Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Acinar Cell Damage and...
3. The Role of Immune Cell...
4. Metabolic Reprogramming in...
5. Metabolic Reprogramming as a...
6. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(14):3528-3542. doi:10.7150/ijms.118338 This issue Cite
Review
Metabolism and Targeted Therapy of Fibrosis in Chronic Pancreatitis: A Review
Hongqing Luo, Shan Guo, Yuning Chu, Yiping Xin, Xiaoyan Yin, Xiaoyu Li ![]()
Department of Gastroenterology, The Affiliated Hospital of Qingdao University, Qingdao, China.
Received 2025-5-27; Accepted 2025-7-11; Published 2025-7-28
Abstract

Chronic pancreatitis (CP) is a progressive condition characterized by persistent pancreatic inflammation, tissue destruction, and fibrosis. Recent studies have highlighted the crucial role of metabolic processes in the pathogenesis of pancreatic fibrosis, particularly the metabolic reprogramming of pancreatic stellate cells (PSCs) and immune cells. Disruptions in glucose, lipid, and amino acid metabolism have been shown to play a key role in the progression of CP fibrosis, exacerbating disease severity. Activated PSCs exhibit enhanced glycolysis and lipid metabolism, which promote excessive extracellular matrix (ECM) production and tissue remodeling. Simultaneously, immune cells such as macrophages and T cells undergo metabolic reprogramming, further intensifying inflammation and fibrosis. This review discusses the role of metabolic reprogramming in pancreatic fibrosis and proposes potential therapeutic strategies targeting metabolic pathways, including glycolysis inhibitors, lipid metabolism modulators, and amino acid metabolism regulators. These strategies offer promising prospects for mitigating the progression of CP fibrosis and provide new therapeutic avenues for clinical applications.
Keywords: chronic pancreatitis, pancreatic fibrosis, metabolic reprogramming, pancreatic stellate cells, glycolysis, therapeutic strategies
1. Introduction
Chronic pancreatitis (CP) is characterized by prolonged inflammation in the pancreas, resulting in tissue damage and fibrosis. The disease has diverse etiologies, including alcohol abuse, biliary obstruction, and—in a small subset of cases—hereditary mutations such as PRSS1, SPINK1, or CFTR [1]. Regardless of etiology, sustained pancreatic injury leads to chronic inflammation and progressive fibrosis, which are closely linked to immune-metabolic dysregulation.
Early in the disease, repeated inflammatory episodes in pancreatic tissue gradually lead to fibrosis, severely impairing pancreatic structure and function [2]. This fibrosis eventually results in the decline of both endocrine and exocrine functions, affecting digestion and glucose regulation in patients and potentially leading to more severe complications such as pancreatic cancer [3].
Mechanistically, injured acinar cells release signals that prompt the recruitment of immune cells, triggering inflammatory responses and differentiation processes [4]. Due to their significant plasticity and heterogeneity, macrophages originate from bone marrow-derived monocytes, and their diverse phenotypes play crucial roles in both the onset and persistence of tissue damage and fibrosis [5]. Activated T cells have different cell subsets and can secrete various cytokines, which play a key role in the inflammation and fibrosis of CP [6]. Pancreatic stellate cells (PSCs) are a type of mesenchymal cell located within the pancreatic tissue, where they help preserve normal physiological structure and function. Signals stemming from injury and immune cells activate PSCs, transforming them into a myofibroblast-like phenotype, which is a critical step in the progression of pancreatic fibrosis [7]. Upon activation, PSCs proliferate extensively and produce excessive extracellular matrix (ECM) components. Consequently, macrophages, T cells, and PSCs adapt their metabolism to meet the increased energy demands and the need for raw materials to sustain these physiological activities. The excessive ECM accumulation and cross-linking that ensue eventually compromise the cellular microenvironment, leading to the disruption of structural integrity and homeostasis within the pancreas. Ultimately, pancreatic fibrosis is associated with high morbidity and mortality due to pancreatic failure and its potential progression to pancreatic cancer [8].
Pancreatic fibrosis stands out as a hallmark of chronic pancreatitis and is a significant contributor to the long-term complications of the disease [9, 1]. Various therapeutic strategies have been explored in recent years, including antioxidant therapy, gene therapy, and immunotherapy [6, 10]. For a small subset of CP cases that is genetically determined, targeting the underlying disease-predisposing or -causing mutations may be a crucial therapeutic strategy [10]. However, most of these therapeutic approaches have had limited efficacy in halting or reversing fibrosis. Therefore, greater attention has been directed toward antifibrotic therapies, particularly those that target the underlying cellular and molecular mechanisms of fibrosis.
This article emphasizes the connection between metabolic regulation and pancreatic fibrosis. The metabolic states of immune cells and PSCs are closely intertwined with their functional capacities [11]. Factors such as nutrient availability, redox balance, gut-derived metabolites and microbiota, and circadian rhythms modulate cellular metabolism, ultimately determining cellular function. Similarly, alterations in cellular functions driven by immune or growth factor signals necessitate adjustments in metabolism to fulfill these demands [12]. Therefore, targeting immune cell and PSC metabolism represents a promising therapeutic strategy in addressing pancreatic fibrosis.
While existing reviews have discussed immune cell function or PSC activation in CP, few have specifically focused on the metabolic reprogramming shared across immune and stromal cells and its role in driving fibrogenesis [13, 6]. This review aims to fill this gap by providing an integrative perspective on how metabolism links inflammation and fibrosis in CP and how these insights may inform more precise anti-fibrotic interventions. Recognizing the influence of metabolic reprogramming on pancreatic fibrosis may pave the way for new treatments for chronic pancreatitis.
2. Acinar Cell Damage and Oxidative Stress
Metabolic disturbances play a fundamental role in the pathogenesis of fibrosis during chronic pancreatitis. Normally, pancreatic cells utilize various metabolic pathways, including glucose, lipid, and amino acid metabolism, to support their functions. However, in the presence of chronic inflammation and changes in the immune microenvironment, both recruited and resident immune cells, alter their metabolic activity and prioritize specific pathways to adapt to the demands of survival, proliferation, and function [14, 15]. Acinar cell damage is recognized as a key trigger in chronic pancreatitis. In patients with CP, the expression of interferon-γ-inducible protein 10 (CXCL10) in pancreatic tissue is upregulated. CXCL10 can induce apoptosis and DNA damage through C-X-C motif chemokine receptor 3 (CXCR3) signaling. Upon stimulation by CXCL10, the expression of cytochrome C, Apaf-1, and caspase 3/9 is upregulated in acinar cells, leading to apoptosis. Concurrently, there is a change in membrane potential, mitochondrial dysfunction occurs, and ATP is significantly depleted [16]. Mitochondrial dysfunction leads to ATP depletion, which in turn causes an accumulation of Ca2+ in the cytoplasm. Pathological Ca2+ signaling activates the calcium-dependent phosphatase calcineurin, resulting in the premature activation of trypsinogen [17]. Trypsinogen, through a non-ATPase proteasome pathway, mediates the degradation of glutathione peroxidase 4, thereby increasing the sensitivity of acinar cells to ferroptosis and triggering pancreatitis [18]. Estrogen-related receptor γ (ERRγ) is vital for maintaining mitochondrial oxidative phosphorylation (OXPHOS) in pancreatic acinar cells, and its downregulation can lead to mitochondrial dysfunction, energy depletion, increased reactive oxygen species (ROS) accumulation, and progressive pancreatic atrophy, thereby exacerbating pancreatitis. In patients with CP, ERRγ expression is downregulated, and polymorphisms in the ESRRG gene, which encodes ERRγ, as well as single nucleotide variants, have been linked to CP [19].
ROS are byproducts of oxidative metabolism. An imbalance between ROS production and degradation can lead to oxidative stress, which may trigger chronic pancreatitis [20]. ROS regulate the expression of nuclear factor kappa B (NF-κB), which in turn induces the expression of transforming growth factor β (TGF-β) and fibrosis-related genes [21]. TGF-β activates PSCs, leading to the production of extracellular matrix proteins and the promotion of pancreatic fibrosis [22]. Excessive consumption of carbohydrates and fats enhances mitochondrial oxidative respiration, significantly elevating ROS levels. This increase in ROS triggers lipid peroxidation, resulting in the substantial accumulation of metabolites like malondialdehyde (MDA) within the body, which in turn disrupts lipid metabolism in the pancreas [23, 24]. Elevated glucose concentrations enhance the production of ROS in PSCs, thereby driving their activation and contributing to the progression of fibrosis [25]. Therefore, it is essential to acknowledge the pivotal role of metabolic pathways in driving the progression of chronic pancreatitis.
3. The Role of Immune Cell Metabolic Reprogramming in Fibrosis
3.1 Macrophage Metabolic Reprogramming
Macrophages, monocytes derived from bone marrow monocytes or yolk sac cells during embryonic development, are responsible for the removal of dead or harmful pathogens and play an important role in inflammation, tissue repair, and homeostasis [26] [27]. Macrophages can be divided into M1 macrophages (classical activated type) and M2 macrophages (alternative activated type) according to their different phenotypic functions [28]. Under normal physiological conditions, resident macrophages are sparse in the pancreas, proliferating, polarizing, and repairing tissue only when pathological changes occur. Initially, they respond to the local environment in a pro-inflammatory manner, but they rapidly transition to a wound repair phenotype. The immune characteristics of macrophages differ across CP subtypes. For example, idiopathic CP presents a higher prevalence of CD68+ macrophages compared to hereditary CP. Idiopathic CP also shows increased expression of interleukin-4 (IL-4) and IL-13, whereas hereditary CP shows elevated tumor necrosis factor-α (TNF-α) and IL-6 levels [29].
