Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(14):3511-3527. doi:10.7150/ijms.114372 This issue Cite
Review
Association Between Gut Microbiota and Pneumonia Risk: A Systematic Review and Mendelian Randomization
Qingping Deng1#, Yuanyuan Liu2#, Hui Rong2 ![]() , Qing Liu1
, Qing Liu1 ![]() , Rongyuan Yang1
, Rongyuan Yang1 ![]()
1. State Key Laboratory of Traditional Chinese Medicine Syndrome, Guangdong Provincial Hospital of Chinese Medicine, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, 111 Dade Road, Guangzhou, Guangdong 510120, China.
2. Hubei University of Traditional Chinese Medicine, Wuhan Hospital of Traditional Chinese Medicine, Wuhan 430065, China.
# These authors contribute equally to this work.
Received 2025-3-24; Accepted 2025-7-14; Published 2025-7-28
Abstract

Background: The gut-lung axis represents a critical pathway potentially modulating COVID-19 pathogenesis. We employed meta-analysis to investigate the Mendelian randomization (MR) studies for the putative causal relationships between gut microbiota composition/metabolites and COVID-19 severity.
Methods: Adhering to PRISMA 2020 guidelines, we conducted a systematic review of MR studies (PubMed/Web of Science/Embase/Scopus/Cochrane; inception to June 2024). Data from 11 studies (aggregating 32,748,274 participants; 1,487 SNPs) underwent meta-analysis across four COVID-19 severity strata including susceptibility, infection, hospitalization, and critical disease. Study quality was evaluated using a validated MR framework assessing 32 core assumptions.
Results: Elevated COVID-19 susceptibility risk was associated with Actinobacteria (OR 1.16, 95% CI 1.06-1.26) and Negativicutes (1.06, 1.03-1.09), whereas protective effects emerged for Oxalobacter (0.84, 0.71-0.99) and Ruminococcaceae UCG014 (0.88, 0.78-0.99). For COVID-19 infection, Negativicutes conferred increased risk (1.13, 1.02-1.26), while the Ruminococcus torques group (0.54, 0.39-0.74) and Parasutterella (0.90, 0.83-0.97) demonstrated protection. Hospitalization risk elevated with MollicutesRF9 (1.13, 1.04-1.22) and Alloprevotella (1.25, 1.07-1.45), contrasting with butyrate (0.97, 0.94-0.99) and Ruminiclostridium6 (0.81, 0.69-0.94) showing protective associations. Severe COVID-19 risk increased with Actinobacteria (1.20, 1.01-1.42), Bifidobacterium (2.09, 1.15-3.81), and Alloprevotella (1.66, 1.36-2.01), while Oxalobacter (0.84, 0.76-0.92) and Subdoligranulum (0.82, 0.76-0.89) exhibited protection. Notably, Actinobacteria, Negativicutes, and Alloprevotella constituted consistent risk factors across severity strata, whereas Oxalobacter and Parasutterella showed trans-stage protective effects. Butyrate production specifically attenuated hospitalization risk, and Bifidobacterium demonstrated strikingly elevated critical disease risk, contrasting with typical probiotic associations.
Conclusions: This meta-analysis of MR studies provides robust evidence for severity-specific causal effects of the gut microbiota on COVID-19 outcomes. The identified microbial taxa and metabolites provide potential biomarkers for clinical risk stratification and targets for novel adjuvant therapeutic strategies.
Keywords: COVID-19, gut microbiota, meta-analysis, Mendelian randomization
Introduction
Human coronaviruses periodically emerge as significant global health threats. The most recent and impactful example is severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19. Global surveillance data indicate a substantial disease burden: as of July 7, 2024, worldwide cumulative confirmed SARS-CoV-2 infections exceeded 775 million cases, with reported fatalities surpassing 7.05 million [1]. The United States (103,436,829 cases), China (99,365,162 cases), and India (45,040,752 cases) reported the highest national cumulative caseloads [1]. While the exponential growth observed during initial pandemic peaks has plateaued, transmission rates remain persistently elevated [2-4]. The pandemic has generated profound health, economic, and societal consequences globally. Its prolonged duration and extensive spread underscore the critical need for continued investigation into its multifaceted health and socioeconomic impacts.
The gastrointestinal tract constitutes the body's largest immune organ, where resident microbiota critically modulate host immunity and nutritional metabolism [5]. Dominated by Firmicutes, Bacteroidetes, Actinobacteria, Fusobacteria, Proteobacteria, Verrucomicrobia, and Cyanobacteria [6], this microbial consortium co-evolves with the host to provide pathogen defense. Clinical evidence links specific gut bacterial populations to pneumonia severity [7-10]. COVID-19 patients exhibit significant depletion of anti-inflammatory butyrate-producing bacteria during acute infection compared to healthy controls [9, 10] SARS-CoV-2 invasion via respiratory epithelium damages mucosal barriers in both pulmonary and gastrointestinal systems. This compromise facilitates viral dissemination and secondary bacterial infections, inducing intestinal dysbiosis. Characteristic alterations include reduced commensal bacteria abundance with concurrent expansion of opportunistic pathogens, fundamentally disrupting gut ecological homeostasis [11].
Mendelian Randomization (MR) represents a robust method for investigating causal relationships between variables. This approach employs genetic variants as instrumental variables, assigning individuals to exposure groups based on naturally occurring genetic differences. By leveraging Mendel's second law of inheritance, MR minimizes confounding and reverse causation biases that limit conventional observational studies. The method consequently enables rigorous causal inference in complex biological systems.
