Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Mechanisms by which Metabolic...
3. Interventions that Target...
4. Clinical Perspectives:...
5. Summary and Future Outlook
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(14):3543-3555. doi:10.7150/ijms.114382 This issue Cite
Review
Role of Metabolic Abnormalities During the Progression of Chronic Kidney Disease and Preventive Strategies
Dongqing Zha, Ping Gao ![]() , Xiaoyan Wu
, Xiaoyan Wu ![]()
Division of Nephrology, Zhongnan Hospital of Wuhan University, Wuhan 430070, China.
Received 2025-3-24; Accepted 2025-7-8; Published 2025-7-28
Abstract

Chronic kidney disease (CKD) is characterized by persistent renal impairment or dysfunction that lasts for at least 3 months, and typically has a progressive and irreversible trajectory. The increasing prevalence of metabolic disorders, such as hyperuricemia, dyslipidemia, obesity, and type 2 diabetes mellitus, have contributed to the increasing incidence of CKD, and it is now a significant public health concern worldwide. Accumulating evidence underscores the intricate relationships of the different metabolic disorders and how they promote the initiation and progression of CKD, and ultimately lead to end-stage renal disease (ESRD). Metabolic abnormalities promote CKD progression by various mechanisms, including oxidative stress, chronic inflammation, dysregulation of autophagy, glomerular hyperfiltration and disruption of hemodynamics, endothelial dysfunction, and dysbiosis of gut microbiota. Ectopic lipid deposition and lipid peroxidation-induced redox imbalance lead to mitochondrial dysfunction, excessive production of reactive oxygen species (ROS), and activation of the p38 MAPK, ERK, and JNK signaling pathways. Metabolic dysregulation activates NF-κB signaling pathways and NLRP3 inflammasomes, leading to increased production of pro-inflammatory factors, lysosomal dysfunction, and impaired autophagic clearance, followed by accumulation of metabolic waste and podocyte injury. Obesity and hyperlipidemia can cause excessive activation of the renin-angiotensin-aldosterone system (RAAS), which then causes glomerular hyperfiltration, endothelial and mesangial cell injury and proliferation, and ultimately glomerulosclerosis. Multiple interventions that target these mechanisms have shown therapeutic potential, and these include pharmacological treatments (xanthine oxidase inhibitors to reduce uric acid levels, statins for lipid regulation, and SGLT2 inhibitors and GLP-1 receptor agonists to improve renal and cardiovascular outcomes), lifestyle interventions (low-salt and low-protein diets, weight management, smoking cessation, and alcohol limitation), intermittent fasting, and microbiome-targeted therapies. This review analyzes the pathways by which metabolic abnormalities affect the onset and progression of CKD, identifies strategies that have potential use for prevention or treatment, and offers a robust theoretical foundation for the future development of effective clinical interventions.
Keywords: metabolic abnormalities, chronic kidney disease, inflammation, oxidative stress, autophagy
1. Introduction
Chronic kidney disease (CKD) is defined by impaired renal function that is sustained for 3 months or longer, irrespective of a decreased glomerular filtration rate (GFR), and by distinctive structural or functional abnormalities of the kidneys that are evident on ultrasound. The global prevalence of CKD is greater than 10%, and recent studies projected that CKD will be the fifth leading cause of mortality worldwide by 2040 [1-3]. Metabolic dysregulation, diabetes mellitus, arterial hypertension, and cellular senescence are the major risk factors for CKD [4-6]. However, due to the asymptomatic nature of early-stage CKD, many patients receive delayed diagnosis and treatment, and experience accelerated disease progression. As a result, many patients have advanced-stage CKD at the time of diagnosis, leading to significant healthcare costs and a substantial burden for patients, families, and healthcare systems [7].
The specific types of metabolic dysregulation that lead to CKD include arterial hypertension, hyperuricemia, obesity, insulin resistance, and disruptions of carbohydrate and lipid metabolism (Table 1). All of these conditions are affected by the complex interplay among gut microbiota, genetic factors, and nutrition [8, 9]. Hypertensive patients frequently exhibit endothelial dysfunction, and chronic endothelial damage can exacerbate glomerulosclerosis and accelerate the decline of renal function and the pathogenesis of CKD [8]. Recent researchers have investigated genetic factors related to CKD susceptibility using biobank datasets and clinical repositories, with a focus on the role of hyperuricemia. The findings underscore hyperuricemia as a critical risk factor for the development of CKD [10]. A growing body of evidence shows that obesity can drive the progression of CKD and that the pathophysiological mechanisms are multifaceted, including altered hemodynamics, systemic inflammation, oxidative stress, and activation of the renin-angiotensin-aldosterone system (RAAS). Interventions, such as weight reduction and RAAS inhibitors, therefore have important nephroprotective effects [11]. Furthermore, studies of rat models of insulin resistance demonstrated that untreated rats experienced significant declines in renal function, along with increased oxidative stress, chronic inflammation, apoptosis, profibrotic remodeling, aggravated renal histopathological damage, and excessive collagen deposition. However, treatment of insulin-resistant rats with vildagliptin (an anti-hyperglycemic drug) led to substantial improvements in renal function, thus emphasizing the critical link between insulin resistance and the pathogenesis of CKD [12]. Dysregulation of lipid metabolism also induces renal tubular epithelial cell injury, and this can trigger the release of pro-inflammatory cytokines and the onset of CKD [13].
Impact of diseases that cause metabolic abnormalities in CKD.
| Disease | Influence on CKD | References |
|---|---|---|
| Hypertension | Damages glomerular endothelial cells and mesangial cells, leading to filtration disorders and accelerating the progression of CKD | Ambroselli et al. (8) Frąk et al. (16) Jia et al. (17) |
| Hyperuricemia | Induces inflammatory responses, intensifies oxidative stress, and promotes glomerular sclerosis and fibrosis | Gherghina et al. (19) |
| Obesity | Triggers an inflammatory response and accelerates the progression of CKD | Czaja-Stolc et al. (22) Aamir et al. (23) |
| Insulin resistance | Leads to oxidative stress, inflammation, and apoptosis, accelerating the progression of CKD | Wahba et al. (12) |
| Lipid metabolism disorder | Injury of renal tubular epithelial cells accelerates the progression of CKD | Chen et al. (13) |
Given the complex and interdependent relationship between metabolic dysregulation and CKD, prompt treatment of metabolic abnormalities is essential for slowing the progression of CKD, improving patient outcomes, and reducing healthcare expenditures. Diseases related to metabolic abnormalities can accelerate the progression of CKD by activating multiple pathways, such as those that function in inflammation, oxidative stress, inhibition of autophagy, and accumulation of metabolic waste.
2. Mechanisms by which Metabolic Abnormalities Promote CKD
2.1 Oxidative stress
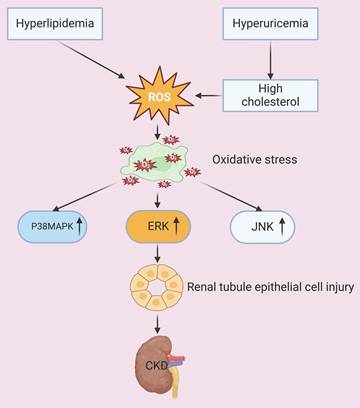
The ectopic deposition of excessive lipids in visceral tissues and organs, coupled with a redox imbalance caused by lipid peroxidation, culminates in mitochondrial dysfunction. The resultant overproduction of reactive oxygen species (ROS) induces oxidative damage, which then activates different apoptosis pathways. The peroxidized lipids also disrupt cell membrane integrity and function, and the subsequent oxidative stress causes further damage of membrane lipids, nucleic acids, and proteins, culminating in impaired cellular and organ-level homeostasis [14, 15]. Research has also demonstrated that oxidative stress increases the apoptosis of renal tubular epithelial cells by activation of different intracellular signaling cascades, including the p38 mitogen-activated protein kinase (p38 MAPK), extracellular regulated protein kinase (ERK), and c-Jun N-terminal kinase (JNK) pathways. Oxidative stress also exacerbates glomerular hypertension, resulting in dysfunctional endothelial and mesangial cells, disruption of the glomerular filtration barrier, and accelerated progression of CKD [16, 17]. An in vitro study of human endothelial cells demonstrated that the cytotoxic effects of uric acid can be attributed to an increased production of ROS and angiotensin II [18]. The occurrence of this pathological process in vivo is associated with increased blood pressure, which promotes the onset and progression of CKD; the co-occurrence of hyperuricemia can also lead to the deposition of monosodium urate crystals within renal tissues, which triggers inflammatory responses, exacerbates oxidative stress, and promotes glomerulosclerosis and fibrosis [19]. Additional evidence showed that an elevated level of cholesterol and hyperlipidemia lead to lipid accumulation in renal tubular cells, and this increases the generation of ROS, apoptosis, and inflammatory responses. All of these processes contribute to structural and functional damage of the renal tubules and glomeruli, and also promote the progression of CKD [20, 21]. Thus, abundant evidence indicates that certain metabolic derangements, especially hyperuricemia and hyperlipidemia, exacerbate oxidative stress and can lead to renal injury and dysfunction (Figure 1).