M1 macrophages predominantly infiltrate during the early stages of chronic pancreatitis. M0 macrophages migrating from the bone marrow polarize into M1 macrophages, releasing TGF-α/β, IL-6, and matrix metalloproteinase-10 (MMP-10), contributing to pancreatic inflammation and injury [30]. In CP, M2 macrophages become the predominant infiltrating cells, mediating pancreatic fibrosis. Both infiltrating macrophages and PSCs secrete IL-6, which induces PSCs to produce TGF-β1 through IL-6R/STAT3 signaling, promoting PSC activation and fibrosis [31]. Dachaihu decoction, a traditional Chinese medicine formula, has been shown to inhibit pancreatic macrophage infiltration, reduce IL-6 and chemokine expression (such as monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1α (MIP-1α)), and improve pancreatic fibrosis [32]. Moreover, M2 macrophages interact with PSCs, promoting pancreatic fibrosis by secreting TGF-β and platelet-derived growth factor β (PDGFβ), which further activate PSCs [26]. When bone marrow-derived macrophages are co-cultured with PSCs, they exhibit increased expression of CD206, IL-10, TGF-β, and PDGFβ, while nitric oxide (NO) synthase expression decreases, indicating that PSCs can promote macrophage polarization to the M2 phenotype [33]. PSCs secrete chemokine MCP-1, inducing alternative macrophage activation via NF-Κb [34]. Pirfenidone reduces M2 macrophage infiltration in the pancreas and inhibits PSC cytokine release, decreasing both M2 macrophage and PSC activation, thereby alleviating pancreatic fibrosis [35]. The physiological function of macrophages is closely related to fibrosis in chronic pancreatitis. Therefore, understanding macrophage metabolism can better explore the relationship between macrophages and CP [Figure 1].
Macrophage metabolic reprogramming in chronic pancreatitis. Macrophages exhibit distinct metabolic phenotypes in chronic pancreatitis (CP). Classically activated macrophages (M1) primarily rely on glycolysis, even in the presence of oxygen, a phenomenon similar to the Warburg effect. This shift leads to the production of lactate and the interruption of the tricarboxylic acid (TCA) cycle at two key nodes, resulting in the accumulation of citrate and succinate. Succinate stabilizes hypoxia-inducible factor 1-alpha (HIF-1α), which in turn promotes the transcription of proinflammatory cytokines such as IL-1β. Additionally, pyruvate kinase M2 (PKM2) enhances HIF-1α and IL-1β expression, reinforcing the inflammatory phenotype of M1 macrophages. In contrast, alternatively activated macrophages (M2) utilize oxidative phosphorylation (OXPHOS) and fatty acid oxidation to support anti-inflammatory functions and tissue repair. In M2 macrophages, glutamine-derived α-ketoglutarate (α-KG) fuels the TCA cycle and serves as a substrate for epigenetic regulation via histone demethylation and UDP-GlcNAc synthesis, facilitating M2 polarization. Arginine metabolism also differs between subtypes: M1 macrophages convert arginine to nitric oxide (NO) to maintain their inflammatory role, whereas M2 macrophages convert arginine to proline, contributing to collagen synthesis and fibrosis progression. HIF-1α, hypoxia-inducible factor 1-alpha; OXPHOS, oxidative phosphorylation; PKM, pyruvate kinase M; α-KG, α-ketoglutarate; TCA, tricarboxylic acid cycle; NO, nitric oxide; UDP-GlcNAc, uridine diphosphate N-acetylglucosamine.
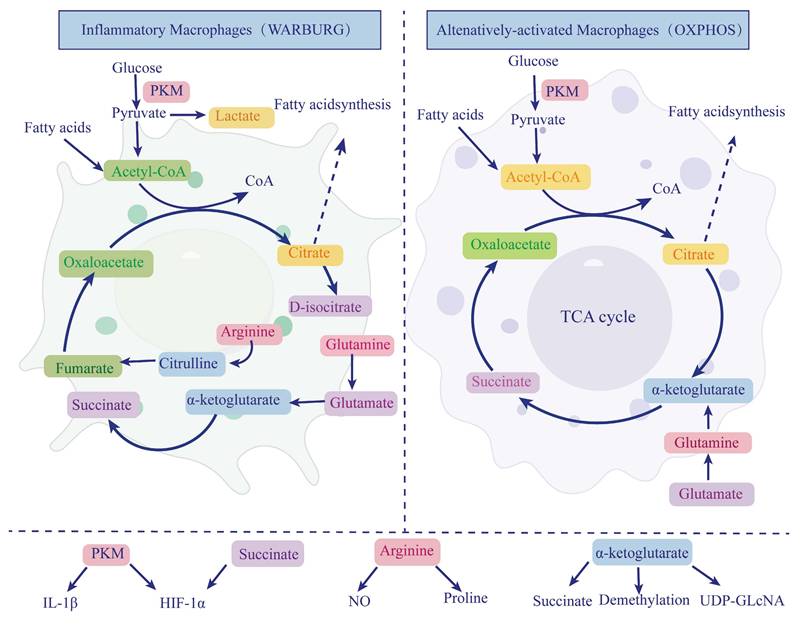
3.1.1 Glucose Metabolism
Macrophage glucose metabolism is central to their activation and function, particularly in different immune responses, where glucose metabolic pathways regulate macrophage phenotypes [36]. In M1 macrophages, glycolysis serves as the primary energy source. Even under aerobic conditions, macrophages tend to favor anaerobic glycolysis, a pathway that provides quick energy while also enhancing the expression of MMP-1, IL-1β, and IL-6 in U937 macrophage-like cells via NF-κB and mitogen-activated protein kinase (MAPK) signaling [37, 38]. This metabolic shift exacerbates inflammation in CP, as increased glycolysis is closely linked to maintaining and intensifying pro-inflammatory responses. The Warburg effect observed in M1 macrophages is similar to that in tumor cells, demonstrating a high dependence on hexokinase and glucose-6-phosphate dehydrogenase, further supporting their heightened metabolic activity and sustained inflammatory responses [39]. In contrast, M2 macrophages rely more on OXPHOS for energy production [40]. OXPHOS occurs through the mitochondrial electron transport chain, generating ATP more efficiently, giving M2 macrophages an advantage in tissue repair and anti-inflammatory responses [41].
In M1 macrophages, HIF-1α is upregulated, while in M2 macrophages, HIF-2α activation induces the expression of arginase 1 (ARG1), which suppresses NO production [42]. Neddylation, a key post-translational modification, can be targeted with small-molecule inhibitors such as MLN4924, which inactivates the neddylation pathway, promoting HIF-1α-mediated chemokine ligand 5 (CCL5) secretion and enhancing M2 macrophage infiltration in chronic pancreatitis [43]. Pyruvate kinase M2 (PKM2) is a key regulator of the Warburg effect in LPS-activated macrophages, modulating HIF-1α activity and IL-1β induction [44]. PKM2 interacts with Smad7, interfering with Smad7 and TGFβR1 interaction and affecting the activation of the TGFβ1 signaling pathway. The tetrameric form of PKM2 helps regulate the TGFβ1 signaling pathway balance, promoting fibrosis [45]. PKM2 also acts as a co-activator of HIF-1α, stimulated by prolyl hydroxylases 3 (PHD3) [46]. Additionally, succinate accumulation inhibits PHDs, preventing them from hydroxylating HIF-1α, leading to HIF-1α stabilization [47]. PKM2 further regulates glycolysis in LPS-activated macrophages by controlling lactate-induced K62 lactylation, facilitating the transition from a pro-inflammatory phenotype to a reparative phenotype, and accelerating wound healing [48].
In summary, macrophage glucose metabolism regulates energy production and biosynthesis, finely tuning their functions and phenotypes in different immune environments. Modulating macrophage glucose metabolism could potentially control the excessive activation of M1 macrophages, alleviating pancreatic inflammation and slowing disease progression.
3.1.2 Amino Acid Metabolism
Amino acid metabolism is also crucial for macrophage function. Glutamine is one of the most important amino acids in macrophage metabolism, serving not only as a substrate for protein synthesis but also influencing macrophage activation and function through various pathways [49]. In M1 macrophages, glutamine is converted into α-ketoglutarate, entering the Krebs cycle to support succinate production. Glutamine metabolism generates succinate, which can inhibit HIF-1α degradation, stabilizing its expression and promoting the pro-inflammatory functions of M1 macrophages.
Conversely, M2 macrophages utilize glutamine for oxidative phosphorylation and epigenetic reprogramming. The α-ketoglutarate (α-KG) produced from glutamine catabolism supports the anti-inflammatory phenotype of M2 macrophages and regulates gene expression through demethylation, promoting tissue repair functions [50, 40]. Additionally, glutamine serves as a precursor for UDP-GlcNAc synthesis, participating in protein glycosylation in M2 macrophages, further enhancing their reparative functions [51]. In chronic pancreatitis, imbalances in glutamine metabolism may lead to dysregulation of M1 and M2 macrophage functions, exacerbating inflammation or promoting fibrosis.
Other amino acids, such as arginine, also play critical roles in regulating macrophage phenotypes [52]. M1 macrophages metabolize arginine to produce NO, enhancing bactericidal and anti-infection capabilities. Meanwhile, M2 macrophages utilize arginine to promote proline synthesis and collagen production, contributing to tissue repair and fibrosis [53-55]. Macrophage amino acid metabolism regulates functional states through multiple pathways and is key to maintaining immune homeostasis and tissue health. Understanding and regulating macrophage amino acid metabolism, particularly glutamine metabolism, may offer new therapeutic approaches for CP.