Current literature on COVID-19 and gut microbiota includes numerous reviews, though many provide limited mechanistic analysis of intestinal microorganisms' involvement [12-14]. While Mendelian randomization (MR) studies have investigated gut microbiota-COVID-19 relationships [15], a comprehensive synthesis of this evidence remains unavailable. This study addresses this gap through meta-analysis of existing MR investigations on COVID-19-gut microbiota associations. We further stratify analyses by COVID-19 severity to establish an evidence-synthesis framework for disease-microbiota interactions.
Methods
Study design
This study followed the Preferred Reporting Items for Systematic Evaluation and Meta-Analysis Protocols (PRISMA) 2020 guidelines. The study protocol is registered with PROSPERO under the registration number CRD42024570240.
Literature search strategies
This Mendelian randomization (MR) study investigated the causal effects of gut microbiota and metabolites on pneumonia risk. We systematically searched PubMed, Web of Science, Embase, Scopus, and the Cochrane Library for English-language publications published from database inception until June 25, 2024. The search strategy combined Medical Subject Headings (MeSH) and free-text terms (e.g., "Mendelian randomization","COVID-19","covid-19 virus disease","2019-ncov diseases","gut microbiota","microflora intestinal","gut microbiota metabolites"). Studies on other pneumonias identified under these search terms were also included; the full search strategy is detailed in Supplementary Table S1. Supplementary Table S1 is used to present the search strategies and results in each database.
Study selection
Literature records were imported into NoteExpress. Two reviewers independently screened titles, abstracts, and full-text articles against uniform eligibility criteria. Disagreements were resolved through consensus with a third reviewer.
Quality assessment
The quality evaluation of this article was conducted using the methodological framework for MR studies developed by Mengyuan Wang et al. [16]. This framework comprises six key components: the completeness of instrumental variable analysis, validation of the assumptions of association, independence, and exclusivity, implementation of sensitivity analyses, consideration of population stratification, and examination of nonlinear associations. The criteria for assessing each component are detailed in Table 1. By systematically applying these principles, we ensured a comprehensive and rigorous evaluation of the article's methodological quality.
Methods to assess the quality of MR studies
| Item | Grade | Criteria |
|---|---|---|
| A. Full IV analysis | Good | Full IV analyses, such as two-stage least-squares regression for one-sample MR study andinverse-variance weighted method for two-sample MR study. |
| Poor | Without full IV analyses; only uses other approaches, such as an association analysis betweerthe genetic variant and outcome. | |
| B.Relevance assumption | Good | For one-sample R study, the assumption is tested by reporting an F-statistic (F>10); for twosample MR study, strongly and robustly associated SNPs from GWAS are selected (P<5x10-8). |
| Moderate | Associated SNPs are selected using a P value threshold not satisfying Bonferroni correction | |
| Poor | Failure to describe whether the assumption is satisfied. | |
| C. Independence assumption and exclusion restriction assumption | Good | Assumption is tested using MR-Egger regression, MR-Pleiotropy Residual Sum and Outlier, and other noyel methods |
| Moderate | Full IV analysis is selected based on literature research and without testing the assumption. | |
| Poor | Failure to describe whether the assumption is satisfied. | |
| D. Sensitivity analysis | Good | Sensitivity analysis is conducted, and results that are consistent with primary analyses are reported. |
| Poor | Failed sensitivity analysis or inconsistent results are reported. | |
| E.Population stratification | Good | Absence of population stratification. |
| Poor | Presence of population stratification or failure to report population information. | |
| F. Non-linearity correlation | Good | Potential non-linearity correlations of exposure and outcome variables are explored. |
| Poor | Failure to describe potential non-linearity correlations. |
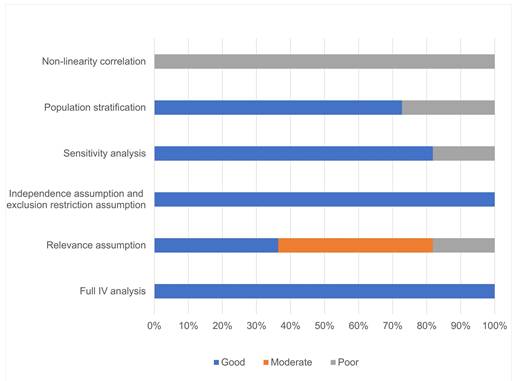
MR studies necessitate full IV analysis to ensure quality (Table 1A). Simultaneous fulfillment of the 3 core assumptions of association (genetic variants are associated with the exposure phenotype), independence (genetic variants are independent of confounders affecting the association), and exclusivity (genetic variants affect the outcome only through exposure) is necessary for reliable results. Single-sample and two-sample MR studies often rely on different methods to test the hypothesis of association of genetic variants with exposure phenotypes (Table 1 B). Multiplicity of effects of genetic variation is prevalent, and the independence and exclusivity hypotheses may be violated if genetic tools influence outcomes through factors other than the exposure of interest, and the two hypotheses can generally be tested together (Table 1 C).MR studies assessed the robustness of the results through sensitivity analyses (Table 1 D). Given the heterogeneity of genetic susceptibility across races, attention also needs to be paid to the potential impact of population stratification (Table 1 E). In addition, we were concerned about how well studies explored and rated potential nonlinear associations between exposure and outcomes (Table 1 F). Given that there is no MR methodology that uses summary statistics to explore nonlinear associations, and that most MR studies focus only on linear associations between exposures and outcomes, we judged studies with "good" ratings on all five items as high-quality MR studies, relying primarily on entries A to E in Table 1.