Hyperlipidemia and hyperuricemia increase the production of ROS and oxidative stress. Oxidative stress activates the P38MAPK, ERK, and JNK signaling pathways, which then leads to injury of tubular epithelial cells and accelerated progression of CKD.
2.2 Chronic inflammation
The proinflammatory alterations that occur during metabolic dysregulation contribute to the progression of CKD via multiple mechanisms. For example, individuals with obesity or excessive adiposity have an increased risk for upregulation of inflammatory cascades. Clinical investigations have demonstrated that inflammatory mediators, including C-reactive protein (CRP), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6), play pivotal roles in impairing the renal function of patients with metabolic dysfunction. Renal injury also disrupts the production and secretion of adipokines, thereby promoting the pathogenesis and progression of CKD [22, 23]. Proteins in the NF-κB family are pivotal pro-inflammatory transcription factors, and their upregulation leads to mitochondrial dysfunction. The resultant excessive production of ROS activates NLRP3 inflammasomes and other inflammasomes, thereby inducing the production of interleukin-1β (IL-1β). IL-1β then binds to its receptor, and further stimulates the NF-κB signaling pathway, creating a self-amplifying inflammatory feedback loop. This inflammatory cascade also activates leukocytes and resident cells, thus driving the additional production of ROS and reactive nitrogen species. The resulting oxidative stress induces apoptosis, necrosis, and fibrosis, leading to inflamed and dysfunctional kidneys (Figure 2) [24, 25].
Metabolic abnormalities increase the level of TNF-α and also damage mitochondria and endothelial cells. Mitochondrial dysfunction leads to the production of ROS, activation of NLRP3 inflammasomes, increased expression of IL-1β, and then activation of the NF-κB pathway. These mitochondria-mediated responses, concurrent with the increased levels of TNF-α and MCP-1, lead to injury of tubular epithelial cells and accelerated progression of CKD.
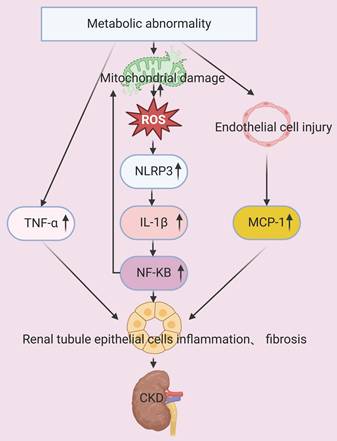
Thus, chronic inflammation directly impairs renal tissues and contributes to tubular damage, interstitial fibrosis, and impaired renal function. There is clinical evidence that an elevated serum uric acid level has cytotoxic effects on vascular endothelial cells, and the manifestations of endothelial dysfunction include renal vasoconstriction and ischemia-related exacerbations of renal injury. The dysfunction of endothelial cells also activates inflammatory mediators, such as monocyte chemoattractant protein-1 (MCP-1), and this intensifies renal inflammation and promotes renal fibrosis in patients with diabetic nephropathy [26]. A study of OLETF (hyperglycemic) rats demonstrated that a high-sugar diet increased renal expression of IL-1β and macrophage infiltration, and that decreasing the serum uric acid level ameliorated albuminuria, tubular injury, macrophage infiltration, and renal secretion of IL-1β. Hyperuricemia in these rats also activated NLRP3 inflammasomes within macrophages, increased chemokine signaling in proximal tubular cells, and promoted diabetic nephropathy due to the interactions of macrophages and proximal tubular cells [27]. The chronic inflammatory responses triggered by metabolic dysregulation are therefore critical to the pathogenesis and progression of CKD (Figure 2).
2.3 Dysregulation of autophagy
Metabolic abnormalities that increase inflammation and oxidative stress also increase the proliferation of glomerular cells, necrosis, glomerulosclerosis, renal interstitial fibrosis, and albuminuria [28]. Autophagy is a critical cellular process that facilitates the recycling and removal of damaged proteins, organelles, and pathogens within phagocytic cells. During this process, autophagosomes first encapsulate damaged cell products within their double-membranes; the autophagosomes then fuse with lysosomes, and the resulting phagolysosomes perform the degradation [29]. AMP-activated protein kinase (AMPK) is a highly conserved sensor of energy and nutrients in eukaryotic cells that regulates cellular energy and metabolism, and thereby affects diverse biochemical and physiological processes [30]. AMPK is also a critical sensor of mitochondrial dynamics, and transmits signals to lysosomes that modulate autophagy. Dual-specificity phosphatase 5 (DUSP5), a protein in the serine-threonine phosphatase family, dephosphorylates ERK and is a pivotal regulator of metabolic signaling, inflammatory responses, and the progression of cancer. Recent studies demonstrated that DUSP5 knockdown reduced the production of inflammatory mediators in renal tubular epithelial cells and also attenuated apoptosis by enhancing AMPK/ULK1-mediated autophagy, leading to improved renal function [31, 32].
These findings demonstrate that an appropriate level of autophagy can prevent renal inflammatory damage, including acute, chronic, metabolic, and age-related insults [33]. However, the abnormal accumulation of metabolites that results from metabolic dysregulation leads to lysosome dysfunction in autophagy-associated effector organelles, and this can cause or aggravate diabetes and its complications [34]. For example, Yasuda-Yamahara et al. demonstrated that dysregulation of podocyte autophagy contributed to the pathogenesis of diabetic nephropathy. Diabetic patients with severe proteinuria have insufficient podocyte autophagy and significantly decreased podocyte function; similarly, diabetic mice with deficient podocyte autophagy due to a high-fat diet develop extensive proteinuria and marked loss of functional podocytes. The dysregulation of podocyte autophagy therefore plays an important role in the progression of CKD [35].
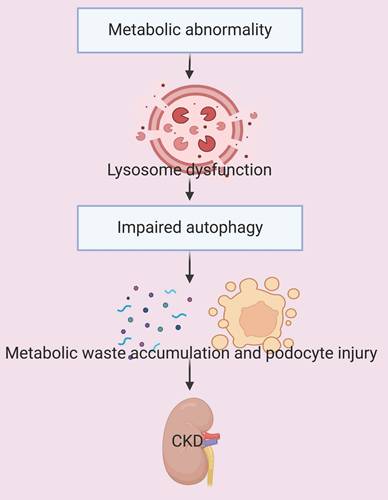
There is also evidence that the dysregulation of autophagy due to lysosome dysfunction and impaired autophagic clearance is a hallmark of the accumulation of white adipose tissues in obese mice [36]. One of the key mechanisms by which hyperuricemia induces renal tubular injury is by disruption of Na-K-ATPase (NKA) signaling. This disruption triggers inflammation, dysregulation of autophagy, dysfunction of mitochondria, and ultimately renal damage. Notably, activation of autophagy decreases the degradation of NKA in lysosomes, and this decreases inflammation and protects renal tubular cells from hyperuricemia-induced injury [37]. In summary, metabolic abnormalities can lead to dysfunctional autophagy and accelerate the progression of CKD (Figure 3). This suggests that interventions which restore the normal level of autophagy have potential as therapeutic strategies for treatment of CKD.
Metabolic abnormalities lead to lysosome dysfunction and impaired autophagy. The disruption of autophagy leads to the accumulation of metabolic waste, causing podocyte injury and accelerated progression of CKD.
2.4 Glomerular hyperfiltration and disruption of hemodynamics
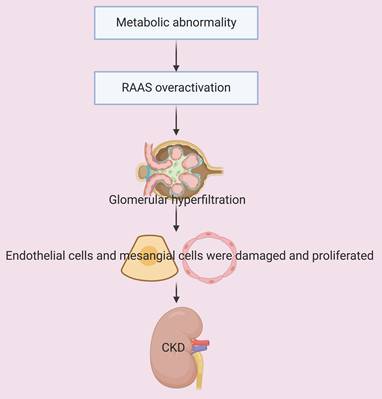
Obesity and hyperlipidemia are frequently associated with overactivation of the RAAS, and this leads to increased blood pressure in the glomerular afferent arterioles and renal hyperfiltration. Obesity-induced insulin resistance increases insulin-like growth factor-1 (IGF-1) activity, and also increases glomerular blood flow and hyperfiltration [38, 39]. Hyperlipidemia and obesity trigger inflammation, a decreased production of nitric oxide (NO), and an increased level of endothelin, culminating in glomerular vasoconstriction, increased glomerular filtration, and renal dysfunction [40, 41]. These responses lead to increased glomerular blood pressure, mechanical stress on the glomerular capillaries, injury and proliferation of endothelial and mesangial cells, and then glomerulosclerosis and the loss of functional nephron units [42]. In addition, hyperfiltration leads to excessive excretion of proteins through the glomeruli and proteinuria, and the excess secretion of these proteins has cytotoxic effects, in that it activates inflammatory responses in tubular epithelial cells and induces oxidative stress. This cascade of responses to hyperfiltration increases the production of pro-inflammatory cytokines (TNF-α and IL-6) and free radicals, and decreases the structural and functional integrity of tubular and glomerular cells, thereby accelerating the decline in renal function [43, 44]. In summary, metabolic dysregulation injures tubular and glomerular cells and the resulting hyperfiltration and hemodynamic alterations accelerate the progression of CKD (Figure 4).