3.1.3 Lipid Metabolism
Lipid metabolism plays multiple roles in macrophage function, particularly in the polarization of M1 and M2 macrophages. Lipids serve not only as energy sources but also participate in cell signaling and gene regulation [56]. In M1 macrophages, lipid metabolism is typically associated with pro-inflammatory responses. Excess lipid accumulation, particularly when fatty acid β-oxidation is enhanced, promotes the production of inflammatory cytokines such as TNF-α and IL-6, potentially amplifying the inflammatory response in CP [56].
Studies show that IL-4-activated M2 macrophages exhibit significantly upregulated fatty acid uptake and oxidation, while these functions are suppressed in M1 macrophages [57]. M2 macrophages rely on lipid synthesis and storage to support their anti-inflammatory and tissue repair functions [57]. In these cells, OXPHOS depends on the oxidation of fatty acids and glutamine, activating peroxisome proliferator-activated receptor-gamma (PPARγ), which regulates gene expression in M2 macrophages [58, 59]. The natural ligand of PPARγ, 15-deoxy-Δ12,14-prostaglandin J2, inhibits NO, TNF-α, IL-1, and IL-6 production in monocytes and macrophages [60]. PPARγ expression in macrophages is crucial for counteracting inflammation. It also inhibits NF-κB activity by physically interacting with NF-κB or inducing the expression of NF-κB inhibitors (IκB). For example, in the spontaneous CP model of WBN/Kob mice, PPARγ is highly expressed in macrophages. After treatment with PPARγ ligands, such as thiazolidinedione derivatives (TZD), the severity of pancreatic morphological damage is reduced, fibrosis is significantly improved, pancreatic amylase content decreases, serum IL-8 and TNF-α levels are reduced, and NF-κB binding activity decreases, preventing the exacerbation of CP [61, 62]. In CP, M2 macrophages may contribute to tissue repair and fibrosis through lipid metabolism regulation. Therefore, targeting lipid metabolism pathways could become an effective strategy for controlling inflammation and promoting tissue repair in CP.
3.2 T Cell Metabolic Reprogramming
T cells play a vital role in defending against infections and in the immune response against tumors [63]. T cells consist of various subtypes, each with distinct functions. During T cell activation, metabolic pathways undergo changes to provide sufficient energy and nutrients to support T cells' physiological functions. Once the immune response is completed, surviving T cells persist as memory T cells and revert to a quiescent oxidative metabolism state [64].
Regardless of the cause, CP is characterized by inflammatory cell infiltration, including lymphocytes and macrophages [65]. Therefore, multiple studies suggest that the adaptive immune system plays a crucial role in CP pathogenesis, both in humans and animal models. Local chemokines recruit CD4+ and CD8+ T cells to CP lesions [66]. Single-cell RNA sequencing of pancreatic immune cells from CP patients revealed that genetic CP is dominated by CD4+ helper T cell subpopulations, including CCR6+Th1, regulatory T cells (Tregs), and HLA-DA+CD8+ T cells. In contrast, idiopathic CP samples primarily exhibit FTH1+ CD4+ and BAG3+ CD8+ T cells [29]. Peripheral blood samples from CP patients show an increased proportion of T-helper 1 cells (Th1), Th2, and Th17 compared to controls. Among alcohol-consuming patients, the proportion of Th1 cells is significantly higher than in non-drinkers, with CD4+ helper T cells infiltrating the pancreas mainly as Th1 and Th17 cells, and fewer Th2 cells [67]. T cell response analysis in peripheral blood from CP patients, pancreatic cancer patients, and healthy individuals found that IL-10-specific responses in pancreatitis were mediated by IL-10+ IFN-γ- FoxP3+ regulatory T cells. Compared to pancreatic cancer, IL-10 levels in pancreatitis lesions are elevated, while interferon-γ (IFN-γ) levels are reduced, indicating that Tregs exhibit specific activity in pancreatitis [68].
T cells play a pivotal role in the inflammatory response and the progression of tissue damage in CP. Beyond the distinct distribution and functions of T cell subsets, metabolic changes within the immune microenvironment are also crucial in CP pathogenesis. Upon activation, T cells undergo metabolic reprogramming to meet their heightened energy demands and to support their functional roles. For instance, effector T cells (Teff) rely on glycolysis to sustain rapid proliferation and the production of inflammatory cytokines, while Tregs depend on fatty acid oxidation (FAO) to maintain their immunosuppressive function. The local metabolic environment within the pancreas of CP patients may regulate these metabolic pathways, thereby influencing T cell differentiation and function, and either exacerbating or alleviating the inflammatory response. Additionally, the activity of IL-10+ regulatory T cells in the pancreas could be closely linked to local metabolic signaling, offering new insights into the complex pathophysiology of chronic pancreatitis. Therefore, investigating the interaction between T cell metabolism and the pancreatic microenvironment is essential for developing novel therapeutic strategies.
3.2.1 Glucose Metabolism
Glucose metabolism is critical for T cell activation and function. Studies show that during T cell activation, glucose demand increases significantly, primarily through upregulation of glucose transporters such as glucose transporter 1 (GLUT1) [69]. T cells with specific GLUT1 deletion exhibit impaired CD4+T cell activation, clonal expansion, and survival [70]. When glucose is scarce, CD8+ T cell function is inhibited, leading to reduced production of IFN-γ, granzyme, and perforin [71, 72].
Glycolysis not only provides energy for T cells but also regulates downstream effector functions, such as cytokine production, influencing T cell immune responses [73]. Enhanced glycolysis can promote effector T cells (Teff) to produce more inflammatory cytokines, such as IFN-γ and TNF-α, which are drivers of chronic inflammatory responses [74]. Glycolysis varies among different T cell subtypes: CD4+ T cells differentiate into Th1 and Th17 cells in a glycolysis-dependent manner, while Th2 cells exhibit higher glycolytic activity [75, 76]. In CP and pancreatic fibrosis, T cell glycolysis may exacerbate inflammation, leading to tissue damage and fibrosis progression. This metabolic reprogramming not only affects immune cell activity but also regulates PSC activation through glucose metabolism-related signaling pathways, aggravating pancreatic fibrosis [6]. Additionally, glucose metabolism changes may impact Tregs in the pancreas [77]. Tregs play a vital role in maintaining tissue homeostasis and reducing inflammation, but glucose metabolism disorders may inhibit Treg activity, worsening the condition in CP.
Furthermore, T cell glucose metabolism reprogramming may influence their persistence and effector function in the pancreas. T cells with high glycolytic activity in chronic inflammation may exacerbate pancreatic fibrosis, as lactate promotes fibroblast activation and excessive extracellular matrix deposition [78, 79].
3.2.2 Amino Acid Metabolism
Amino acid metabolism is essential for T cell proliferation and differentiation, particularly glutamine metabolism. Glutamine plays a crucial role in T cell activation by increasing the expression of transport proteins such as alanine-serine-cysteine transporter 2 (ASCT2) to facilitate glutamine uptake [80]. Glutamine metabolism provides energy for T cells and regulates T cell function through the mTORC1 signaling pathway [81]. Changes in amino acid metabolism are equally important in pancreatic diseases. Chronic inflammation leads to imbalances in amino acid metabolism, affecting T cells and other immune cells. For example, glutamine depletion can impair T cell polarization, exacerbating inflammation and fibrosis progression. Intermediate metabolites of glutamine metabolism, such as α-ketoglutarate, play roles in energy metabolism and may also influence gene expression through epigenetic modifications, such as DNA methylation, impacting T cell-mediated immune responses [82]. This metabolic-epigenetic interaction may be crucial for maintaining chronic inflammation and exacerbating pancreatic fibrosis [50].
3.2.3 Lipid Metabolism
T cell lipid metabolism exhibits significant variation in different functional states. Resting T cells primarily rely on fatty acid oxidation (FAO) to maintain energy balance, whereas effector T cells switch to glycolysis and lipid synthesis upon activation to support rapid proliferation and effector functions [83]. Lipid metabolism is essential for T cell function, as activated T cells require large amounts of fatty acid synthesis to support cell membrane construction and signal transduction [75]. In CP, lipid metabolism may influence T cell survival and function in the inflammatory microenvironment, contributing to disease progression. For example, increased FAO may help maintain the suppressive function of Tregs, partially limiting pancreatic inflammation [84]. Further research suggests that changes in lipid metabolism pathways may exacerbate inflammation and fibrosis by affecting lipid deposition in pancreatic tissue [85].
The reprogramming of these metabolic pathways forms the foundation for T cell functional changes and plays a critical role in the inflammatory response and fibrosis progression in CP. This metabolic-immune interaction presents potential targets for developing new therapeutic strategies against chronic pancreatitis.
4. Metabolic Reprogramming in Pancreatic Stellate Cells
Pancreatic fibrosis is a key pathological feature of CP, playing a critical role in the disease's progression [86, 87]. PSCs are mesenchymal cells that account for only 4-7% of all parenchymal cells in a healthy pancreas and exist in a quiescent state. These cells contain lipid droplets rich in vitamin A [88, 89]. The activation of PSCs is a crucial factor in pancreatic fibrosis. Once activated, PSCs transform into a myofibroblast-like phenotype, characterized by increased expression of α-smooth muscle actin (α-SMA), loss of lipid droplets, enhanced secretion of cytokines and ECM components, and increased migration and proliferation capabilities [90, 91]. The sustained activation of PSCs leads to significant ECM overproduction, resulting in an imbalance between ECM synthesis and degradation, ultimately leading to fibrosis [92, 87, 31]. An in-depth exploration of the metabolic reprogramming of PSCs could provide valuable insights into the underlying mechanisms of fibrosis in CP [Figure 2].