Data extraction
Two investigators independently extracted data including: title, authors, publication year, disease phenotype, case/control counts, microbiota/metabolite features, SNP numbers, ORs (95% CIs), and IVW causal estimates. Discrepancies were resolved through iterative discussion. Three researchers implemented this process: two performed literature review and data extraction, with the third overseeing result verification and facilitating consensus discussions when required.
Statistical analysis
Study data were standardized prior to analysis to ensure methodological consistency. Using Review Manager (RevMan v5.4, Cochrane Collaboration), we conducted: (1) risk-of-bias assessments, (2) data harmonization, and (3) meta-analyses. Pooled odds ratios (ORs) with 95% confidence intervals (CIs) quantified associations between gut microbiota metabolites and pneumonia. Heterogeneity was evaluated using Cochran's Q-test and I2 statistics, with Cochrane-recommended significance thresholds (PQ < 0.10 or I2 > 50%). Random-effects models were applied when significant heterogeneity was detected. Publication bias was assessed through funnel plot symmetry examination supplemented by Egger's regression tests. Sensitivity analyses evaluated the robustness of findings. For exposures in studies reporting F-statistics (9 out of 11) [17-25], all F-statistics exceeded 10, meeting the relevance assumption requirement for instrument strength; F-statistics were not reported in the other two studies [26, 27].
Results
Literature search results and study characteristics
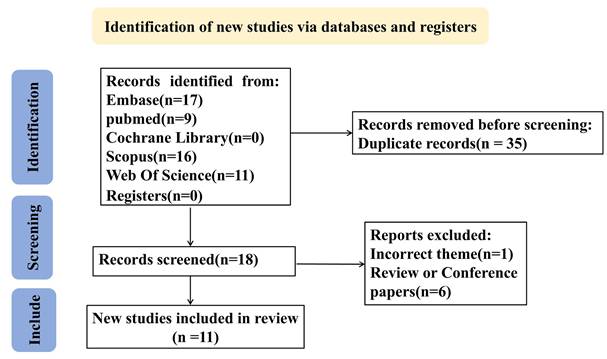
Retrieved records were imported into NoteExpress (Aegean Software, China) for standardized management. The search strategy combined Medical Subject Headings (MeSH) and free-text terms including "Mendelian randomization","COVID-19","covid-19 virus disease", "2019-ncov diseases", "gut microbiota", "microflora intestinal" and "gut microbiota metabolites" with intentional inclusion of pneumted onia-reladisorders through COVID-19 terminology (See Supplementary Table S1 for search strategies and results in each database). During screening, 34 publications were excluded based on: (1) cross-database duplication, (2) supplementary materials, (3) funding announcements, (4) review articles/meta-analyses, or (5) non-pneumonia relevance. Two investigators (YYL and QPD) independently performed quality assessment and data extraction, with discordant evaluations resolved by third-reviewer (QL) arbitration. The final analysis incorporated 11 eligible studies [17-27], encompassing 32,748,274 participants and 1,487 single-nucleotide polymorphisms (SNPs). Study characteristics are summarized in Table 2, while the screening process is depicted in the PRISMA flowchart (Figure 1).
Baseline characteristic of the included literature
| Label | Disease | Case | Sample | Bacterial flora/Metabolites | SNP Quantities | OR (95%CI) IVW Results | Remark |
|---|---|---|---|---|---|---|---|
| Yingjian Liu 2024 [17] | Pneumonia | / | 456348 | genus.Anaerofilum.id.2053 | 7 | 1.34(1.04,1.72) | IVW |
| family.Bifidobacteriaceae.id.433 | 13 | 0.68(0.54,0.85) | IVW | ||||
| family.Coriobacteriaceae.id.811 | 17 | 1.34(1,1.81) | IVW | ||||
| order.Coriobacteriales.id.810 | 17 | 1.34(1,1.81) | IVW | ||||
| class.Coriobacteriia.id.809 | 17 | 1.34(1,1.81) | IVW | ||||
| family.FamilyXI.id.1936 | 8 | 1.18(1.01,1.38) | IVW | ||||
| genus.LachnospiraceaeND3007group.id.11317 | 3 | 2.1(1.17,3.78) | IVW | ||||
| BP(bacterial pneumonia) | / | 456348 | genus.Parasutterella.id.2892 | 12 | 2.75(1.49,5.08) | IVW | |
| phylum.Actinobacteria.id.400 | 16 | 2.09(1.13,3.88) | IVW | ||||
| family.Bifidobacteriaceae.id.433 | 15 | 2.05(1.17,3.58) | IVW | ||||
| genus.Bifidobacterium.id.436 | 16 | 1.93(1.15,3.22) | IVW | ||||