Metabolic abnormalities lead to overactivation of the RAAS. This is followed by glomerular hyperfiltration, damage and proliferation of endothelial and mesangial cells, and accelerated progression of CKD.
2.5 Endothelial dysfunction
Endothelial dysfunction, which is characterized by impaired vasodilation, inflammation, and thrombosis, can trigger cardiovascular diseases and is associated with a decline in the estimated glomerular filtration rate [45], albuminuria, edema, and coagulopathy [46]. The intricate relationships among three factors—renal dysfunction, an altered metabolic profile, and adverse cardiovascular outcomes—are a critical area of concern. Mounting evidence underscores endothelial dysfunction and chronic inflammation as driving forces of this detrimental triad. For instance, recent work by Prabhahar et al. [47] and Borri et al. [48] elucidated the specific pathways of endothelial dysfunction in recipients of kidney transplants and aging kidneys, highlighting its profound impact on renal health and systemic vascular integrity. Moreover, the pathophysiology of vascular aging, particularly in the presence of CKD and diabetes, points to shared mechanisms, suggesting that novel cardio-renal protective medications may offer therapeutic benefits by targeting these common pathways [49]. A study by Liu et al. [50] also contributed to this understanding by detailing the role of metabolic factors, such as uric acid, in promoting atherosclerosis in patients with CKD, a process intrinsically linked with endothelial damage and inflammation. Collectively, these recent findings reinforce the necessity of considering several interconnected pathologies and underscore the importance of incorporating the latest research to fully appreciate that therapeutic targeting of endothelial dysfunction and inflammation can improve cardiovascular outcomes in patients with renal and metabolic disturbances.
2.6 Secondary effects of metabolic abnormalities
Metabolic abnormalities, such as hyperglycemia and hyperlipidemia, increase the oxidative stress of renal cells and lead to the dysfunction of mitochondria. The resulting excessive generation of ROS has numerous secondary effects, such as damage of mitochondrial membranes, proteins, and DNA (Figure 2). Impaired mitochondrial function decreases the production of ATP, so that less energy is available to support the basic functions of renal tubular and glomerular cells, leading to the dysfunction of these cells [51]. Metabolic abnormalities can also suppress activation of AMPK signaling in renal cells, and this pathway plays a pivotal role in regulating glycolysis and fatty acid oxidation (Figure 5). This dysmetabolism of renal lipids leads to lipotoxicity within renal cells and exacerbates cellular injury [52].
Metabolic abnormalities decrease the level of AMPK, lead to overactivation of SREB-1, and increase the level of AGEs. Suppression of AMPK and overactivation of SREB-1 decreases fatty acid oxidation and increases lipid synthesis, leading to lipotoxicity and injury of tubular epithelial cells. Concurrently, AGEs bind with their receptor (RAGE), and this activates the MAPK and NF-κB pathways. The combined effects of these changes lead to accelerated progression of CKD.
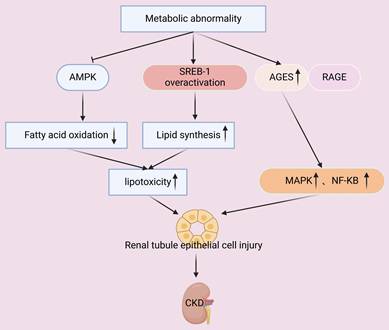
Other studies showed that hyperglycemia leads to the accumulation of advanced glycation end-products (AGEs), and that the binding of these molecules to their receptors (RAGEs) activates the MAPK and NF-κB pathways and increases inflammation and oxidative stress (Figure 5). In particular, the cytotoxic effects of AGEs are attributed to their inhibition of glycolysis and mitochondrial oxidative phosphorylation. AGEs also directly damage cellular membranes and disrupt protein structure, and research indicated that this can damage mouse mesangial cells and accelerate the progression of kidney disease in patients with diabetes [53, 54].
Another secondary effect of metabolic abnormalities is that they can stimulate innate renal cells (mesangial and endothelial cells) to upregulate genes that function in lipid synthesis and downregulate genes that function in lipid catabolism, leading to abnormal lipid accumulation and cellular dysfunction [55]. In particular, sterol regulatory element-binding protein 1 (SREBP-1) is a critical transcription factor that regulates the synthesis of fatty acids and cholesterol. Metabolic dysregulation leads to over-activation of SREBP-1, and the consequences are increased lipid synthesis and decreased lipid clearance in renal cells, manifesting as lipotoxicity (Figure 5). This disrupts cellular membrane integrity and triggers apoptosis and inflammation [52, 56]. Disruption of glomerular lipid metabolism also compromises the integrity of the glomerular filtration barrier, leading to proteinuria and impaired renal function. Lipid accumulation within the glomerulus promotes the deposition of extracellular matrix (ECM) and glomerulosclerosis, contributing to the progression of CKD [57, 58]. Therefore, further elucidation of the molecular mechanisms responsible for metabolic dysregulation and the abnormal metabolism of renal lipids is essential for developing targeted metabolic therapies for CKD.
2.7 Dysbiosis of gut microbiota
Many recent studies have examined the potential impact of gut microbiota on lipid homeostasis of the host and the pathogenesis of CKD. Gut microbiota metabolize a broad spectrum of bioactive lipid metabolites, including short-chain fatty acids (SCFAs) and secondary bile acids, that modulate lipid metabolism and immune responses in the host, and these can affect the progression of CKD [59]. Intestinal microbiota ferment dietary fiber and produce SCFAs, such as acetate, propionate, and butyrate. These metabolites are an energy source for intestinal epithelial cells and can also regulate lipid metabolism and inflammatory pathways by activation of G-protein-coupled receptors (GPR41 and GPR43) and inhibition of histone deacetylases [60-63]. SCFAs also lower the serum levels of cholesterol and lipids, and this could potentially slow the progression of CKD [64, 65]. Furthermore, gut microbiota transforms primary bile acids into secondary bile acids. This suppresses hepatic synthesis of cholesterol and increases cholesterol metabolism and excretion via pathways regulated by the farnesoid X receptor (FXR) and the G protein-coupled bile acid receptor 1 (TGR5) [66]. Notably, the activation of TGR5 decreases oxidative stress, modulates lipid metabolism, and protects renal function [67]. In addition, CKD patients often present with an imbalanced intestinal microflora, i.e., with fewer beneficial bacteria (such as Bifidobacterium and Lactobacillus) and more harmful bacteria (such as toxin-producing Clostridium), and this imbalance can lead to the accumulation of harmful metabolites (such as urinary toxins) that aggravate kidney damage and systemic inflammation [65, 68]. Gut dysbiosis can increase intestinal permeability, leading to the translocation of lipopolysaccharides and other bacterial components into the bloodstream, which then trigger a chronic low-grade inflammatory state and contribute to endothelial dysfunction [69-71]. Therefore, restoration of the gut microbiota and normalization of lipid metabolism is a potential strategy for slowing the progression of CKD and increasing immune function.
2.8 Cross-talk among pathways
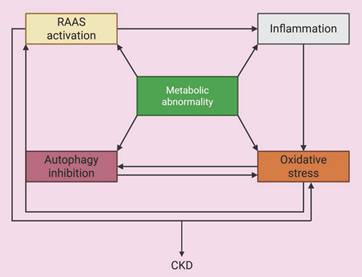
The above pathologic processes do not exist in isolation, but are intricately interrelated, forming a complex network of interactions that together drive the development of CKD (Figure 6). For example, chronic inflammation is a potent trigger of oxidative stress because inflammatory cells release ROS and the excessive production of ROS activates NLRP3 inflammasomes and other inflammasomes, leading to a self-amplifying inflammatory feedback loop. Both inflammation and oxidative stress severely impair the autophagic process, and the inefficient clearance of damaged organelles and protein aggregates exacerbates cellular stress, inflammation, and renal fibrosis [72]. Moreover, stimulation of mitophagy by the mTOR/PINK1/Parkin pathway ameliorates renal inflammation [73]. In addition, RAAS dysregulation, which is commonly associated with glomerular hyperfiltration and hemodynamic disturbances, can drive inflammation and oxidative stress within the kidney [43, 44]. Furthermore, endothelial dysfunction can be considered a consequence and a trigger of oxidative stress and the inflammatory milieu, and it further compromises vascular health and renal perfusion. Finally, the secondary effects of metabolic abnormalities, such as lipotoxicity and glucotoxicity, can each trigger or exacerbate inflammation, oxidative stress, and autophagy dysfunction. These interactions create a vicious cycle, in which each pathological pathway triggers or exacerbates the others, leading to persistent renal injury, fibrosis, and the progressive decline in renal function, culminating in CKD.