Metabolic reprogramming of pancreatic stellate cells in chronic pancreatitis-associated fibrosis. During chronic pancreatitis (CP), pancreatic stellate cells (PSCs) are activated by inflammatory and fibrogenic stimuli, undergoing a profound metabolic shift to support their transformation into myofibroblast-like cells. These activated PSCs exhibit enhanced glycolysis, lipid β-oxidation, and glutaminolysis to meet elevated bioenergetic and biosynthetic demands. Hypoxia, a common feature of fibrotic pancreatic tissue, induces the stabilization of hypoxia-inducible factor 1-alpha (HIF-1α), which promotes glycolytic flux and facilitates glycine biosynthesis for collagen production. Pyruvate kinase M2 (PKM2) activity further supports this glycolytic shift, resulting in increased lactate accumulation. Glutamine uptake and metabolism via glutaminase-1 (GLS1) generate glutamate and α-ketoglutarate (α-KG), fueling the tricarboxylic acid (TCA) cycle and driving proline synthesis, a critical amino acid for collagen assembly. Concurrently, fatty acid uptake and β-oxidation contribute acetyl-CoA to the TCA cycle, further amplifying metabolic flux. Additionally, lipid signaling plays a role in PSC activation. Very-low-density lipoproteins (VLDL) bind to their receptors (VLDLR) on PSC membranes, upregulating early B-cell factor 2 (EBF2) and stimulating the release of IL-33, a cytokine that activates type 2 immune responses. TRPV4 expression is also enhanced, linking lipid metabolism to mechanotransduction and promoting fibrogenic gene expression. These coordinated metabolic programs endow PSCs with sustained proliferative and fibrotic capacity, highlighting potential therapeutic targets to disrupt fibrosis progression in CP. PKM, pyruvate kinase M; HIF-1α, hypoxia-inducible factor-1 alpha; α-KG, α-ketoglutarate; TCA, tricarboxylic acid cycle; VLDL, very low-density lipoprotein; VLDLR, very low-density lipoprotein receptor; TRPV4, transient receptor potential vanilloid type 4; EBF2, early B-cell factor 2; GLS1, glutaminase 1.
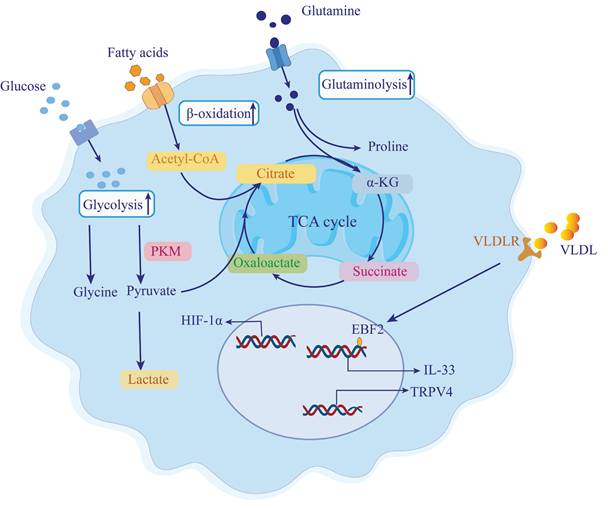
4.1 Increased Glycolysis
PSCs are activated in response to pancreatic tissue injury and stress, acquiring a myofibroblast-like state. This activation alters PSC metabolism, elevating glycolysis levels in CP and enhancing the Warburg effect through HIF-1α, thus promoting CP progression [93]. During fibroblast differentiation into myofibroblasts, aerobic glycolysis is the primary energy source [94]. In fibrosis, aerobic glycolysis provides the necessary ATP, amino acids, and nucleotides for ECM synthesis [95]. Inhibiting aerobic glycolysis can suppress fibroblast activation and fibrosis [96]. For example, myofibroblasts produce excess glycine during fibrosis to meet the demand for collagen production and secretion, and this glycine resynthesis depends on glycolysis. Glycolytic intermediates such as 3-phosphoglycerate are converted into serine, which is then converted into glycine [97, 98]. PKM converts phosphoenolpyruvate (PEP) into pyruvate during glycolysis, and PKM2 reduces pyruvate kinase activity, promoting the accumulation of glycolytic intermediates, inducing glycine synthesis, and enhancing collagen secretion and expression [99]. Using the PKM2 activator TEPP46 to convert PKM2 dimers into tetramers reduces serine and glycine expression in fibrosis models, decreasing collagen deposition in the pancreas and reversing cerulein-induced fibrosis [99]. Resveratrol (RSV) inhibits PSC activation by suppressing miR-21, reducing lactate, Glut1, hexokinase 2 (HK2), PKM2, and lactate dehydrogenase A (LDHA) expression, thus blocking ROS-induced glycolysis in PSCs [100].
4.2 Amino Acid Metabolism
The proliferation of PSCs relies on glutamine catabolism, which provides the necessary bioenergetic and biosynthetic resources for activated PSCs. These activated PSCs exhibit elevated levels of glutaminase 1 (GLS1), a key enzyme in glutamine metabolism. High GLS1 expression enhances glutamine catabolism, resulting in increased production of α-KG [101]. α-KG enters the tricarboxylic acid (TCA) cycle, boosting ATP production and supporting the synthesis of lipids, amino acids, and nucleic acids [99]. Additionally, α-KG can be enzymatically converted into succinate, and the elevated levels of succinate promote glycolysis and stabilize HIF-1α, which facilitates myofibroblast differentiation [102]. Glutamine also contributes to collagen synthesis by providing proline, thereby promoting fibrogenesis [103]. It is essential for maintaining the proliferative phenotype of PSCs, with Yap-Myc signaling regulating PSC transdifferentiation by modulating glutamine metabolism. Inhibition of mitochondrial respiration and PSC growth was observed with the Yap inhibitor verteporfin, which reduced the expression of GLS1, Col1α1, and MMP2 in PSCs. Similarly, the Myc inhibitor MYCi975 also impaired PSC growth and mitochondrial respiration [101].
4.3 Lipid Metabolism
Lipid metabolism plays an important role in PSC activation and pancreatic fibrosis. Triglycerides (TGs) are lipid components that are transported to tissue cells via very-low-density lipoproteins (VLDLs), where they undergo catabolism to produce glycerol and fatty acids. These fatty acids are further oxidized to produce ATP, carbon dioxide, and water [104]. Compared to glycolysis, lipid metabolism generates a large amount of ATP. Studies have shown that lipid metabolism disorders occur in pancreatitis, with lipid accumulation exacerbating inflammation [105, 106]. However, the precise mechanism between lipid metabolism and pancreatic fibrosis remains unclear.
Consuming a high-fat diet can elevate pancreatic free fatty acids (FFAs) and trigger lipid peroxidation processes [107]. In mice fed a long-term high-fat diet, metabolites promote TRPV4 expression and enhance PSC activation [108]. In mice specifically expressing the R122H mutant, a high-fat diet induces more severe pancreatitis, including greater DNA damage, apoptosis, and collagen deposition [109]. Wistar rats fed special feed (MB-3, a high-protein, high-fat diet) for 12 weeks develop fibrosis [110]. Recent studies have shown that increased VLDLR expression in PSCs and the uptake of TG-rich lipoproteins upregulate lipid metabolism, increase IL-33 secretion, and activate type 2 immune responses, thereby inducing PSC activation and ultimately causing pancreatic fibrosis [111].
In conclusion, abnormal glucose, amino acid, and lipid metabolism provide energy and nutrients to PSCs, promoting pancreatic fibrosis. Therefore, further research into energy metabolism irregularities in CP and identifying metabolic pathway targets could enhance treatment options.
5. Metabolic Reprogramming as a Potential Target for Fibrosis Therapy
Currently, there are no effective drugs or therapies for fibrosis associated with chronic pancreatitis. However, research has revealed that aberrant reprogramming of metabolic pathways not only contributes to persistent damage to pancreatic cells but also exacerbates the fibrosis process. Consequently, targeting metabolic reprogramming presents a promising new strategy for treating fibrosis in chronic pancreatitis. By modulating specific metabolic pathways, it may be possible to slow down or even reverse fibrosis progression, ultimately improving clinical outcomes for patients [Figure 3].
Metabolism-targeted anti-fibrotic therapies in chronic pancreatitis. This schematic highlights four therapeutic strategies targeting metabolic pathways in chronic pancreatitis (CP), applied across three key cell types involved in fibrosis: T cells, macrophages, and pancreatic stellate cells (PSCs). Mitochondrial-targeted antioxidants such as mitoquinone reduce oxidative stress and mitochondrial dysfunction, limiting ROS production, acinar cell death, and PSC activation. Glycolysis inhibitors like melatonin and resveratrol modulate macrophage and T cell phenotypes, suppressing inflammation (M1 to M2 polarization in macrophages) and reducing fibrosis-related PSC activation. Glutamine metabolism inhibitors target PSC metabolic reprogramming, limiting their activation, proliferation, and migration by disrupting key biosynthetic pathways such as α-ketoglutarate and proline synthesis. Finally, lipid metabolism modulation reduces fatty acid oxidation in PSCs and macrophages, limiting M2 macrophage polarization and decreasing PSC activation and ECM production. These therapies target distinct metabolic pathways but collectively address the metabolic vulnerabilities contributing to fibrogenesis in CP, with cell-specific targeting strategies offering the potential to minimize off-target effects and improve therapeutic efficacy. Abbreviations: PSC, pancreatic stellate cell; ROS, reactive oxygen species; M1, classically activated macrophage; M2, alternatively activated macrophage; α-KG, α-ketoglutarate; ECM, extracellular matrix.