| family.Enterobacteriaceae.id.3469 | 7 | 3.39(1.35,8.48) | IVW | ||||
| order.Enterobacteriales.id.3468 | 7 | 3.39(1.35,8.48) | IVW | ||||
| order.Gastranaerophilales.id.1591 | 9 | 1.67(1.02,2.74) | IVW | ||||
| family.Rhodospirillaceae.id.2717 | 14 | 0.55(0.33,0.91) | IVW | ||||
| order.Rhodospirillales.id.2667 | 13 | 0.56(0.33,0.95) | IVW | ||||
| BLA(bronchopneumonia、lung abscess) | / | 456348 | genus.Odoribacter.id.952 | 3 | 0.17(0.03,0.97) | IVW | |
| genus.Paraprevotella.id.962 | 12 | 0.54(0.3,0.94) | IVW | ||||
| phylum.Bacteroidetes.id.905 | 11 | 0.32(0.11,0.92) | IVW | ||||
| genus.ChristensenellaceaeR.7group.id.11283 | 9 | 0.25(0.09,0.73) | IVW | ||||
| genus.Fusicatenibacter.id.11305 | 18 | 2.2(1.03,4.7) | IVW | ||||
| genus.Marvinbryantia.id.2005 | 9 | 0.41(0.17,1) | IVW | ||||
| class.Methanobacteria.id.119 | 10 | 1.58(1,2.5) | IVW | ||||
| family.Methanobacteriaceae.id.121 | 10 | 1.58(1,2.5) | IVW | ||||
| order.Methanobacteriales.id.120 | 10 | 1.58(1,2.5) | IVW | ||||
| genus.Methanobrevibacter.id.123 | 6 | 1.91(1.05,3.47) | IVW | ||||
| family.Peptococcaceae.id.2024 | 10 | 2.03(1.01,4.07) | IVW | ||||
| family.Porphyromonadaceae.id.943 | 9 | 4.93(1.2,20.15) | IVW | ||||
| PP(pneumococcal pneumonia) | / | 456348 | genus.Adlercreutzia.id.812 | 5 | 0.74(0.56,0.97) | IVW | |
| genus.Holdemanella.id.11393 | 10 | 1.2(1.02,1.41) | IVW | ||||
| genus.Lachnospira.id.2004 | 6 | 0.66(0.47,0.93) | IVW | ||||
| genus.LachnospiraceaeNC2004group.id.11316 | 9 | 0.77(0.65,0.91) | IVW | ||||
| family.Rikenellaceae.id.967 | 21 | 1.31(1.1,1.57) | IVW | ||||
| Tian, Siyu 2024 [18] | COVID-19 infection | 112612 | 2474079 | ||||
| COVID-19 hospitalization | 24274 | 2061529 | Bifidobacterium.id.436 | 13 | 1.126(1.021-1.242) | IVW | |
| LachnospiraceaeUCG010.id.11330 | 10 | 1.139(1.009-1.287) | IVW | ||||
| RikenellaceaeRC9gutgroup.id.11191 | 13 | 1.081(1.019-1.147) | IVW | ||||
| RuminococcaceaeUCG014.id.11371 | 11 | 0.822(0.782-0.995) | IVW | ||||
| COVID-19 severity | 8779 | 1001875 | Intestinimas.id.2062 | 16 | 1.179(1.006-1.383) | IVW | |
| Yuxin Zou 2024 [19] | COVID-19 severity | 13769 | 1072442 | Victivallaceae, | 12 | 0.888 (0.801-0.984) | IVW |
| Weifeng Shang 2023 [20] | COVID-19 susceptibility | 159 840.00 | 2782977 | class Negativicutes | 12 | 1.05(1.01-1.10) | IVW |
| class Gammaproteobacteria | 7 | 0.94( 0.89-0.99) | IVW | ||||
| order Selenomonadales | 12 | 1.05(1.01-1.10) | IVW | ||||
| family Streptococcaceae | 14 | 0.95(0.92-1.00) | IVW | ||||
| family Bacteroidaceae | 9 | 1.06( 1.01-1.12) | IVW | ||||
| genus Bacteroides | 9 | 1.06(1.01-1.12) | IVW | ||||
| COVID-19 severity | 18 152.00 | 1145546 | phylum Cyanobacteria | 8 | 0.85(0.76-0.96) | IVW | |
| order Lactobacillales | 15 | 0.87(0.76-0.98) | IVW | ||||
| family Christensenellaceae | 11 | 0.87(0.77-0.99) | IVW | ||||
| genus Subdoligranulum | 11 | 0.8(0.69-0.92) | IVW | ||||
| genus Tyzzerella3 | 13 | 0.89(0.81-0.97) | IVW | ||||
| genus RuminococcaceaeUCG011 | 8 | 0.91(0.83-0.99) | IVW | ||||
| order MollicutesRF9 | 13 | 1.14(1.01-1.29) | IVW | ||||
| genus RikenellaceaeRC9 | 11 | 1.09(1.01-1.17) | IVW | ||||
| genus LachnospiraceaeUCG008 | 11 | 1.12(1.00-1.26) | IVW | ||||
| Meng-Mei Zhong 2023 [21] | COVID-19 susceptibility | 159840 | 2782977 | class Gammaproteobacteria | 6 | 0.933(0.879-0.991) | IVW |
| family Streptococcaceae | 14 | 0.955(0.916-0.995) | IVW | ||||
| class Negativicutes | 13 | 1.054(1.005-1.105) | IVW | ||||
| order Selenomonadales | 12 | 1.054(1.005-1.105) | IVW | ||||
| family Bacteroidaceae | 9 | 1.06(1.007-1.125) | IVW | ||||
| genus Bacteroides | 9 | 1.064(1.007-1.125) | IVW | ||||
| COVID-19 severity | 18152 | 1145546 | phylum Cyanobacteria | 8 | 0.852(0.760-0.955) | IVW | |
| order Lactobacillales | 15 | 0.867(0.764-0.983) | IVW | ||||