Interactions among pathogenic mechanisms related to metabolic abnormalities. Metabolic abnormalities cause oxidative stress, inflammation, inhibition of autophagy and activation of RAAS, and the interaction of these processes jointly increase the progression of CKD.
Gender differences also have critical roles in CKD, in that they can affect patient susceptibility, disease progression, and patient prognosis [74]. Studies have consistently shown that males develop ESRD more frequently and rapidly than females, and that this difference is partly attributable to differences in sex hormones [75]. For example, estrogen can affect cell proliferation, programmed cell death, immune responses, and metabolic regulation [74]. An animal study by Ren et al. showed that supplementation with 17β-estradiol (E2), one of the main circulating estrogens in females, protected the kidneys from damage by decreasing inflammation and collagen synthesis [76]. Therefore, possible mechanisms responsible for gender differences in the progression of CKD could be differences in the direct effects of sex steroids on the kidneys, differences in nitric oxide metabolism and levels of oxidative stress, and differences in comorbidities and lifestyle-related risk factors [77]. Therefore, attention should be paid to gender differences and the above mechanisms.
3. Interventions that Target Metabolic Abnormalities
3.1 Drugs
A dysregulated metabolism can lead to many adverse effects, and CKD is one of the most consequential because it has a profound impact on physical and psychological well-being. Pharmacological interventions are one of the most common and efficacious approaches for mitigating and managing CKD. For example, clinical investigations have demonstrated that xanthine oxidase inhibitors (allopurinol and febuxostat) lower the serum level of urate, attenuate the progression of renal dysfunction, and reduce the incidence of cardiovascular complications [78-80]. Additionally, an animal study showed that febuxostat ameliorated renal interstitial fibrosis by increasing the signaling from bone morphogenetic protein 7 (BMP-7), which inhibits transforming growth factor β (TGF-β) signaling and expression of uterine sensitization-associated gene-1 (USAG-1) [81]. Clinical evidence indicates that statins are efficacious and well-tolerated when used to lower the level of serum lipids, and that they can decrease the risk of end-stage CKD and cardiovascular complications following renal transplantation [82]. For example, lovastatin slowed the progression of CKD by attenuating oxidative stress, modulating the TGF-β1/Smad signaling, and ameliorating the glomerular endothelial-to-mesenchymal transition in rats with diabetic nephropathy [83].
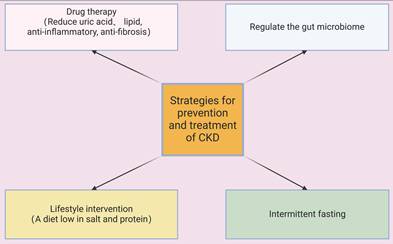
Alirocumab and evolocumab are monoclonal antibodies that target hepatic proprotein convertase subtilisin/kexin type 9 (PCSK9), decrease the circulating levels of atherogenic low-density lipoprotein cholesterol (LDL-C), alleviate endothelial inflammation and atherogenesis, and slow the progression of CKD [84]. TGF-β increases fibroblast activation and the accumulation of ECM by activating canonical Smad-dependent signaling and non-canonical pathways, including the mitogen-activated protein kinase (MAPK) and PI3K-Akt cascades, and this accelerates renal fibrosis in CKD [85]. TGF-β antagonists (although not yet clinically approved) can slow the progression of CKD by neutralizing TGF-β ligands, inhibiting receptor kinase activity, and disrupting downstream signaling in animal models and patients [86]. TGF-β inhibitors also suppress fibroblast transdifferentiation, slow the deposition of ECM, decrease inflammatory responses, enhance tubular function, and reduce proteinuria and glomerulosclerosis [87, 88]. Thus, many studies indicated that different therapeutic interventions which decrease the levels of urate and lipids and increase anti-inflammatory and antifibrotic pathways have potential as strategies for the prevention and management of CKD (Figure 7).
Multiple interventions have the potential to prevent, treat, or slow the progression of CKD. These include medications that lower the levels of uric acid and lipids, and those that have anti-inflammatory and anti-fibrotic effects; lifestyle modifications, such a low-salt and low-protein diet; intermittent fasting; and restoration of the balance of the gut microbiome.
Metabolic modulation, especially for targeting of uremic toxins, is an important approach that can address metabolic abnormalities and renal dysfunction at the same time (Table 2). Sodium-glucose cotransporter 2 (SGLT2) inhibitors and glucagon-like peptide-1 receptor agonists (GLP-1RAs) are two large classes of drugs that can markedly improve renal and cardiovascular outcomes in patients with diabetes and CKD [89]. A study of dapagliflozin (an SGLT2 inhibitor) examined its effect on hyperuricemic nephropathy (HN) by examining human biopsy samples, mice with HN, and human proximal tubule cells. The results showed that this drug targeted estrogen-related receptor α (ERRα), thereby activating the ERRα-organic anion transporter 1 (OAT1) axis, promoting the excretion of urate, and inhibiting renal interstitial fibrosis [90]. Treatment with tirzepatide, a dual agonist of the GLP-1 receptor and Glucose-dependent insulinotropic polypeptide (GIP) receptor [91], induced significant reduction in body weight in multiple trials, and did not increase the risks of adverse renal events, nephrolithiasis, and acute kidney injury, when compared to a placebo and insulin [92]. In addition, tirzepatide is a promising therapeutic option for the treatment of heart failure that provides significant metabolic and cardiovascular benefits [93], including a reduced risk of death from cardiovascular diseases and deterioration of heart failure [94].
New therapies for slowing the progression of CKD.
| Metabolism-related Therapy | Influence on CKD | Mechanism | Reference |
|---|---|---|---|
| Xanthine oxidase inhibitors | Reduce uric acid level and slow the progression of CKD | Specifically inhibits xanthine oxidase, thereby reducing the production of uric acid | Siu et al. (53) Goicoechea et al. (54) Kohagura et al. (55) |
| SGLT2/GLP1 dual therapy | Promote the excretion of urate and delay the progression of CKD | Activates the ERRα-OAT1 axis | Alicic et al. (60) Hu et al. (61) |
| Microbiome transplantation | Regulates lipid metabolism and immune response, and slows the progression of CKD | Activates protein-coupled receptors and inhibits histone deacetylases | Zhang et al. (77) Wang et al. (78) Seljeset et al. (80) Andrade et al. (81) |
Mitochondria-targeted antioxidants, including mitoQ, mitoTEMPO, mitoE, mitoCP, SkQ1, SkQR1, and SS-31, can directly eliminate mitochondrial ROS, alleviate the damage of oxidative stress to DNA, lipids and proteins, and protect the functions of renal tubular epithelial cells and podocytes [95]. MitoQ conjugates triphenyl alkyl phosphonium cation with coenzyme-Q, and its administration to mice with diabetes dramatically reversed renal tubular injury, downregulated the oxidative stress in tubular cells, and had a protective effect on podocytes by maintaining mitochondrial fitness [96, 97]. The results of a randomized controlled trial showed that a 4-week MitoQ supplement was well tolerated in patients with stage 3-4 CKD, and it also improved large vessel endothelial function, arterial hemodynamics, and microvascular function [98]. MitoTEMPO significantly improved renal function and decreased podocyte damage in a rat model of CKD through the inhibition of NLRP3 inflammasomes via PINK1/Parkin pathway-mediated mitochondrial autophagy [99]. SS-31 promotes oxidative phosphorylation, alleviates mitochondrial oxidative stress, and prevents damage to renal tubules and podocytes [100, 101].
3.2 Lifestyle interventions
Although pharmacological treatments can be used to treat or prevent CKD, a multifaceted approach that integrates these treatments with lifestyle modifications can provide even more benefit. Firstly, it is important for susceptible or affected patients to adopt a low-sodium and low-protein diet to alleviate the hemodynamic load on the glomeruli [102]. The consumption of potassium- and magnesium-enriched foods should be adjusted according to a patient's residual renal function, and phosphorus intake should be restricted to prevent hyperphosphatemia and secondary hyperparathyroidism [103]. Secondly, weight management and structured exercise regimens can also help to slow the progression of CKD. In particular, obesity is a well-established risk factor for CKD, and moderate-intensity aerobic exercise can increase insulin sensitivity, decrease blood pressure and systemic inflammation, and improve cardiorespiratory fitness and overall quality-of-life [104, 105]. Thirdly, smoking cessation and avoidance of excessive alcohol consumption are especially important for patients with CKD. Tobacco use exacerbates glomerular sclerosis and microvascular injury, and excessive alcohol consumption increases metabolic stress; discontinuing smoking and limiting alcohol intake significantly lowers the risk of cardiovascular complications [106]. In summary, adopting a healthy lifestyle is an effective complementary strategy that can help slow CKD progression and improve overall well-being (Figure 7).