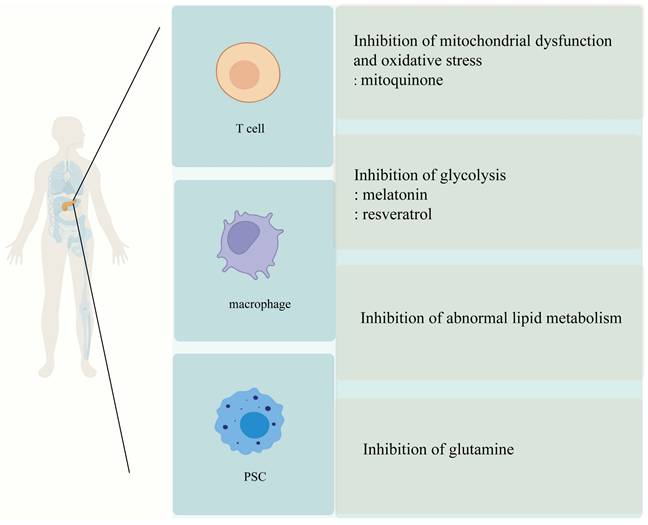
5.1 Regulation of Glucose Metabolism
Glycolysis is upregulated in both pancreatic stellate cells (PSCs) and pro-inflammatory immune cells under fibrotic conditions. Targeting glucose metabolism can disrupt the energy supply and biosynthetic pathways necessary for cell activation and ECM production, making it a common and viable antifibrotic strategy.
Glucose metabolism is central to the energy demands of activated PSCs in fibrotic tissues. Under hypoxic conditions, PSCs exhibit enhanced glycolysis, characterized by elevated expression of Glut1, phosphofructokinase (PFK), and lactate dehydrogenase (LDH). Melatonin, an indoleamine synthesized by the mammalian pineal gland at night, has protective effects in improving inflammation, oxidative stress, and pancreatic fibrosis [112]. In PSCs treated with melatonin, glycolysis and glucose metabolism decrease. Melatonin regulates the MAPK and PI3K/AKT/mTOR signaling pathways, reducing PSC proliferation and viability. It also regulates the expression of mitochondrial fusion proteins responsible for cellular energy and metabolic performance [113]. During pancreatic fibrosis, low oxygen utilization creates a hypoxic environment. Melatonin activates the p44/42/mTOR/p70S6K signaling pathway, inhibiting PSC proliferation and reducing α-SMA and type I collagen expression. Under hypoxic conditions, PSC glycolysis and glucose metabolism significantly increase, with elevated expression of Glut1, phosphofructokinase (PFK), and lactate dehydrogenase (LDH). After melatonin treatment, the expression of Glut-1, PFK, and LDH decreases. Therefore, melatonin inhibits PSC adaptation to mitochondrial energy supply under hypoxic conditions [114]. Melatonin induces ROS production in mitochondria and the cytoplasm in a concentration-dependent manner, reducing PSC viability in a time- and concentration-dependent manner, leading to oxidative state changes in PSCs [115]. Simultaneously, pharmacological concentrations of melatonin regulate the cell cycle, promote apoptosis, and reduce PSC proliferation under hypoxic conditions by activating caspase-3 and altering the expression of cyclins A and D [116]. The melatonin receptor antagonist Luzindole regulates the p44/42/p38MAPKs signaling pathway, inducing endoplasmic reticulum stress and reducing the viability of rat pancreatic stellate cells [117]. RSV, a natural polyphenol with multiple pharmacological effects, effectively inhibits H2O2-induced PSC activation, invasion, and migration by regulating miR-21 and suppressing ROS-induced PSC glycolysis. RSV also inhibits pancreatic cancer cell invasion and migration by suppressing ROS/miR-21 in PSCs [100]. Hypoxia activates PSCs through HIF-1, and activated PSCs release IL-6, vascular endothelial growth factor A, and stromal cell-derived factor 1, promoting pancreatic cancer invasion, epithelial-mesenchymal transition, and inhibiting apoptosis. In the KPC mouse model, RSV inhibits hypoxia-induced PSC activation, blocking the interaction between PSCs and pancreatic cancer cells and disrupting the malignant evolution of pancreatic cancer [118].
Targeting glucose metabolism effectively limits PSC activation and reduces profibrotic signaling. Melatonin and RSV represent potent modulators that act on both energy production and inflammatory feedback, validating glycolysis as a therapeutic axis in fibrotic progression.
5.2 Targeting Amino Acid Metabolism
Amino acid metabolism supports redox balance, biosynthesis, and signaling in both PSCs and immune cells. Dysregulation in glutamine and cysteine pathways contributes to oxidative stress and fibrogenic transformation. Intervening in these pathways provides opportunities to disrupt multiple profibrotic mechanisms.
Amino acid imbalance is common in CP due to impaired exocrine function. Among amino acids, glutamine and cysteine metabolism are particularly relevant to PSC activation and redox homeostasis. In patients with CP, serum levels of essential and aromatic amino acids are reduced, possibly due to exocrine pancreatic dysfunction. The mechanisms linking amino acid metabolism to CP development are still under investigation. N-acetylcysteine (NAC) serves as a precursor for the biosynthesis of glutathione (GSH), an antioxidant with properties that can reduce oxidative stress. NAC supplementation increases glutathione levels and reduces oxidative stress. Additionally, NAC can inhibit PSCs activation, and when combined with pioglitazone, it helps maintain PSCs in a quiescent-like state [119]. Glutathione, composed of glutamate, glycine, and cysteine, is an antioxidant that inhibits PSC activation and proliferation by blocking the ROS/TGFβ/SMAD signaling pathway, thereby reducing pancreatic fibrosis [120]. Up-regulation of glutamine catabolism is important for PSC activation, proliferation, and migration, so inhibiting glutamine catabolism may be an effective way to inhibit pancreatic fibrosis. Yap-Myc signaling regulates glutamine catabolism to activate PSCs, and inhibition of glutamine catabolism inhibits cell growth and fibrosis formation [101]. Glutamine in PSC promotes the biological function of cancer cells, and treatment with palmatine (PMT) for 48 h reduced the expression of GLI1 and GLI2 in PSC and downstream target COL1A1 expression, disrupting glutamine-mediated PCC action. PMT in combination with gemcitabine inhibited stellate cell and cancer cell growth [121].
Amino acid metabolism is essential to maintaining the redox state and biosynthetic demand of activated PSCs. Agents such as NAC and PMT interfere with glutamine and GSH pathways, providing both antifibrotic and anti-inflammatory benefits.
5.3 Targeting Lipid Metabolism
Lipid metabolism affects PSC activation through nuclear receptors like PPARγ and modulates immune cell polarization via fatty acid oxidation. Regulating lipid metabolism bridges stromal and immune axes in pancreatic fibrosis.
Lipid metabolic reprogramming influences both PSC activation and macrophage polarization in CP. PPARγ is a transcription factor that plays a critical role in adipogenesis. Exosomes derived from acinar cells, containing miR-13a-3p, act on PSCs by targeting PPARγ, leading to PSC activation and collagen synthesis, thus driving pancreatic fibrosis [122]. Pioglitazone, a PPARγ agonist, reduces cerulein-induced pancreatitis, inhibits IL-1β production and release, and enhances pancreatic HSP70 expression [123]. During the early stages of chronic pancreatitis, saturated fatty acids may inhibit fibrosis through the PERK pathway [124]. Fatty acid metabolism provides energy for M2 macrophages. Short-chain fatty acids (SCFAs) produced by G+ bacteria help maintain intestinal barrier integrity, regulate M2 macrophage infiltration and polarization, and alleviate pancreatic fibrosis [125]. Under oxidative stress conditions, fatty acid peroxidation increases MDA levels. Intervening in lipid metabolic disorders offers another therapeutic route to protect pancreatic tissue from fibrotic remodeling.
Lipid metabolic interventions—via PPARγ agonists, SCFAs, or saturated fatty acids—have pleiotropic effects on PSCs and immune cells. Their ability to suppress inflammation and ECM production highlights lipid regulation as a versatile antifibrotic approach.
5.4 Antioxidant Therapies
Oxidative stress is a shared upstream driver of PSC and immune cell activation. Targeting mitochondrial ROS and redox imbalance is a foundational strategy to disrupt fibrotic signaling.
Mitochondria are the primary source of ROS within cells [126]. Using mitochondria-targeted antioxidants can significantly reduce oxidative stress (OS) [127], preventing OS from activating PSCs through the AMPK signaling pathway. This leads to the overexpression of α-SMA, enhanced cell proliferation, and migration [128, 129].
MitoQ is a well-studied mitochondria-targeted antioxidant and one of the most extensively researched compounds of its kind. Clinical studies on liver disease models have consistently shown that MitoQ is highly effective in treating various liver diseases [130-132]. In a cerulein-induced acute pancreatitis mouse model, two intraperitoneal injections of MitoQ inhibited ROS production, preventing protective apoptosis and promoting pancreatic acinar cell death, worsening vacuole damage [133]. Recently, another study demonstrated that oral administration of MitoQ downregulated fibrosis-related genes in CP mice, including type I collagen (Col I), type III collagen (Col III), and tissue inhibitor of metalloproteinases 1 (TIMP1). Additionally, SOD enzyme mRNA expression levels and activity were elevated in CP mice and HPSCs [134]. Superoxide dismutase (SOD) catalyzes the reduction of superoxide ions to H2O2, which can be decomposed into water by peroxidases. Decreased SOD activity exacerbates ROS-induced damage, leading to increased levels of DNA, lipid, and protein damage caused by free radicals [126, 135]. Therefore, when present in appropriate concentrations, MitoQ is expected to alleviate pancreatic fibrosis by reducing ROS levels and inhibiting PSC activation. Moreover, it reduces intracellular OS by enhancing SOD expression and activity, accelerating the clearance of oxygen free radicals. By reducing ROS levels, enhancing antioxidant defenses, and inhibiting PSC activation, MitoQ and related compounds hold potential as adjunct therapies for fibrosis prevention.
