merlin is part of the Ezrin/Radixin/Moesin (ERM) family of membrane cytoskeletal junction proteins. Furthermore, studies on skin tumors, vestibular schwannomas, and meningiomas in NF2-SWN patients have suggested that loss of heterozygosity due to NF2 allele deletion and the inactivation of NF2 gene transcription through hypermethylation may represent additional mechanisms of tumorigenesis[21]. However, these mechanisms have not been fully elucidated, indicating that the genetic study of NF2-SWN remains incomplete. In this study, genomic sequencing was performed on blood and tumor tissue samples, accompanied by an analysis of familial genetic history, leading to the identification of several genes of notable research interest. In this study, Whole-exome sequencing was conducted on tumor tissue samples using matched blood samples and the 1000 Genomes Project as control datasets. Integrated with family genetic history analysis, several genes of notable research significance were identified. These include TTN, which ranked among the top mutated genes in both analytical contexts; CR2, CREBBP, and FSIP2, which exhibited a 100% mutation frequency across both controls; and genes such as TTN, FLG, and HUWE1, which displayed a relatively high proportion of multi-hit mutations. These mutated genes may hold significant research implications for understanding the pathogenesis and progression of the disease.
The discovery of highly mutated genes such as TTN and FLG in this study warrants preliminary biological speculation. Currently, there are no studies have specifically investigated the involvement of TTN or FLG in NF2-SWN. TTN encodes a giant structural protein essential for sarcomere assembly in muscle cells and has been well-established as a key player in cardiomyopathy and heart failure[26, 27]. While its direct role in neuro-oncogenesis remains incompletely understood, a recent genomic analysis of craniospinal axis malignant peripheral nerve sheath tumors reported TTN mutations in up to 61% of cases[28]. This independent observation in nervous system tumors suggests that TTN mutations may potentially affect the biological behavior of schwannomas through mechanisms such as influencing cellular mechanical stability, signaling, or as yet unknown functions in Schwann cells. FLG (filaggrin) is mainly associated with skin barrier function and plays a significant role especially in skin diseases such as atopic dermatitis[29]. Its association with neuro-oncology is less direct. However, its recurrent appearance in our analysis prompts us to consider potential off-target effects or alterations in cell differentiation pathways that may interact with merlin loss of function. Future functional studies are crucial for validating these preliminary observations and elucidating any potential mechanistic links.
This study has several limitations that should be acknowledged. The most significant is the small sample size, particularly the limited number of familial cases with matched blood-tumor pairs (n=3), which is inherent to research on rare diseases like NF2-SWN. For the remaining four patients, we utilized the 1000 Genomes Project database as a control for somatic mutation analysis. Although this is a common strategy in genetic studies, it is theoretically less accurate than using patient-matched samples and may affect the precision of mutation screening. Consequently, the mutation frequencies reported here (e.g., in TTN, FLG) must be interpreted with caution. These findings are preliminary and hypothesis-generating, highlighting candidate genes that may contribute to NF2-SWN pathogenesis but require validation in a larger, independent cohort through future multi-center collaborations.
In conclusion, the specific genes discovered in this study provide potential avenues for further research. Genes such as TTN, CR2, FSIP2 and FLG may play a crucial role in specific aspects of disease progression, and it is worth exploring their specific functions and mechanisms in more detail.
Abbreviations
NF2: Neurofibromatosis type 2
NF2-SWN: NF2-related schwannomatosis
SNV: Single nucleotide variation
SNPs: Single nucleotide polymorphisms
INDELs: Insertions/deletions
WES: Whole-exome sequencing
ERM: Ezrin/Radixin/Moesin
Supplementary Material
Supplementary tables.
Acknowledgements
Thanks to the Neurosurgery Department of Xiangya Hospital for its support.
Financial support and sponsorship
This research was funded by one grant from the National Key R&D Program of China (No. 2022YFE0133400), three grants from National Natural Science Foundation of China (No. 82203397, No. 82303253 and No. 82172834), two grants from the Nature Science Foundation of Hunan Province (No. 2022JJ40814 and No. 2024JJ6686), one grant from the Nature Science Foundation of Changsha (No. kq2202375), one grant from the Youth Foundation of Xiangya Hospital (No. 2021Q06) and two grants from the China Postdoctoral Science Foundation (No. 2022M723560 and No. 2023M733960).
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Medical Ethics Committee of Xiangya Hospital of Central South University (Approval number: 202407141) and written informed consent was provided by all of the patients.
Data availability statement
All data is available from the corresponding author upon reasonable request. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA011281) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human[30, 31].