3.3 Special types of lifestyle interventions
There has been recent interest in the use of intermittent fasting (IF) to modulate diverse metabolic and cellular signaling pathways, including pathways that promote renal dysfunction. IF is associated with certain benefits and risks. For example, it can improve metabolic homeostasis by increasing insulin sensitivity and lipid catabolism, and decreasing chronic inflammation, processes intricately linked to the activation of AMPK and inhibition of the mammalian target of rapamycin (mTOR) [107, 108]. Moreover, IF decreases oxidative stress and inflammation in the kidneys by upregulating the nuclear factor erythroid 2-related factor 2 (Nrf2)/Kelch-like ECH-associated protein 1 (Keap1) antioxidant axis and downregulating the nuclear factor-κB (NF-κB) inflammatory pathway. Animal studies also showed that IF modulated the sirtuin 1 (SIRT1) pathway, leading to improved mitochondrial bioenergetics and increased cellular resilience to stress [109, 110].
IF appears to provide nephroprotective effects in CKD due to its induction of autophagy, and this is primarily due to its inhibition of mTOR and promotion of LC3-II (the membrane-associated form of LC3), effects that attenuate glomerulosclerosis and interstitial fibrosis [111]. Additionally, IF alleviates inflammatory and oxidative stress by suppressing the formation of AGEs and downregulating RAGE, and this decreases renal tubular injury and interstitial fibrosis [112]. Collectively, the multifaceted effects of IF on energy metabolism and autophagy, as well as its anti-inflammatory and antioxidant effects, suggest it has promise as an intervention for management of CKD. However, many studies of IF have focused on animal models, so its effects and precise molecular mechanisms on CKD in humans require further exploration.
3.4 Microbiome-targeted therapies
Microbiome-targeted therapies are those that treat diseases by altering the composition and function of the microbiome [113]. There are several types of microbiome-targeted therapies that directly affectthe microbiome, such as antibiotics, probiotics, prebiotics, synbiotics, oral absorbents, and fecal microbial transplantation [68], and others that have indirect effects such as enzyme inhibitors, receptor agonists, and antagonists [114]. Microbiome-targeted therapies have shown potential therapeutic value in a variety of diseases, including intestinal diseases, metabolic diseases, and kidney disease [115].
Probiotics, prebiotics, and synbiotics modulate the gut microbiome by focusing on increasing glycolytic bacteria rather than proteolytic bacteria [116]. Briefly, probiotics are living microorganisms that [117], when consumed in moderation, can provide health benefits to their hosts. Typical strains include Lactobacillus and Bifidobacterium, both of which support gut health and alleviate dysbiosis in patients with CKD [118]. The use of probiotics to treat patients with CKD may be accompanied by cardiovascular benefits, an important consideration because patients with CKD often have concurrent cardiovascular problems [119]. Prebiotics are primarily non-digestible components that can have a beneficial effect on the health of the host by selectively stimulating the growth or activity of certain genera of microorganisms in the colon. Fermentation of prebiotics, such as inulin, fructooligosaccharides, resistant starch, and galactooligosaccharides, in the gut produces SCFAs, which can regulate lipid metabolism and inflammatory pathways and potentially slow the progression of CKD [120]. Synbiotics are made with various formulations of probiotics and prebiotics that work synergistically to restore intestinal ecology [68]. Fecal microbial transplantation involves the transfer of feces from a healthy donor to a recipient [121]. To normalize the integrity of the intestinal barrier and reduce the levels of pro-inflammatory metabolites, thereby reducing kidney injury and inflammation [122]. However, fecal microbial transplantation entails potential risks, such as transmission of an infection and unforeseen immune responses, and future studies are needed to evaluate its safety.
Future studies should also examine the effect of altering the intestinal microbiota on lipid metabolism as a strategy for the prevention or treatment of CKD [123].
4. Clinical Perspectives: Integrating Research into Practice
Nephrologists have a clear road map for implementing targeted therapies that can significantly improve the outcomes of patients with CKD, and this review provided insights into the mechanisms of these different therapies. However, instead of treating specific risk factors separately, a holistic clinical strategy is required due to interactions among different metabolic abnormalities, which include oxidative stress, chronic inflammation, dysregulation of autophagy, and hemodynamic alterations.
4.1 Early detection and risk stratification
Nephrologists should prioritize routine screening for metabolic markers including uric acid level, lipid profile, and inflammatory biomarkers (CRP, IL-6, TNF-α) in patients with early-stage CKD or those at risk of CKD. This proactive approach can enable identification of patients most likely to experience rapid disease progression and enable timely interventions to improve therapeutic efficacy.
4.2 Personalized therapeutic strategies
Significant evidence supports the use of xanthine oxidase inhibitors (allopurinol, febuxostat) for management of hyperuricemia, SGLT2 inhibitors for control of glucose, and statins for dyslipidemia, and nephrologists therefore have specific pharmacological tools that target the metabolic pathways which drive CKD progression. Rather than treating these conditions separately, an integrated approach that addresses multiple metabolic abnormalities simultaneously is likely to provide the most benefit. The demonstrated benefits of dietary modifications, structured exercise programs, and emerging strategies, such as intermittent fasting, offer nephrologists evidence-based lifestyle interventions to recommend alongside pharmacotherapy. The emerging knowledge of the role of gut microbiota in CKD progression also presents nephrologists with novel therapeutic opportunities. Recommendations for dietary fiber supplementation to promote the beneficial production of SCFAs and administration of probiotics to restore the microbial balance of the gut are two practical applications of this research that can be implemented in clinical practice.
By systematically addressing these interconnected metabolic pathways, nephrologists can move beyond simple management of symptoms toward modifying the course of disease, reducing cardiovascular complications, and improving patient quality-of-life. The key to success lies in early intervention, comprehensive metabolic assessment, and patient education that emphasizes the critical role of lifestyle factors in kidney health.
5. Summary and Future Outlook
Metabolic abnormalities can aggravate kidney injury and promote the progression of CKD by altering multiple interrelated pathways that can lead to increased inflammation and oxidative stress, dysregulation of autophagy, glomerular hyperfiltration and disruption of hemodynamics, endothelial dysfunction, and dysbiosis of the gut microbiota. Drug interventions, changes in lifestyle, and altering the microbial composition of the gut can slow the progression of CKD by targeting specific abnormalities in the metabolism of lipids, carbohydrates, proteins, and minerals. Future studies that combine multi-omics technologies (metabolomics, genomics, transcriptomics, and microbiomics) may provide a more thorough understanding of the molecular alterations responsible for CKD and a basis for the development of more effective targeted therapies. Future investigations should also examine the metabolic profiles of patients with different stages and subtypes of CKD, and integrate this information with data about genetic predispositions, lifestyle factors, and metabolomics to formulate more personalized therapeutic strategies. However, because CKD is a chronically progressive disease, studying the long-term impact of metabolic dysregulation on disease trajectory will require substantial resources. Therefore, designing efficient research methodologies and optimizing the allocation of resources remain significant challenges. Although animal studies have identified numerous potential therapeutic targets, translating these findings into clinical applications remains challenging because of the need to consider drug safety and efficacy in humans, as well as cost-effectiveness. We anticipate that future research will surmount these obstacles and achieve significant advancements and identification of interventions that improve the prevention and treatment of CKD, and deliver promising outcomes for patients.
Abbreviations
CKD: Chronic kidney disease; ESRD: end-stage renal disease; GFR: glomerular filtration rate; RAAS: renin-angiotensin-aldosterone system; ROS: reactive oxygen species; p38 MAPK: p38 mitogen-activated protein kinase; ERK: extracellular regulated protein kinase; JNK: Jun N-terminal kinase; CRP: C-reactive protein; TNF-α: tumor necrosis factor-alpha; IL-6: interleukin-6; IL-1β: interleukin-1β; MCP-1: monocyte chemoattractant protein-1; AMPK: AMP-activated protein kinase; DUSP5: Dual-specificity phosphatase 5; NKA: Na-K-ATPase; IGF-1: insulin-like growth factor-1; NO: nitric oxide; AGEs: advanced glycation end-products; SREBP-1: Sterol regulatory element-binding protein 1; TGF-β: transforming growth factor β; USAG-1: uterine sensitization-associated gene-1; PCSK9: proprotein convertase subtilisin/kexin type 9; LDL-C: low-density lipoprotein cholesterol; SGLT2: Sodium-glucose cotransporter 2; GLP-1RAs: glucagon-like peptide-1 receptor agonists; GIP: Glucose-dependent insulinotropic polypeptide; HN: hyperuricemic nephropathy; ERRα: estrogen-related receptor α; OAT1: organic anion transporter 1; MAPK: mitogen-activated protein kinase; IF: intermittent fasting; mTOR: mammalian target of rapamycin; NF-κB: nuclear factor-κB; SCFAs: short-chain fatty acids; FXR: farnesoid X receptor.