Mitochondria-targeted antioxidants like MitoQ, by restoring redox homeostasis, inhibit fibrotic pathways common to PSCs and immune cells. Antioxidant therapy serves as a critical upstream intervention in fibrosis prevention.
6. Conclusion
CP is marked by a complex interplay of cellular and molecular mechanisms that lead to pancreatic fibrosis, significantly impacting patients' quality of life and increasing their risk of developing pancreatic cancer [136]. The recent focus on metabolic reprogramming in PSCs and immune cells offers promising avenues for therapeutic intervention. By understanding how glucose, lipid, and amino acid metabolism contribute to fibrosis, we can identify potential targets to slow or reverse disease progression.
The reprogramming of glucose metabolism, particularly the enhancement of glycolysis in PSCs, is a critical factor in fibrosis. Targeting key glycolytic enzymes such as PKM2 could reduce excessive extracellular matrix production and fibrosis. Similarly, modulating lipid metabolism, which provides energy for PSCs and immune cells during chronic inflammation, presents another potential strategy. The role of fatty acid oxidation and the activation of PPARγ in reducing fibrosis highlights the therapeutic potential of targeting lipid metabolism.
Amino acid metabolism, especially glutamine metabolism, also plays a crucial role in PSC and immune cell function, influencing fibrosis through the provision of essential metabolites and the regulation of signaling pathways. Inhibiting glutamine metabolism may help reduce fibrosis and improve clinical outcomes.
As we advance in the exploration of metabolic reprogramming in CP, we open up promising avenues for the development of innovative therapeutic strategies. Future research should focus on elucidating the molecular mechanisms that drive metabolic alterations in PSCs and immune cells, with an emphasis on translating these insights into effective clinical interventions. Combining targeted metabolic inhibitors with existing anti-inflammatory and antioxidant therapies could offer a more comprehensive approach to managing CP, potentially decelerating disease progression and enhancing patient outcomes.
Furthermore, as we deepen our understanding of the intricate metabolic changes in CP, there is a significant opportunity to discover biomarkers that could predict disease progression and therapeutic response. These biomarkers could be instrumental in advancing personalized medicine, allowing for more tailored and effective treatment strategies for patients suffering from chronic pancreatitis.
In summary, integrating metabolic insights into the treatment of CP holds immense potential to revolutionize the therapeutic landscape of this challenging disease, providing new hope for better management and an improved quality of life for patients.
Acknowledgements
Funding
This study was supported by the National Natural Science Foundation of China (Grant NO.:82270676), the Taishan Young Scholars Program of Shandong Province (NO. tsqn202306395), and the 2023 Qingdao Technology Benefiting Demonstration Project (Grant No.:23-2-8-smjk-8-nsh).
Author contributions
HL and SG drafted the manuscript and prepared all the figures. YC and YX critically revised the manuscript. XY and XL conceived the study, supervised the work, and critically revised the manuscript. All authors approved the final submitted version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Beyer G, Habtezion A, Werner J, Lerch MM, Mayerle J. Chronic pancreatitis. Lancet. 2020Aug15;396(10249):499-512
2. Vege SS, Chari ST. Chronic Pancreatitis. N Engl J Med. 2022Mar3;386(9):869-78
3. Hines OJ, Pandol SJ. Management of chronic pancreatitis. Bmj. 2024Feb26;384:e070920
4. An J, Jiang T, Qi L, Xie K. Acinar cells and the development of pancreatic fibrosis. Cytokine Growth Factor Rev. 2023 Jun-Aug;71-72:40-53
5. Hu F, Lou N, Jiao J, Guo F, Xiang H, Shang D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. Biomed Pharmacother. 2020Nov;131:110693
6. Zhang Y, Zhang WQ, Liu XY, Zhang Q, Mao T, Li XY. Immune cells and immune cell-targeted therapy in chronic pancreatitis. Front Oncol. 2023;13:1151103
7. Apte M, Pirola RC, Wilson JS. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr Opin Gastroenterol. 2015Sep;31(5):416-23
8. Jaster R, Emmrich J. Crucial role of fibrogenesis in pancreatic diseases. Best Pract Res Clin Gastroenterol. 2008;22(1):17-29
9. Bynigeri RR, Jakkampudi A, Jangala R, Subramanyam C, Sasikala M, Rao GV. et al. Pancreatic stellate cell: Pandora's box for pancreatic disease biology. World J Gastroenterol. 2017Jan21;23(3):382-405
10. Wang YC, Mao XT, Sun C, Wang YH, Zheng YZ, Xiong SH. et al. Pancreas-directed AAV8-hSPINK1 gene therapy safely and effectively protects against pancreatitis in mice. Gut. 2024Jun6;73(7):1142-55
11. O'Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016Jan11;213(1):15-23
12. Krejčí A. Metabolic sensors and their interplay with cell signalling and transcription. Biochem Soc Trans. 2012Apr;40(2):311-23
13. Hart PA, Conwell DL. Chronic Pancreatitis: Managing a Difficult Disease. Am J Gastroenterol. 2020Jan;115(1):49-55
14. Møller SH, Hsueh PC, Yu YR, Zhang L, Ho PC. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging. Cell Metab. 2022Mar1;34(3):378-95
15. Cortés M, Brischetto A, Martinez-Campanario MC, Ninfali C, Domínguez V, Fernández S. et al. Inflammatory macrophages reprogram to immunosuppression by reducing mitochondrial translation. Nat Commun. 2023Nov17;14(1):7471
16. Singh L, Arora SK, Bakshi DK, Majumdar S, Wig JD. Potential role of CXCL10 in the induction of cell injury and mitochondrial dysfunction. Int J Exp Pathol. 2010Jun;91(3):210-23
17. Saluja A, Dudeja V, Dawra R, Sah RP. Early Intra-Acinar Events in Pathogenesis of Pancreatitis. Gastroenterology. 2019May;156(7):1979-93
18. Liu K, Liu J, Zou B, Li C, Zeh HJ, Kang R. et al. Trypsin-Mediated Sensitization to Ferroptosis Increases the Severity of Pancreatitis in Mice. Cell Mol Gastroenterol Hepatol. 2022;13(2):483-500
19. Choi J, Oh TG, Jung HW, Park KY, Shin H, Jo T. et al. Estrogen-Related Receptor γ Maintains Pancreatic Acinar Cell Function and Identity by Regulating Cellular Metabolism. Gastroenterology. 2022Jul;163(1):239-56
20. Tandon RK, Garg PK. Oxidative stress in chronic pancreatitis: pathophysiological relevance and management. Antioxid Redox Signal. 2011Nov15;15(10):2757-66
21. Huang H, Liu Y, Daniluk J, Gaiser S, Chu J, Wang H. et al. Activation of nuclear factor-κB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology. 2013Jan;144(1):202-10
22. Kong F, Pan Y, Wu D. Activation and Regulation of Pancreatic Stellate Cells in Chronic Pancreatic Fibrosis: A Potential Therapeutic Approach for Chronic Pancreatitis. Biomedicines. 2024 Jan 4;12(1)
23. Teodoro JS, Duarte FV, Gomes AP, Varela AT, Peixoto FM, Rolo AP. et al. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: a possible role for SirT3 activation. Mitochondrion. 2013Nov;13(6):637-46
24. Rahal A, Kumar A, Singh V, Yadav B, Tiwari R, Chakraborty S. et al. Oxidative stress, prooxidants, and antioxidants: the interplay. Biomed Res Int. 2014;2014:761264
25. Ryu GR, Lee E, Chun HJ, Yoon KH, Ko SH, Ahn YB. et al. Oxidative stress plays a role in high glucose-induced activation of pancreatic stellate cells. Biochem Biophys Res Commun. 2013Sep20;439(2):258-63
26. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020Jun15;877:173090
27. Bian Z, Gong Y, Huang T, Lee CZW, Bian L, Bai Z. et al. Deciphering human macrophage development at single-cell resolution. Nature. 2020Jun;582(7813):571-76
28. Luo M, Zhao F, Cheng H, Su M, Wang Y. Macrophage polarization: an important role in inflammatory diseases. Front Immunol. 2024;15:1352946
29. Lee B, Namkoong H, Yang Y, Huang H, Heller D, Szot GL. et al. Single-cell sequencing unveils distinct immune microenvironments with CCR6-CCL20 crosstalk in human chronic pancreatitis. Gut. 2022Sep;71(9):1831-42
30. Hashimoto A, Karim MR, Kuramochi M, Izawa T, Kuwamura M, Yamate J. Characterization of Macrophages and Myofibroblasts Appearing in Dibutyltin Dichloride-Induced Rat Pancreatic Fibrosis. Toxicol Pathol. 2020Apr;48(3):509-23
31. Zheng M, Li H, Sun L, Brigstock DR, Gao R. Interleukin-6 participates in human pancreatic stellate cell activation and collagen I production via TGF-β1/Smad pathway. Cytokine. 2021Jul;143:155536
32. Duan LF, Xu XF, Zhu LJ, Liu F, Zhang XQ, Wu N. et al. Dachaihu decoction ameliorates pancreatic fibrosis by inhibiting macrophage infiltration in chronic pancreatitis. World J Gastroenterol. 2017Oct28;23(40):7242-52
33. Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ. et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. 2015May18;6:7158