| genus RuminococcaceaeUCG011 | 11 | 0.907(0.832-0.988) | IVW | ||||
| genus Subdoligranulum | 11 | 0.807(0.699-0.932) | IVW | ||||
| genus Tyzzerella3 | 13 | 0.885(0.810-0.967) | IVW | ||||
| order MollicutesRF9 | 13 | 1.141(1.009-1.291) | IVW | ||||
| genus RikenellaceaeRC9 | 8 | 1.085(1.009-1.167) | IVW | ||||
| COVID-19 hospitalization | 44986 | 2356386 | genus Marvinbryantia | 10 | 0.886(0.812-0.967) | IVW | |
| genus Olsenella | 11 | 0.942(0.897-0.990) | IVW | ||||
| family Veillonellaceae | 19 | 1.069(1.002-1.140) | IVW | ||||
| genus Eubacteriumruminantiumgroup | 18 | 1.065(1.010-1.123) | IVW | ||||
| genus Dorea | 10 | 1.162(1.055-1.279) | IVW | ||||
| Zengbin Li 2023 [22] | COVID-19 infection | 38984 | 1644784 | phylum Lentisphaerae | 9 | 0.93(0.87-0.99) | IVW |
| family Alcaligenaceae | 12 | 0.87(0.78-0.96) | IVW | ||||
| family Lachnospiraceae | 17 | 0.91(0.84-1.00) | IVW | ||||
| genus Dialister | 11 | 0.91(0.82-1.00) | IVW | ||||
| genus Parasutterella | 14 | 0.89(0.83-0.97) | IVW | ||||
| genus Ruminococcaceae UCG003 | 12 | 0.90(0.82-0.99) | IVW | ||||
| genus Ruminococcaceae UCG014 | 11 | 0.88(0.80-0.97) | IVW | ||||
| class Negativicutes | 12 | 1.13(1.02-1.26) | IVW | ||||
| order Selenomonadales | 12 | 1.13(1.02-1.26) | IVW | ||||
| genus Phascolarctobacterium | 9 | 1.13(1.02-1.25) | IVW | ||||
| COVID-19 hospitalization | 9986 | 1877672 | genus Alistipes | 14 | 0.78(0.63-0.96) | IVW | |
| genus Parasutterella | 14 | 0.84(0.72-0.98) | IVW | ||||
| genus Ruminiclostridium6 | 15 | 0.80(0.69-0.94) | IVW | ||||
| genus Ruminococcaceae UCG014 | 11 | 0.79(0.65-0.97) | IVW | ||||
| family Family XIII | 10 | 1.30(1.03-1.64) | IVW | ||||
| family Victivallaceae | 12 | 1.11(1.00-1.24) | IVW | ||||
| genus Alloprevotella | 5 | 1.25(1.07-1.45) | IVW | ||||
| genus Prevotella9 | 14 | 1.21(1.04-1.41) | IVW | ||||
| COVID-19 severity | 5101 | 1383241 | genus Ruminococcus gnavus group | 12 | 0.77(0.62-0.95) | IVW | |
| genus Oxalobacter | 11 | 0.84(0.71-1.00) | IVW | ||||
| genus Ruminiclostridium6 | 16 | 0.78(0.62-0.98) | IVW | ||||
| genus Alloprevotella | 5 | 1.67(1.32-2.11) | IVW | ||||
| Han Chen 2023 [23] | COVID-19 susceptibility | 38984 | 1644784 | Genus Butyricimonas | 13 | 0.919(0.847-0.998) | IVW |
| Genus Parasutterella | 16 | 0.902(0.836-0.973) | IVW | ||||
| Genus Ruminococcaceae UCG014 | 9 | 0.878(0.777-0.992) | IVW | ||||
| Genus Oxalobacter | 13 | 0.842(0.712-0.994) | IVW | ||||
| Class Actinobacteria | 21 | 1.156(1.062-1.258) | IVW | ||||
| Class Alphaproteobacteria | 9 | 1.102(1.004-1.211) | IVW | ||||
| Genus Alloprevotella | 7 | 1.088(1.021-1.160) | IVW | ||||
| Genus Coprococcus | 10 | 1.159(1.030-1.304) | IVW | ||||
| Genus Erysipelatoclostri-dium | 13 | 1.083(1.001-1.172) | IVW | ||||
| COVID-19 severity | 5101 | 1383241 | Genus Oxalobacter | 13 | 0.842 (0.712-0.994) | IVW | |
| Wanqiang Lv 2023 [24] | COVID-19 hospitalization | 6406 | 902088 | Gut production of the SCFA butyrate | 8 | 0.96832539912073(0.94416699163366-0.993101947950892) | IVW |
| Fecal propionate | 3 | 0.941469863248343(0.808083479895778-1.09687368379214) | IVW | ||||
| COVID-19 severity | 3886 | 622265 | Gut production of the SCFA butyrate | 7 | 1.00845602976638(0.96356320994773-1.05544042515628) | IVW | |
| Fecal propionate | 3 | 0.968794000309792(0.857951748844421-1.09395640990347) | IVW | ||||
| Jukun Song 2023 [25] | COVID-19 susceptibility | / | 159840 | class.Gammaproteobacteria.id.3303 | 10 | 0.943826(0.898701-0.991217) | IVW |
| phylum.Lentisphaerae.id.2238 | 15 | 1.021896(1.00006-1.044209) | IVW | ||||
| genus.Eisenbergiella.id.11304 | 12 | 1.027563(1.000392-1.055472) | IVW | ||||
| genus.unknowngenus.id.2041 | 13 | 1.03016(1.000408-1.060798) | IVW | ||||
| genus.Bifidobacterium.id.436 | 21 | 1.030965(1.000158-1.062721) | IVW | ||||