Acknowledgements
We thank Medjaden Inc. for scientific editing of this manuscript.
Funding
This work was supported by Hubei Provincial Natural Science Foundation (Grant No. 2023AFB732) and Scientific Research Project of Hubei Provincial Health Commission (Grant No. WJ2023M053).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gluba-Sagr A, Franczyk B, Rysz-Górzyńska M. et al. The Role of miRNA in Renal Fibrosis Leading to Chronic Kidney Disease. Biomedicines. 2023;11:2358
2. Chevalier RL. Bioenergetics: the evolutionary basis of progressive kidney disease. Physiol Rev. 2023;103:2451-506
3. Amini Khiabani S, Asgharzadeh M, Samadi Kafil H. Chronic kidney disease and gut microbiota. Heliyon. 2023;9:e18991
4. Ameer OZ. Hypertension in chronic kidney disease: What lies behind the scene. Front Pharmacol. 2022;13:949260
5. Braga PC, Alves MG, Rodrigues AS, Oliveira PF. Mitochondrial Pathophysiology on Chronic Kidney Disease. Int J Mol Sci. 2022;23:1776
6. Brennan E, Kantharidis P, Cooper ME, Godson C. Pro-resolving lipid mediators: regulators of inflammation, metabolism and kidney function. Nat Rev Nephrol. 2021;17:725-39
7. Ebert T, Neytchev O, Witasp A. et al. Inflammation and Oxidative Stress in Chronic Kidney Disease and Dialysis Patients. Antioxid Redox Signal. 2021;35:1426-48
8. Ambroselli D, Masciulli F, Romano E. et al. New Advances in Metabolic Syndrome, from Prevention to Treatment: The Role of Diet and Food. Nutrients. 2023;15:640
9. Fahed G, Aoun L, Bou Zerdan M. et al. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int J Mol Sci. 2022;23:786
10. Kim Y, Jo J, Ji Y. et al. Impact of hyperuricemia on CKD risk beyond genetic predisposition in a population-based cohort study. Sci Rep. 2024;14:18466
11. Jiang Z, Wang Y, Zhao X. et al. Obesity and chronic kidney disease. Am J Physiol Endocrinol Metab. 2023;324:E24-e41
12. Wahba NS, Abdel-Ghany RH, Ghareib SA. et al. Vitamin D3 potentiates the renoprotective effects of vildagliptin in a rat model of fructose/salt-induced insulin resistance. Eur J Pharm Sci. 2020;144:105196
13. Chen S, Chen J, Li S. et al. High-Fat Diet-Induced Renal Proximal Tubular Inflammatory Injury: Emerging Risk Factor of Chronic Kidney Disease. Front Physiol. 2021;12:786599
14. Stepien KM, Heaton R, Rankin S. et al. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. J Clin Med. 2017;6:71
15. Alberici LC, Oliveira HCF. Mitochondrial Adaptive Responses to Hypertriglyceridemia and Bioactive Lipids. Antioxid Redox Signal. 2022;36:953-68
16. Frąk W, Dąbek B, Balcerczyk-Lis M. et al. Role of Uremic Toxins, Oxidative Stress, and Renal Fibrosis in Chronic Kidney Disease. Antioxidants (Basel). 2024;13:687
17. Jia K, Shi P, Zhang L. et al. Trans-cinnamic acid alleviates high-fat diet-induced renal injury via JNK/ERK/P38 MAPK pathway. J Nutr Biochem. 2025;135:109769
18. Yu MA, Sánchez-Lozada LG, Johnson RJ, Kang DH. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens. 2010;28:1234-42
19. Gherghina ME, Peride I, Tiglis M. et al. Uric Acid and Oxidative Stress-Relationship with Cardiovascular, Metabolic, and Renal Impairment. Int J Mol Sci. 2022;23:3188
20. Chen Z, Shrestha R, Yang X. et al. Oxidative Stress and Lipid Dysregulation in Lipid Droplets: A Connection to Chronic Kidney Disease Revealed in Human Kidney Cells. Antioxidants (Basel). 2022;11:1387
21. Giardini E, Moore D, Sadlier D. et al. The dual role of lipids in chronic kidney disease: Pathogenic culprits and therapeutic allies. Atherosclerosis. 2024;398:118615
22. Czaja-Stolc S, Potrykus M, Stankiewicz M. et al. Pro-Inflammatory Profile of Adipokines in Obesity Contributes to Pathogenesis, Nutritional Disorders, and Cardiovascular Risk in Chronic Kidney Disease. Nutrients. 2022;14:1457
23. Aamir AB, Kumari R, Latif R. et al. Effects of intermittent fasting and caloric restriction on inflammatory biomarkers in individuals with obesity/overweight: A systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2025;26:e13838
24. Ren N, Wang WF, Zou L. et al. The nuclear factor kappa B signaling pathway is a master regulator of renal fibrosis. Front Pharmacol. 2023;14:1335094
25. Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210-5
26. Alex R, Press E, Sanchez L. et al. Comparative Levels of Urinary Biomarkers of Renal Injury and Inflammation Among Patients With Diabetic Nephropathy With or Without Hyperuricemia. J Clin Rheumatol. 2024: Online ahead of print.
27. Kim SM, Lee SH, Kim YG. et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am J Physiol Renal Physiol. 2015;308:F993-f1003
28. Xu Z, Tao L, Su H. The Complement System in Metabolic-Associated Kidney Diseases. Front Immunol. 2022;13:902063
29. Biasizzo M, Kopitar-Jerala N. Interplay Between NLRP3 Inflammasome and Autophagy. Front Immunol. 2020;11:591803
30. Cui Y, Chen J, Zhang Z. et al. The role of AMPK in macrophage metabolism, function and polarisation. J Transl Med. 2023;21:892
31. Agostini F, Bisaglia M, Plotegher N. Linking ROS Levels to Autophagy: The Key Role of AMPK. Antioxidants (Basel). 2023;12:1406
32. Bai F, Wang C, Wang S. et al. DUSP5 deficiency suppresses the progression of acute kidney injury by enhancing autophagy through AMPK/ULK1 pathway. Transl Res. 2024;274:1-9
33. Kimura T, Isaka Y, Yoshimori T. Autophagy and kidney inflammation. Autophagy. 2017;13:997-1003
34. Arden C, Park SH, Yasasilka XR. et al. Autophagy and lysosomal dysfunction in diabetes and its complications. Trends Endocrinol Metab. 2024;35:1078-90
35. Yasuda-Yamahara M, Kume S, Tagawa A. et al. Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy. 2015;11:2385-6
36. Mizunoe Y, Sudo Y, Okita N. et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy. 2017;13:642-53
37. Xiao J, Zhu S, Guan H. et al. AMPK alleviates high uric acid-induced Na(+)-K(+)-ATPase signaling impairment and cell injury in renal tubules. Exp Mol Med. 2019;51:1-14
38. Sharma I, Liao Y, Zheng X, Kanwar YS. New Pandemic: Obesity and Associated Nephropathy. Front Med (Lausanne). 2021;8:673556
39. Bancu I, Navarro Díaz M, Serra A. et al. Low Insulin-Like Growth Factor-1 Level in Obesity Nephropathy: A New Risk Factor? PLoS One. 2016;11:e0154451
40. Vaziri ND, Rodríguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:582-93
41. Aly R, Dogan YE, Bala N. et al. Dysregulation of kidney proteases in the pathogenesis of hypertension following unilateral nephrectomy in juvenile mice. Am J Transl Res. 2024;16:544-56
42. Hostetter TH, Olson JL, Rennke HG. et al. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol. 1981;241:F85-93
43. Donate-Correa J, Martín-Núñez E, Muros-de-Fuentes M. et al. Inflammatory cytokines in diabetic nephropathy. J Diabetes Res. 2015;2015:948417
44. Fan L, Gao W, Nguyen BV. et al. Impaired renal hemodynamics and glomerular hyperfiltration contribute to hypertension-induced renal injury. Am J Physiol Renal Physiol. 2020;319:F624-f35
45. Sprague SM, Martin KJ, Coyne DW. Phosphate Balance and CKD-Mineral Bone Disease. Kidney Int Rep. 2021;6:2049-58
46. Zhang M, Liu W, Dai H. et al. Heterogeneity of Renal Endothelial Cells, Interact with Neighboring Cells, and Endothelial Injury in Chronic Kidney Disease: Mechanisms and Therapeutic Implications. Int J Med Sci. 2025;22:2103-18
47. Prabhahar A, Batta A, Hatwal J. et al. Endothelial dysfunction in the kidney transplant population: Current evidence and management strategies. World J Transplant. 2025;15:97458