34. Wu N, Xu XF, Xin JQ, Fan JW, Wei YY, Peng QX. et al. The effects of nuclear factor-kappa B in pancreatic stellate cells on inflammation and fibrosis of chronic pancreatitis. J Cell Mol Med. 2021Feb;25(4):2213-27
35. Palathingal Bava E, George J, Iyer S, Sahay P, Tarique M, Jain T. et al. Pirfenidone ameliorates chronic pancreatitis in mouse models through immune and cytokine modulation. Pancreatology. 2022Jun;22(5):553-63
36. Kolliniati O, Ieronymaki E, Vergadi E, Tsatsanis C. Metabolic Regulation of Macrophage Activation. J Innate Immun. 2022;14(1):51-68
37. Nareika A, He L, Game BA, Slate EH, Sanders JJ, London SD. et al. Sodium lactate increases LPS-stimulated MMP and cytokine expression in U937 histiocytes by enhancing AP-1 and NF-kappaB transcriptional activities. Am J Physiol Endocrinol Metab. 2005Oct;289(4):E534-42
38. Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P. et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010Jul1;185(1):605-14
39. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J. 1986Oct1;239(1):121-5
40. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol. 2019;10:1462
41. Yin M, O'Neill LAJ. The role of the electron transport chain in immunity. Faseb j. 2021Dec;35(12):e21974
42. Takeda N, O'Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C. et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010Mar1;24(5):491-501
43. Lin Y, Chen Y, Feng W, Hua R, Zhang J, Huo Y. et al. Neddylation pathway alleviates chronic pancreatitis by reducing HIF1α-CCL5-dependent macrophage infiltration. Cell Death Dis. 2021Mar15;12(3):273
44. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE. et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015Jan6;21(1):65-80
45. Gao S, Li X, Jiang Q, Liang Q, Zhang F, Li S. et al. PKM2 promotes pulmonary fibrosis by stabilizing TGF-β1 receptor I and enhancing TGF-β1 signaling. Sci Adv. 2022Sep23;8(38):eabo0987
46. Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R. et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011May27;145(5):732-44
47. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G. et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013Apr11;496(7444):238-42
48. Wang J, Yang P, Yu T, Gao M, Liu D, Zhang J. et al. Lactylation of PKM2 Suppresses Inflammatory Metabolic Adaptation in Pro-inflammatory Macrophages. Int J Biol Sci. 2022;18(16):6210-25
49. Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients. 2018 Oct 23;10(11)
50. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S. et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017Sep;18(9):985-94
51. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E. et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015Mar17;42(3):419-30
52. Pekarova M, Lojek A. The crucial role of l-arginine in macrophage activation: What you need to know about it. Life Sci. 2015Sep15;137:44-8
53. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW. et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J Immunol. 2001Dec1;167(11):6533-44
54. Hu X, Liu G, Hou Y, Shi J, Zhu L, Jin D. et al. Induction of M2-like macrophages in recipient NOD-scid mice by allogeneic donor CD4(+)CD25(+) regulatory T cells. Cell Mol Immunol. 2012Nov;9(6):464-72
55. Sun C, Sun L, Ma H, Peng J, Zhen Y, Duan K. et al. The phenotype and functional alterations of macrophages in mice with hyperglycemia for long term. J Cell Physiol. 2012Apr;227(4):1670-9
56. Yan J, Horng T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020Dec;30(12):979-89
57. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275-97
58. Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S. et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007Aug;6(2):137-43
59. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L. et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007Jun28;447(7148):1116-20
60. Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE. et al. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002Mar1;168(5):2508-15
61. Shimizu K, Shiratori K, Hayashi N, Kobayashi M, Fujiwara T, Horikoshi H. Thiazolidinedione derivatives as novel therapeutic agents to prevent the development of chronic pancreatitis. Pancreas. 2002Mar;24(2):184-90
62. Hisada S, Shimizu K, Shiratori K, Kobayashi M. Peroxisome proliferator-activated receptor gamma ligand prevents the development of chronic pancreatitis through modulating NF-kappaB-dependent proinflammatory cytokine production and pancreatic stellate cell activation. Rocz Akad Med Bialymst. 2005;50:142-7
63. Madden MZ, Rathmell JC. The Complex Integration of T-cell Metabolism and Immunotherapy. Cancer Discov. 2021Jul;11(7):1636-43
64. Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev. 2010Jul;236:190-202
65. Xue J, Sharma V, Habtezion A. Immune cells and immune-based therapy in pancreatitis. Immunol Res. 2014May;58(2-3):378-86
66. Hunger RE, Mueller C, Z'Graggen K, Friess H, Büchler MW. Cytotoxic cells are activated in cellular infiltrates of alcoholic chronic pancreatitis. Gastroenterology. 1997May;112(5):1656-63
67. Jupp J, Mansour S, Johnson CD, Sanderson J, Fine D, Gadola S. T-cell populations in chronic pancreatitis. Pancreatology. 2015 Jul-Aug;15(4):311-2
68. Schmitz-Winnenthal H, Pietsch DH, Schimmack S, Bonertz A, Udonta F, Ge Y. et al. Chronic pancreatitis is associated with disease-specific regulatory T-cell responses. Gastroenterology. 2010Mar;138(3):1178-88
69. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ. et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008Apr1;180(7):4476-86
70. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D. et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014Jul1;20(1):61-72
71. Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. 2005Apr15;174(8):4670-7
72. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008Sep;38(9):2438-50
73. Liu S, Liao S, Liang L, Deng J, Zhou Y. The relationship between CD4(+) T cell glycolysis and their functions. Trends Endocrinol Metab. 2023Jun;34(6):345-60
74. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015Aug24;212(9):1345-60
75. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011Dec23;35(6):871-82
76. Shehade H, Acolty V, Moser M, Oldenhove G. Cutting Edge: Hypoxia-Inducible Factor 1 Negatively Regulates Th1 Function. J Immunol. 2015Aug15;195(4):1372-6
77. Soto-Heredero G, Gómez de Las Heras MM, Gabandé-Rodríguez E, Oller J, Mittelbrunn M. Glycolysis - a key player in the inflammatory response. Febs j. 2020Aug;287(16):3350-69
78. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y. et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019Oct;574(7779):575-80
79. Cui H, Xie N, Banerjee S, Ge J, Jiang D, Dey T. et al. Lung Myofibroblasts Promote Macrophage Profibrotic Activity through Lactate-induced Histone Lactylation. Am J Respir Cell Mol Biol. 2021Jan;64(1):115-25
80. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X. et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014May15;40(5):692-705
81. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC. et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell. 2018Dec13;175(7):1780-95.e19
82. Wilfahrt D, Delgoffe GM. Metabolic waypoints during T cell differentiation. Nat Immunol. 2024Feb;25(2):206-17
83. Sharabi A, Tsokos GC. T cell metabolism: new insights in systemic lupus erythematosus pathogenesis and therapy. Nat Rev Rheumatol. 2020Feb;16(2):100-12
84. Soriano-Baguet L, Brenner D. Metabolism and epigenetics at the heart of T cell function. Trends Immunol. 2023Mar;44(3):231-44
85. Wu L, Huang X, Ouyang Q, Liu W, Liu S, Huang Y. et al. Serum metabolomics study for acute attack of chronic pancreatitis. Clin Chim Acta. 2023Feb15;541:117251
86. Witt H. Chronic pancreatitis and cystic fibrosis. Gut. 2003 May;52 Suppl 2(Suppl 2):ii31-41
87. Kanikovskiy OE, Pavlyk IV, Oliinyk IV, Mosondz VV. The key role of pancreatic fibrosis severity in the surgical treatment algorithm of patients with chronic pancreatitis. Wiad Lek. 2020;73(2):235-38
88. Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA. et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998Jul;43(1):128-33
89. Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A. et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998Aug;115(2):421-32
90. Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol. 2019;10:1835
91. Hamada S, Matsumoto R, Masamune A. Pancreatic Stellate Cells and Metabolic Alteration: Physiology and Pathophysiology. Front Physiol. 2022;13:865105
92. Jin G, Hong W, Guo Y, Bai Y, Chen B. Molecular Mechanism of Pancreatic Stellate Cells Activation in Chronic Pancreatitis and Pancreatic Cancer. J Cancer. 2020;11(6):1505-15
93. Tao Y, Shao F, Cai M, Liu Z, Peng Y, Huang Q. et al. Activated Pancreatic Stellate Cells Enhance the Warburg Effect to Cause the Malignant Development in Chronic Pancreatitis. Front Oncol. 2021;11:714598