| genus.unknowngenus.id.2001 | 11 | 1.039753(1.002465-1.078429) | IVW | ||||
| genus.Flavonifractor.id.2059 | 10 | 1.044181(1.002618-1.087467) | IVW | ||||
| genus.Dorea.id.1997 | 14 | 1.048128(1.006111-1.091899) | IVW | ||||
| order.Selenomonadales.id.2165 | 15 | 1.053979(1.010557-1.099266) | IVW | ||||
| class.Deltaproteobacteria.id.3087 | 13 | 1.055903(1.003375-1.11118) | IVW | ||||
| genus.Bacteroides.id.918 | 12 | 1.059099(1.010079-1.110498) | IVW | ||||
| class.Negativicutes.id.2164 | 11 | 1.069307(1.016779-1.124549) | IVW | ||||
| family.Bacteroidaceae.id.917 | 8 | 1.072539(1.011616-1.137131) | IVW | ||||
| COVID-19 hospitalization | / | 44986 | family.Christensenellaceae.id.1866 | 13 | 0.918613(0.846107-0.997332) | IVW | |
| genus.Eubacteriumoxidoreducensgroup.id.11339 | 9 | 0.934016(0.87255-0.999811) | IVW | ||||
| genus.Olsenella.id.822 | 13 | 0.938009(0.896461-0.981482) | IVW | ||||
| genus.Anaerofilum.id.2053 | 12 | 0.945835(0.896604-0.997769) | IVW | ||||
| genus.Tyzzerella3.id.113.35 | 18 | 0.951996(0.910977-0.994862) | IVW | ||||
| family.FamilyXI.id.1936 | 12 | 0.95861(0.919248-0.999658) | IVW | ||||
| order.Bacteroidales.id.913 | 16 | 1.092775(1.01484-1.176694) | IVW | ||||
| genus.unknowngenus.id.1000005472 | 15 | 1.101318(1.028715-1.179046) | IVW | ||||
| class.Actinobacteria.id.419 | 21 | 1.104484(1.031225-1.182947) | IVW | ||||
| family.unknownfamily.id.1000005471 | 12 | 1.11355(1.026157-1.208386) | IVW | ||||
| phylum.Actinobacteria.id 400 | 20 | 1.121487(1.028754-1.22258) | IVW | ||||
| order.MollicutesRF9.id.11579 | 12 | 1.126898(1.044468-1.215834) | IVW | ||||
| order.Selenomonadales.id.2165 | 15 | 1.134561(1.026603-1.253872) | IVW | ||||
| class.Negativicutes.id.2164 | 11 | 1.234846(1.112774-1.37031) | IVW | ||||
| COVID-19 severity | / | 18152 | genus.Subdoligranulum.id.2070 | 13 | 0.855125(0.749524-0.975604) | IVW | |
| genus.Tyzzerella3.id.11335 | 14 | 0.896826(0.82266-0.97768) | IVW | ||||
| genus.RuminococcaceaeUCG011.id.11368 | 8 | 0.906709(0.832425-0.987621) | IVW | ||||
| genus.Prevotella9.id.11183 | 19 | 1.108017(1.015604-1.208839) | IVW | ||||
| genus.LachnospiraceaeUCG008.id.11328 | 12 | 1.110538(1.000231-1.233009) | IVW | ||||
| family.BacteroidalesS24.7group.id.11173 | 10 | 1.149522(1.02712-1.28651) | IVW | ||||
| genus.unknowngenus.id.1000005479 | 6 | 1.173132(1.004522-1.370044) | IVW | ||||
| order.Selenomonadales.id.2165 | 12 | 1.188812(1.01203-1.396475) | IVW | ||||
| phylum.Actinobacteria.id.400 | 17 | 1.202516(1.015075-1.42457) | IVW | ||||
| family.unknownfamily.id.1000005471 | 11 | 1.23367(1.047721-1.452621) | IVW | ||||
| genus.unknowngenus.id.1000005472 | 11 | 1.237166(1.064543-1.437781) | IVW | ||||
| class.Negativicutes.id.2164 | 8 | 1.292966(1.075591-1.554273) | IVW | ||||
| order.MollicutesRF9.id 11579 | 15 | 1.168451(1.017332-1.342018) | IVW | ||||
| Hanyu Zhang 2023 [26] | COVID-19 infection | 38984 | 1644784 | genus Ruminococcustorquesgroup | 1 | 0.537(0.391-0.738) | IVW |
| genus Ruminococcaceae UCG013 | 1 | 1.38206616435633(1.025-1.863) | IVW | ||||
| genus Ruminococcus1 | 1 | 0.734645873967539(0.545-0.99) | IVW | ||||
| genus Allisonella | 1 | 0.999477974302852(0.879-1.137) | IVW | ||||
| genus Eubacteriumcoprostanoligenesgroup | 1 | 0.839701170368478(0.615-1.146) | IVW | ||||
| genus Oxalobacter | 1 | 0.872561011613187(0.736-1.035) | IVW | ||||
| genus Erysipelatoclostridium | 1 | 0.925967316791888(0.737-1.163) | IVW | ||||
| genus Faecalibacterium | 1 | 0.947829609211915(0.69-1.303) | IVW | ||||
| genus Peptococcus | 1 | 0.957340664124295(0.805-1.138) | IVW | ||||
| family Oxalobacteraceae | 1 | 0.973361241524337(0.818-1.159) | IVW | ||||
| genus Romboutsia | 1 | 0.991370771376931(0.755-1.302) | IVW | ||||
| genus RuminococcaceaeUCG009 | 1 | 0.997685345952551(0.774-1.285) | IVW | ||||