Author contributions
Conceptualization, Jun Tan, Changwu Wu and Qing Liu; Funding acquisition, Jun Tan, Changwu Wu and Qing Liu; Investigation, Liangqi Jiang and Fushu Luo; Methodology, Jun Tan, Changwu Wu and Haoyu Li; Supervision, Qing Liu; Writing - original draft, Fushu Luo and Liangqi Jiang; Writing - review & editing, Fushu Luo, Liangqi Jiang and Yimin Pan. Final approval of manuscript: All authors. Accountable for all aspects of work: All authors. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Plotkin SR, Messiaen L, Legius E, Pancza P, Avery RA, Blakeley JO. et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022;24:1967-77
2. Evans DG, Huson SM, Donnai D, Neary W, Blair V, Teare D. et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29:841-6
3. McClatchey AI. Neurofibromatosis. Annu Rev Pathol. 2007;2:191-216
4. Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z. et al. Neurofibromatosis type 2. Lancet. 2009;373:1974-86
5. Bachir S, Shah S, Shapiro S, Koehler A, Mahammedi A, Samy RN. et al. Neurofibromatosis Type 2 (NF2) and the Implications for Vestibular Schwannoma and Meningioma Pathogenesis. Int J Mol Sci. 2021 22
6. Evans DG, Bowers N, Huson SM, Wallace A. Mutation type and position varies between mosaic and inherited NF2 and correlates with disease severity. Clin Genet. 2013;83:594-5
7. Vlashi R, Sun F, Zheng C, Zhang X, Liu J, Chen G. The molecular biology of NF2/Merlin on tumorigenesis and development. Faseb j. 2024;38:e23809
8. Cooper J, Giancotti FG. Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett. 2014;588:2743-52
9. Forde C, Smith MJ, Burghel GJ, Bowers N, Roberts N, Lavin T. et al. NF2-related schwannomatosis and other schwannomatosis: an updated genetic and epidemiological study. J Med Genet. 2024;61:856-60
10. Vitte J, Gao F, Coppola G, Judkins AR, Giovannini M. Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nat Commun. 2017;8:300
11. Coy S, Rashid R, Stemmer-Rachamimov A, Santagata S. An update on the CNS manifestations of neurofibromatosis type 2. Acta Neuropathol. 2020;139:643-65
12. Woods R, Friedman JM, Evans DG, Baser ME, Joe H. Exploring the "two-hit hypothesis" in NF2: tests of two-hit and three-hit models of vestibular schwannoma development. Genet Epidemiol. 2003;24:265-72
13. Xue L, He W, Zhang Y, Wang Z, Chen H, Chen Z. et al. Origins of biallelic inactivation of NF2 in neurofibromatosis type 2. Neuro Oncol. 2022;24:903-13
14. Sleiman K, Allam S, Akiki D, Megarbane A, Bleik J. Neurofibromatosis type-2-related schwannomatosis presenting as peripapillary hamartoma: report on a novel NF2 mutation. Ophthalmic Genet. 2025;46:65-8
15. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO. et al. A global reference for human genetic variation. Nature. 2015;526:68-74
16. Wu C, Tan J, Wang X, Qin C, Long W, Pan Y. et al. Pan-cancer analyses reveal molecular and clinical characteristics of cuproptosis regulators. Imeta. 2023;2:e68
17. Peng H, Wang Y, Wang P, Huang C, Liu Z, Wu C. A Risk Model Developed Based on Homologous Recombination Deficiency Predicts Overall Survival in Patients With Lower Grade Glioma. Front Genet. 2022;13:919391
18. Gu Z. Complex heatmap visualization. Imeta. 2022;1:e43
19. Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA. et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell. 2022;185:3426-40.e19
20. Hardin HM, Dinh CT, Huegel J, Petrilli AM, Bracho O, Allaf AM. et al. Cotargeting Phosphoinositide 3-Kinase and Focal Adhesion Kinase Pathways Inhibits Proliferation of NF2 Schwannoma Cells. Mol Cancer Ther. 2023;22:1280-9
21. Blakeley JO, Plotkin SR. Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro Oncol. 2016;18:624-38
22. Vasudevan HN, Payne E, Delley CL, John Liu S, Mirchia K, Sale MJ. et al. Functional interactions between neurofibromatosis tumor suppressors underlie Schwann cell tumor de-differentiation and treatment resistance. Nat Commun. 2024;15:477
23. Evans DG, Kalamarides M, Hunter-Schaedle K, Blakeley J, Allen J, Babovic-Vuskanovic D. et al. Consensus recommendations to accelerate clinical trials for neurofibromatosis type 2. Clin Cancer Res. 2009;15:5032-9
24. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015; 519: 223-8.
25. Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L. et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758-67
26. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D. et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619-28
27. Fullenkamp DE. Regulation of TTN as a mechanism of and treatment for heart failure. J Clin Invest. 2025 135
28. Li H, Yu Y, Zheng D, Dong G, Lin S, Liu X. et al. Clinical and genomic profiles of malignant peripheral nerve sheath tumors in the craniospinal axis. J Neurooncol. 2025;173:59-70
29. Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. 2018;4:1
30. Chen T, Chen X, Zhang S, Zhu J, Tang B, Wang A. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics Bioinformatics. 2021;19:578-83
31. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022; 50: D27-d38.
Author contact
![]() Corresponding authors: Jun Tan, M.D. & Ph.D., Department of Neurosurgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008, P.R. China. Email: tanjunseaedu.cn. Changwu Wu, M.D. & Ph.D., Department of Neurosurgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008, P.R. China. Email: wuchangwuedu.cn. Qing Liu, M.D. & Ph.D., Department of Neurosurgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008, P.R. China. Email: liuqingdredu.cn.
Corresponding authors: Jun Tan, M.D. & Ph.D., Department of Neurosurgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008, P.R. China. Email: tanjunseaedu.cn. Changwu Wu, M.D. & Ph.D., Department of Neurosurgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008, P.R. China. Email: wuchangwuedu.cn. Qing Liu, M.D. & Ph.D., Department of Neurosurgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008, P.R. China. Email: liuqingdredu.cn.