94. Chen Z, Liu M, Li L, Chen L. Involvement of the Warburg effect in non-tumor diseases processes. J Cell Physiol. 2018Apr;233(4):2839-49
95. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009May22;324(5930):1029-33
96. Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q. et al. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol. 2017Sep1;313(3):F561-f75
97. Reid MA, Allen AE, Liu S, Liberti MV, Liu P, Liu X. et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat Commun. 2018Dec21;9(1):5442
98. Fan TWM, Bruntz RC, Yang Y, Song H, Chernyavskaya Y, Deng P. et al. De novo synthesis of serine and glycine fuels purine nucleotide biosynthesis in human lung cancer tissues. J Biol Chem. 2019Sep6;294(36):13464-77
99. Satyanarayana G, Turaga RC, Sharma M, Wang S, Mishra F, Peng G. et al. Pyruvate kinase M2 regulates fibrosis development and progression by controlling glycine auxotrophy in myofibroblasts. Theranostics. 2021;11(19):9331-41
100. Yan B, Cheng L, Jiang Z, Chen K, Zhou C, Sun L. et al. Resveratrol Inhibits ROS-Promoted Activation and Glycolysis of Pancreatic Stellate Cells via Suppression of miR-21. Oxid Med Cell Longev. 2018;2018:1346958
101. Zhang D, Zhao L, Luo M, Lei J, Shao S. Yap-Myc signaling induces pancreatic stellate cell activation through regulating glutaminolysis. Exp Cell Res. 2022Feb1;411(1):113000
102. Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM. et al. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am J Respir Crit Care Med. 2015Dec15;192(12):1462-74
103. Hu S, He W, Wu G. Hydroxyproline in animal metabolism, nutrition, and cell signaling. Amino Acids. 2022Apr;54(4):513-28
104. Schönfeld P, Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res. 2016Jun;57(6):943-54
105. de Oliveira C, Khatua B, Noel P, Kostenko S, Bag A, Balakrishnan B. et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J Clin Invest. 2020Apr1;130(4):1931-47
106. Hong YP, Yu J, Su YR, Mei FC, Li M, Zhao KL. et al. High-Fat Diet Aggravates Acute Pancreatitis via TLR4-Mediated Necroptosis and Inflammation in Rats. Oxid Med Cell Longev. 2020;2020:8172714
107. Zhang X, Cui Y, Fang L, Li F. Chronic high-fat diets induce oxide injuries and fibrogenesis of pancreatic cells in rats. Pancreas. 2008Oct;37(3):e31-8
108. Zhang LP, Ma F, Abshire SM, Westlund KN. Prolonged high fat/alcohol exposure increases TRPV4 and its functional responses in pancreatic stellate cells. Am J Physiol Regul Integr Comp Physiol. 2013May1;304(9):R702-11
109. Huang H, Swidnicka-Siergiejko AK, Daniluk J, Gaiser S, Yao Y, Peng L. et al. Transgenic Expression of PRSS1(R122H) Sensitizes Mice to Pancreatitis. Gastroenterology. 2020Mar;158(4):1072-82.e7
110. Su SB, Motoo Y, Xie MJ, Miyazono K, Sawabu N. Expression of transforming growth factor-beta in spontaneous chronic pancreatitis in the WBN/Kob rat. Dig Dis Sci. 2000Jan;45(1):151-9
111. Yang X, Chen J, Wang J, Ma S, Feng W, Wu Z. et al. Very-low-density lipoprotein receptor-enhanced lipid metabolism in pancreatic stellate cells promotes pancreatic fibrosis. Immunity. 2022Jul12;55(7):1185-99.e8
112. Tanoğlu EG, Tanoğlu A, Meriçöz Aydın M, Esen MF. Melatonin has favorable preventive effects on experimental chronic pancreatitis rat model. Turk J Med Sci. 2021Oct;51(5):2734-40
113. Estaras M, Ortiz-Placin C, Castillejo-Rufo A, Fernandez-Bermejo M, Blanco G, Mateos JM. et al. Melatonin controls cell proliferation and modulates mitochondrial physiology in pancreatic stellate cells. J Physiol Biochem. 2023Feb;79(1):235-49
114. Estaras M, Peña FJ, Tapia JA, Fernandez-Bermejo M, Mateos JM, Vara D. et al. Melatonin modulates proliferation of pancreatic stellate cells through caspase-3 activation and changes in cyclin A and D expression. J Physiol Biochem. 2020May;76(2):345-55
115. Estaras M, Moreno N, Santofimia-Castaño P, Martinez-Morcillo S, Roncero V, Blanco G. et al. Melatonin induces reactive oxygen species generation and changes in glutathione levels and reduces viability in human pancreatic stellate cells. J Physiol Biochem. 2019Jun;75(2):185-97
116. Estaras M, Gonzalez-Portillo MR, Fernandez-Bermejo M, Mateos JM, Vara D, Blanco-Fernandez G. et al. Melatonin Induces Apoptosis and Modulates Cyclin Expression and MAPK Phosphorylation in Pancreatic Stellate Cells Subjected to Hypoxia. Int J Mol Sci. 2021 May 24;22(11)
117. Estaras M, Marchena AM, Fernandez-Bermejo M, Mateos JM, Vara D, Roncero V. et al. The melatonin receptor antagonist luzindole induces the activation of cellular stress responses and decreases viability of rat pancreatic stellate cells. J Appl Toxicol. 2020Nov;40(11):1554-65
118. Xiao Y, Qin T, Sun L, Qian W, Li J, Duan W. et al. Resveratrol Ameliorates the Malignant Progression of Pancreatic Cancer by Inhibiting Hypoxia-induced Pancreatic Stellate Cell Activation. Cell Transplant. 2020 Jan-Dec;29:963689720929987
119. Feng H, Moriyama T, Ohuchida K, Sheng N, Iwamoto C, Shindo K. et al. N-acetyl cysteine induces quiescent-like pancreatic stellate cells from an active state and attenuates cancer-stroma interactions. J Exp Clin Cancer Res. 2021Apr15;40(1):133
120. Zhang J, Bai J, Zhou Q, Hu Y, Wang Q, Yang L. et al. Glutathione prevents high glucose-induced pancreatic fibrosis by suppressing pancreatic stellate cell activation via the ROS/TGFβ/SMAD pathway. Cell Death Dis. 2022May6;13(5):440
121. Chakravarthy D, Muñoz AR, Su A, Hwang RF, Keppler BR, Chan DE. et al. Palmatine suppresses glutamine-mediated interaction between pancreatic cancer and stellate cells through simultaneous inhibition of survivin and COL1A1. Cancer Lett. 2018Apr10;419:103-15
122. Wang Q, Wang H, Jing Q, Yang Y, Xue D, Hao C. et al. Regulation of Pancreatic Fibrosis by Acinar Cell-Derived Exosomal miR-130a-3p via Targeting of Stellate Cell PPAR-γ. J Inflamm Res. 2021;14:461-77
123. Konturek PC, Dembinski A, Warzecha Z, Burnat G, Ceranowicz P, Hahn EG. et al. Pioglitazone, a specific ligand of peroxisome proliferator-activated receptor-gamma, protects pancreas against acute cerulein-induced pancreatitis. World J Gastroenterol. 2005Oct28;11(40):6322-9
124. Lee L, Ito T, Nakamura T, Jensen RT, Igarashi H, Takayanagi R. Antifibrotic Effect of Saturated Fatty Acids via Endoplasmic Reticulum Stress Response in Rat Pancreatic Stellate Cells. Pancreas. 2017Mar;46(3):385-94
125. Pan LL, Ren ZN, Yang J, Li BB, Huang YW, Song DX. et al. Gut microbiota controls the development of chronic pancreatitis: A critical role of short-chain fatty acids-producing Gram-positive bacteria. Acta Pharm Sin B. 2023Oct;13(10):4202-16
126. Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol Sci. 2017Jul;38(7):592-607
127. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. 2017May;1863(5):1066-77
128. Masamune A, Kikuta K, Satoh M, Satoh A, Shimosegawa T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J Pharmacol Exp Ther. 2002Jul;302(1):36-42
129. Kikuta K, Masamune A, Satoh M, Suzuki N, Satoh K, Shimosegawa T. Hydrogen peroxide activates activator protein-1 and mitogen-activated protein kinases in pancreatic stellate cells. Mol Cell Biochem. 2006Oct;291(1-2):11-20
130. Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM. et al. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010Aug;30(7):1019-26
131. Mukhopadhyay P, Horváth B, Zsengellėr Z, Bátkai S, Cao Z, Kechrid M. et al. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants. Free Radic Biol Med. 2012Sep1;53(5):1123-38
132. Vilaseca M, García-Calderó H, Lafoz E, Ruart M, López-Sanjurjo CI, Murphy MP. et al. Mitochondria-targeted antioxidant mitoquinone deactivates human and rat hepatic stellate cells and reduces portal hypertension in cirrhotic rats. Liver Int. 2017Jul;37(7):1002-12
133. Huang W, Cash N, Wen L, Szatmary P, Mukherjee R, Armstrong J. et al. Effects of the mitochondria-targeted antioxidant mitoquinone in murine acute pancreatitis. Mediators Inflamm. 2015;2015:901780
134. Li M, Yuan Y, Han X, Liu X, Zhang W, Hao J. Antioxidant Mitoquinone Alleviates Chronic Pancreatitis via Anti-Fibrotic and Antioxidant Effects. J Inflamm Res. 2022;15:4409-20
135. Aviello G, Knaus UG. NADPH oxidases and ROS signaling in the gastrointestinal tract. Mucosal Immunol. 2018Jul;11(4):1011-23
136. Chang M, Chen W, Xia R, Peng Y, Niu P, Fan H. Pancreatic Stellate Cells and the Targeted Therapeutic Strategies in Chronic Pancreatitis. Molecules. 2023 Jul 22;28(14)
Author contact
![]() Corresponding author: Xiaoyu Li; lixiaoyuedu.cn. Present address: Qingdao University, No. 308 Ningxia Road, Qingdao, Shandong Province, China, 266071.
Corresponding author: Xiaoyu Li; lixiaoyuedu.cn. Present address: Qingdao University, No. 308 Ningxia Road, Qingdao, Shandong Province, China, 266071.