| family Peptostreptococcaceae | 1 | 1.00904062177387(0.767-1.328) | IVW | ||||
| genus Bifidobacteriaceae | 1 | 1.06235820628227(0.875-1.29) | IVW | ||||
| genus Streptococcus | 1 | 1.11442825766029(0.844-1.471) | IVW | ||||
| family Streptococcaceae | 1 | 1.12075212488415(0.837-1.501) | IVW | ||||
| genus Intestinibacter | 1 | 1.14628731724782(0.846-1.553) | IVW | ||||
| genus Enterorhabdus | 1 | 1.1672334778462(0.958-1.422) | IVW | ||||
| order Gastranaerophilales | 1 | 1.19602074416788(0.999-1.432) | IVW | ||||
| COVID-19 hospitalization | 3159 | 7206 | Eubacteriumcoprostanoligenesgroup | 1 | 0.568684261257267(0.145-2.223) | IVW | |
| Bifidobacteriales | 1 | 0.648560491804976(0.219-1.917) | IVW | ||||
| genus Peptococcus | 1 | 0.675976747986784(0.313-1.459) | IVW | ||||
| Oxalobacter | 1 | 0.706206493883378(0.3-1.661) | IVW | ||||
| Allisonella | 1 | 0.903849603717244(0.499-1.636) | IVW | ||||
| Enterorhabdus | 1 | 1.02698051702283(0.335-3.146) | IVW | ||||
| Gastranaerophilales | 1 | 1.14110831926724(0.47-2.773) | IVW | ||||
| Intestinibacter | 1 | 1.38991028236733(0.301-6.42) | IVW | ||||
| Bifidobacteriaceae | 1 | 1.54120056443399(0.521-4.56) | IVW | ||||
| class Actinobacteria | 1 | 1.57493389505008(0.504-4.92) | IVW | ||||
| family Oxalobacteraceae | 1 | 1.68539507129741(0.716-3.969) | IVW | ||||
| COVID-19 severity | 5101 | 1383241 | order Bifidobacteriales | 2 | 0.471(0.286-0.774) | IVW | |
| genus Ruminococcustorquesgroup | 1 | 0.536877354869706(0.391-0.738) | IVW | ||||
| genus Bifidobacteriaceae | 2 | 2.124(1.152-3.915) | IVW | ||||
| genus Tyzzerella3 | 1 | 2.21142565432121(1.246-3.924) | IVW | ||||
| class Actinobacteria | 1 | 2.53280022758574(1.228-5.224) | IVW | ||||
| genus Faecalibacterium | 1 | 0.545239789689792(0.184-1.614) | IVW | ||||
| genus Erysipelatoclostridium | 1 | 0.66116800731294(0.298-1.468) | IVW | ||||
| genus Peptococcus | 1 | 0.675101722137951(0.419-1.088) | IVW | ||||
| genus Allisonella | 1 | 0.750932133107426(0.464-1.215) | IVW | ||||
| genus Enterorhabdus | 1 | 0.76744596953411(0.447-1.317) | IVW | ||||
| order Gastranaerophilales | 1 | 0.7795799733847(0.45-1.35) | IVW | ||||
| genus Eubacteriumcoprostanoligenesgroup | 1 | 0.792819896331787(0.277-2.271) | IVW | ||||
| family Streptococcaceae | 1 | 0.999972600375377(0.429-2.332) | IVW | ||||
| genus Streptococcus | 1 | 0.999973966838869(0.447-2.235) | IVW | ||||
| genus RuminococcaceaeUCG009 | 1 | 1.09160755405964(0.494-2.414) | IVW | ||||
| genus Oxalobacter | 1 | 1.09557314891857(0.606-1.981) | IVW | ||||
| family Oxalobacteraceae | 1 | 1.23244499853025(0.755-2.012) | IVW | ||||
| genus Intestinibacter | 1 | 1.57226695768584(0.604-4.094) | IVW | ||||
| genus Romboutsia | 1 | 1.78794246889422(0.825-3.874) | IVW | ||||
| family Peptostreptococcaceae | 1 | 1.79858455998767(0.824-3.924) | IVW | ||||
| Han Yan 2023 [27] | COVID-19 severity | 5101 | 1383241 | Ruminiclostridium6 | 14 | 0.708(0.544-0.921) | IVW(if the IV number was more than 1) |
| unknowngenus.id.1000001215 | 5 | 0.72(0.536-0.966) | IVW(if the IV number was more than 1) | ||||
| Oxalobacter | 4 | 0.752(0.578-0.98) | IVW(if the IV number was more than 1) | ||||
| Butyrivibric | 14 | 0.83(0.69-1.000) | IVW(if the IV number was more than 1) | ||||
| Oxalobacter | 11 | 0.842(0.709-1.000) | IVW(if the IV number was more than 1) | ||||
| Howardella | 7 | 1.264(1.009-1.583) | IVW(if the IV number was more than 1) | ||||
| Alloprevotella | 4 | 1.627(1.14-2.323) | IVW(if the IV number was more than 1) | ||||
| Ruminococcus gnavus group | 2 | 1.703(1.018-2.849) | IVW(if the IV number was more than 1) | ||||
| Bifidobacterium | 2 | 2.092(1.149-3.808) | IVW(if the IV number was more than 1) |
Flow diagram of the data collection and analysis in this study.