48. Borri M, Jacobs ME, Carmeliet P. et al. Endothelial dysfunction in the aging kidney. Am J Physiol Renal Physiol. 2025;328:F542-f62
49. Fountoulakis N, Miyamoto Y, Pavkov ME. et al. Pathophysiology of vascular ageing and the effect of novel cardio-renal protective medications in preventing progression of chronic kidney disease in people living with diabetes. Diabet Med. 2025;42:e15464
50. Liu Y, Li Z, Xu Y. et al. Uric Acid and Atherosclerosis in Patients with Chronic Kidney Disease: Recent Progress, Mechanisms, and Prospect. Kidney Dis (Basel). 2025;11:112-27
51. Bao Y, Shan Q, Lu K. et al. Renal tubular epithelial cell quality control mechanisms as therapeutic targets in renal fibrosis. J Pharm Anal. 2024;14:100933
52. Mitrofanova A, Merscher S, Fornoni A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat Rev Nephrol. 2023;19:629-45
53. Adeshara K, Gordin D, Antikainen AA. et al. Protein glycation products associate with progression of kidney disease and incident cardiovascular events in individuals with type 1 diabetes. Cardiovasc Diabetol. 2024;23:235
54. Cai W, He JC, Zhu L. et al. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc Natl Acad Sci U S A. 2006;103:13801-6
55. Kim YJ, Oh SH, Ahn JS. et al. The Crucial Role of Xanthine Oxidase in CKD Progression Associated with Hypercholesterolemia. Int J Mol Sci. 2020;21:7444
56. Proctor G, Jiang T, Iwahashi M. et al. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes. 2006;55:2502-9
57. Feng YL, Chen H, Chen DQ. et al. Activated NF-κB/Nrf2 and Wnt/β-catenin pathways are associated with lipid metabolism in CKD patients with microalbuminuria and macroalbuminuria. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2317-32
58. Mackowiak-Lewandowicz K, Ostalska-Nowicka D, Zaorska K. et al. Chronic kidney disease predictors in obese adolescents. Pediatr Nephrol. 2022;37:2479-88
59. Zhang H, Xie Y, Cao F, Song X. Gut microbiota-derived fatty acid and sterol metabolites: biotransformation and immunomodulatory functions. Gut Microbes. 2024;16:2382336
60. Wang J, Jiang M, Li X. et al. Inulin Supplementation Alleviates Ochratoxin A-Induced Kidney Injury through Modulating Intestinal Microbiota. J Agric Food Chem. 2024;72:18682-96
61. Corte-Iglesias V, Saiz ML, Andrade-Lopez AC. et al. Propionate and butyrate counteract renal damage and progression to chronic kidney disease. Nephrol Dial Transplant. 2024;40:133-50
62. Seljeset S, Siehler S. Receptor-specific regulation of ERK1/2 activation by members of the "free fatty acid receptor" family. J Recept Signal Transduct Res. 2012;32:196-201
63. Andrade-Oliveira V, Amano MT, Correa-Costa M. et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J Am Soc Nephrol. 2015;26:1877-88
64. Zhang L, Zhang S, Zou W. et al. Maternal high-fat diet regulates offspring hepatic ABCG5 expression and cholesterol metabolism via the gut microbiota and its derived butyrate. Clin Sci (Lond). 2024;138:1039-54
65. Liu W, Huang J, Liu T. et al. Changes in gut microbial community upon chronic kidney disease. PLoS One. 2023;18:e0283389
66. Wang J, Zang J, Yu Y. et al. Lingguizhugan oral solution alleviates MASLD by regulating bile acids metabolism and the gut microbiota through activating FXR/TGR5 signaling pathways. Front Pharmacol. 2024;15:1426049
67. Wang XX, Edelstein MH, Gafter U. et al. G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes. J Am Soc Nephrol. 2016;27:1362-78
68. Coll E, Cigarran S, Portolés J, Cases A. Gut Dysbiosis and Its Role in the Anemia of Chronic Kidney Disease. Toxins (Basel). 2024;16:495
69. Matei DE, Menon M, Alber DG. et al. Intestinal barrier dysfunction plays an integral role in arthritis pathology and can be targeted to ameliorate disease. Med. 2021;2:864-83.e9
70. Suresh MG, Mohamed S, Yukselen Z. et al. Therapeutic Modulation of Gut Microbiome in Cardiovascular Disease: A Literature Review. Heart and Mind. 2025;9:68-79
71. Ribeiro FPB, de Luna Freire MO, de Oliveira Coutinho D. et al. Gut Dysbiosis and Probiotic Therapy in Chronic Kidney Disease: A Comprehensive Review. Probiotics Antimicrob Proteins. 2024: Online ahead of print.
72. Stanigut AM, Tuta L, Pana C. et al. Autophagy and Mitophagy in Diabetic Kidney Disease-A Literature Review. Int J Mol Sci. 2025;26:806
73. Wen D, Tan RZ, Zhao CY. et al. Astragalus mongholicus Bunge and Panax notoginseng (Burkill) F.H. Chen Formula for Renal Injury in Diabetic Nephropathy-In Vivo and In Vitro Evidence for Autophagy Regulation. Front Pharmacol. 2020;11:732
74. Ha S, Son M, Kim J. et al. Gender Differences in Adenine Diet-Induced Kidney Toxicity: The Impact of 17β-Estradiol on Renal Inflammation and Fibrosis. Int J Mol Sci. 2025;26:1358
75. Farahmand M, Ramezani Tehrani F, Khalili D. et al. Endogenous estrogen exposure and chronic kidney disease; a 15-year prospective cohort study. BMC Endocr Disord. 2021;21:155
76. Ren L, Li F, Di Z. et al. Estradiol Ameliorates Acute Kidney Ischemia-Reperfusion Injury by Inhibiting the TGF-βRI-SMAD Pathway. Front Immunol. 2022;13:822604
77. Carrero JJ, Hecking M, Chesnaye NC, Jager KJ. Sex and gender disparities in the epidemiology and outcomes of chronic kidney disease. Nat Rev Nephrol. 2018;14:151-64
78. Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51-9
79. Goicoechea M, Garcia de Vinuesa S, Verdalles U. et al. Allopurinol and progression of CKD and cardiovascular events: long-term follow-up of a randomized clinical trial. Am J Kidney Dis. 2015;65:543-9
80. Kohagura K, Kojima S, Uchiyama K. et al. Febuxostat and renal outcomes: post-hoc analysis of a randomized trial. Hypertens Res. 2023;46:1417-22
81. Cao J, Li Y, Peng Y. et al. Febuxostat Prevents Renal Interstitial Fibrosis by the Activation of BMP-7 Signaling and Inhibition of USAG-1 Expression in Rats. Am J Nephrol. 2015;42:369-78
82. Hager MR, Narla AD, Tannock LR. Dyslipidemia in patients with chronic kidney disease. Rev Endocr Metab Disord. 2017;18:29-40
83. Ma Z, Zhu L, Liu Y. et al. Lovastatin Alleviates Endothelial-to-Mesenchymal Transition in Glomeruli via Suppression of Oxidative Stress and TGF-β1 Signaling. Front Pharmacol. 2017;8:473
84. Bandyopadhyay D, Ashish K, Hajra A. et al. Cardiovascular Outcomes of PCSK9 Inhibitors: With Special Emphasis on Its Effect beyond LDL-Cholesterol Lowering. J Lipids. 2018;2018:3179201
85. Park CH, Yoo TH. TGF-β Inhibitors for Therapeutic Management of Kidney Fibrosis. Pharmaceuticals (Basel). 2022;15:1485
86. Ma G, Chen F, Liu Y. et al. Nur77 ameliorates age-related renal tubulointerstitial fibrosis by suppressing the TGF-β/Smads signaling pathway. Faseb j. 2022;36:e22124
87. Wang X, Liu X, Xu L. et al. Targeted delivery of type I TGF-β receptor-mimicking peptide to fibrotic kidney for improving kidney fibrosis therapy via enhancing the inhibition of TGF-β1/Smad and p38 MAPK pathways. Int Immunopharmacol. 2024;137:112483
88. Li Q, Wang Y, Yan J. et al. Osthole ameliorates early diabetic kidney damage by suppressing oxidative stress, inflammation and inhibiting TGF-β1/Smads signaling pathway. Int Immunopharmacol. 2024;133:112131
89. Alicic RZ, Neumiller JJ, Galindo RJ, Tuttle KR. Use of Glucose-Lowering Agents in Diabetes and CKD. Kidney Int Rep. 2022;7:2589-607
90. Hu H, Li W, Hao Y. et al. The SGLT2 inhibitor dapagliflozin ameliorates renal fibrosis in hyperuricemic nephropathy. Cell Rep Med. 2024;5:101690
91. Singh A, Sohal A, Batta A. GLP-1, GIP/GLP-1, and GCGR/GLP-1 receptor agonists: Novel therapeutic agents for metabolic dysfunction-associated steatohepatitis. World J Gastroenterol. 2024;30:5205-11
92. Kamrul-Hasan A, Patra S, Dutta D. et al. Renal effects and safety of tirzepatide in subjects with and without diabetes: A systematic review and meta-analysis. World J Diabetes. 2025;16:101282
93. Packer M, Zile MR, Kramer CM. et al. Interplay of Chronic Kidney Disease and the Effects of Tirzepatide in Patients With Heart Failure, Preserved Ejection Fraction, and Obesity: The SUMMIT Trial. J Am Coll Cardiol. 2025;85:1721-35
94. Packer M, Zile MR, Kramer CM. et al. Tirzepatide for Heart Failure with Preserved Ejection Fraction and Obesity. N Engl J Med. 2025;392:427-37
95. Zhang L, Miao M, Xu X. et al. From Physiology to Pathology: The Role of Mitochondria in Acute Kidney Injuries and Chronic Kidney Diseases. Kidney Dis (Basel). 2023;9:342-57
96. Xiao L, Xu X, Zhang F. et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017;11:297-311
97. Liu X, Murphy MP, Xing W. et al. Mitochondria-targeted antioxidant MitoQ reduced renal damage caused by ischemia-reperfusion injury in rodent kidneys: Longitudinal observations of T(2) -weighted imaging and dynamic contrast-enhanced MRI. Magn Reson Med. 2018;79:1559-67
98. Kirkman DL, Stock JM, Shenouda N. et al. Effects of a mitochondrial-targeted ubiquinol on vascular function and exercise capacity in chronic kidney disease: a randomized controlled pilot study. Am J Physiol Renal Physiol. 2023;325:F448-f56
99. Liu B, Wang D, Cao Y. et al. MitoTEMPO protects against podocyte injury by inhibiting NLRP3 inflammasome via PINK1/Parkin pathway-mediated mitophagy. Eur J Pharmacol. 2022;929:175136
100. Zhao H, Liu YJ, Liu ZR. et al. Role of mitochondrial dysfunction in renal fibrosis promoted by hypochlorite-modified albumin in a remnant kidney model and protective effects of antioxidant peptide SS-31. Eur J Pharmacol. 2017;804:57-67
101. Liu ZR, Chen SQ, Zou YW. et al. Hypochlorite modified albumins promote cell death in the tubule interstitium in rats via mitochondrial damage in obstructive nephropathy and the protective effects of antioxidant peptides. Free Radic Res. 2018;52:616-28
102. Baker LA, March DS, Wilkinson TJ. et al. Clinical practice guideline exercise and lifestyle in chronic kidney disease. BMC Nephrol. 2022;23:75
103. Cases A, Cigarrán-Guldrís S, Mas S, Gonzalez-Parra E. Vegetable-Based Diets for Chronic Kidney Disease? It Is Time to Reconsider. Nutrients. 2019;11:1263
104. Neale EP, Rosario VD, Probst Y. et al. Lifestyle Interventions, Kidney Disease Progression, and Quality of Life: A Systematic Review and Meta-analysis. Kidney Med. 2023;5:100643
105. Chen TK, Knicely DH, Grams ME. Chronic Kidney Disease Diagnosis and Management: A Review. Jama. 2019;322:1294-304
106. Schrauben SJ, Apple BJ, Chang AR. Modifiable Lifestyle Behaviors and CKD Progression: A Narrative Review. Kidney360. 2022;3:752-78
107. Ma YN, Jiang X, Tang W, Song P. Influence of intermittent fasting on autophagy in the liver. Biosci Trends. 2023;17:335-55
108. Joaquim L, Faria A, Loureiro H, Matafome P. Benefits, mechanisms, and risks of intermittent fasting in metabolic syndrome and type 2 diabetes. J Physiol Biochem. 2022;78:295-305
109. Rifaai RA, El-Tahawy NFG, Abozaid SMM, Abdelwahab A. Intermittent Fasting Ameliorates Age-Induced Morphological Changes in Aged Albino Rat Kidney via Autophagy Activation and Reduction of Apoptosis and Inflammation. Microsc Microanal. 2025;31:ozae102
110. Bilen A, Calik I, Yayla M. et al. Does daily fasting shielding kidney on hyperglycemia-related inflammatory cytokine via TNF-α, NLRP3, TGF-β1 and VCAM-1 mRNA expression. Int J Biol Macromol. 2021;190:911-8
111. Yang M, Chen W, He L. et al. Intermittent Fasting-A Healthy Dietary Pattern for Diabetic Nephropathy. Nutrients. 2022;14:3995
112. Aroni A, Detopoulou P, Presvelos D. et al. A One-Month Advanced Glycation End Products-Restricted Diet Improves CML, RAGE, Metabolic and Inflammatory Profile in Patients with End-Stage Renal Disease Undergoing Haemodialysis. Int J Mol Sci. 2024;25:8893
113. Iyengar A, Ramadass B, Venkatesh S, Mak RH. Gut microbiota-targeted therapies in pediatric chronic kidney disease: gaps and opportunities. Pediatr Nephrol. 2025: Online ahead of print.
114. Tian F, Chen T, Xu W. et al. Curcumin Compensates GLP-1 Deficiency via the Microbiota-Bile Acids Axis and Modulation in Functional Crosstalk between TGR5 and FXR in ob/ob Mice. Mol Nutr Food Res. 2023;67:e2300195
115. Habibi A, Letafatkar N, Sattari N. et al. Modulation of inflammatory markers in type 2 diabetes mellitus through gut microbiome-targeted interventions: An umbrella review on meta-analyses. Clin Nutr ESPEN. 2025;65:93-104
116. Cabała S, Ożgo M, Herosimczyk A. The Kidney-Gut Axis as a Novel Target for Nutritional Intervention to Counteract Chronic Kidney Disease Progression. Metabolites. 2024;14:78
117. Cedillo-Flores R, Cuevas-Budhart MA, Cavero-Redondo I. et al. Impact of Gut Microbiome Modulation on Uremic Toxin Reduction in Chronic Kidney Disease: A Systematic Review and Network Meta-Analysis. Nutrients. 2025;17:1247
118. Kalidindi RK, Reddy CP, Pv K, Kompella P. The Efficacy and Safety of Probiotic Combinations Lobun Forte® Versus Renadyl® in Patients With Chronic Kidney Disease: A Comparative, Phase IV, Randomized, Open-Label, Active-Controlled, Parallel Study. Cureus. 2024;16:e67987
119. Liu C, Yang L, Wei W, Fu P. Efficacy of probiotics/synbiotics supplementation in patients with chronic kidney disease: a systematic review and meta-analysis of randomized controlled trials. Front Nutr. 2024;11:1434613
120. Bakhtiary M, Morvaridzadeh M, Agah S. et al. Effect of Probiotic, Prebiotic, and Synbiotic Supplementation on Cardiometabolic and Oxidative Stress Parameters in Patients With Chronic Kidney Disease: A Systematic Review and Meta-analysis. Clin Ther. 2021;43:e71-e96
121. Lou X, Xue J, Shao R. et al. Fecal microbiota transplantation and short-chain fatty acids reduce sepsis mortality by remodeling antibiotic-induced gut microbiota disturbances. Front Immunol. 2022;13:1063543
122. Caggiano G, Cosola C, Di Leo V. et al. Microbiome modulation to correct uremic toxins and to preserve kidney functions. Curr Opin Nephrol Hypertens. 2020;29:49-56
123. Shao B, Nong Y, Lin Y. et al. Study on the Influence and Mechanism of Resveratrol on Cognitive Impairment in Chronic Kidney Disease Rats Through Regulating Gut Microbiota and the TLR4/NFκB Pathway. J Inflamm Res. 2025;18:6049-60
Author contact
![]() Corresponding authors: Xiaoyan Wu, Division of Nephrology, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan 430070, China, Email: wuxiaoyan2k6edu.cn; Tel.: 86-15972935798. Ping Gao, Division of Nephrology, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan 430070, China, Email: gp6680com; Tel.: 86-18971051281.
Corresponding authors: Xiaoyan Wu, Division of Nephrology, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan 430070, China, Email: wuxiaoyan2k6edu.cn; Tel.: 86-15972935798. Ping Gao, Division of Nephrology, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan 430070, China, Email: gp6680com; Tel.: 86-18971051281.