Quality assessment
Our meta-analysis of 52 gut microbiota taxa identified consistent microbial signatures associated with pneumonia severity (Supplementary Table S2 provides a comprehensive summary of the effects of different gut microbiota taxa on COVID-19 pneumonia.), including COVID-19 outcomes. Among the 11 studies, all performed a complete IV analysis, 9 validated the three core hypotheses of MR research, 9 conducted sensitivity analyses, and 8 showed no evidence of population stratification. Ultimately, 4 studies were deemed high-quality MR research, as illustrated in Figure 2.
Quality evaluation results of a Mendelian randomized study on the relationship between pneumonia and intestinal microbiota and its metabolites.
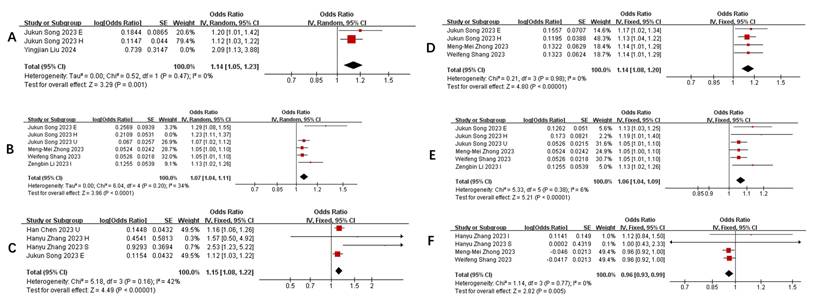
Association between key microbial taxa and pneumonia severity
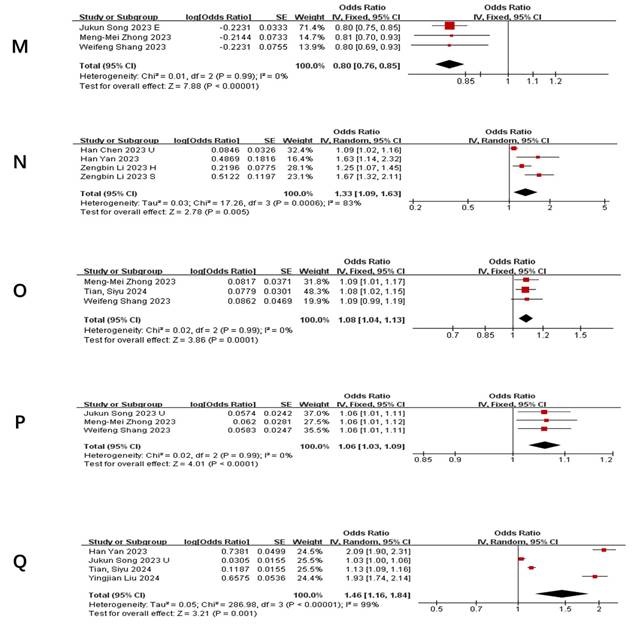
Table 3 summarizes taxa with the strongest and most consistent associations (P<0.01) across ≥3 studies (The complete Summary of Bacterial Flora in COVID-19 Patients with Different Severities is presented in Supplementary Table S3). Notably: Positive associations: Phylum Actinobacteria.id (Figure 3 A), Class Negativicutes (Figure 3 B), Class Actinobacteria (Figure 3 C), Order MollicutesRF9 (Figure 3 D), Order Selenomonadales (Figure 3 E), Family Bacteroidaceae (Figure 3 G), Genus Alloprevotella (Figure 3 N), Genus RikenellaceaeRC9 (Figure 3 O), Genus Bifidobacterium (Figure 3 Q) and Genus Bacteroides (Figure 3 P) were repeatedly linked to increased pneumonia severity. Negative associations: Family Streptococcaceae (Figure 3 F), Genus Tyzzerella3 (Figure 3 H), Oxalobacter (Figure 3 I), Parasutterella (Figure 3 J), RuminococcaceaeUCG014 (Figure 3 K), RuminococcaceaeUCG011 (Figure 3 L), and Subdoligranulum (Figure 3 M) showed protective effects. Heterogeneity adjustments (e.g., exclusion of outlier studies ([19, 23, 27]) strengthened these associations (I² reduced to 0-34%).
Consolidated Effects of Key Taxa on Pneumonia Severity
| Taxon | Studies | WMD (95% CI) | P-value | Heterogeneity (I²) | Direction |
|---|---|---|---|---|---|
| Phylum Actinobacteria.id | 3 | 1.14 (1.05--1.23) | 0.001 | 53% | ↑ Severity |
| Class Negativicutes | 6 | 1.07 (1.04--1.11) | <0.0001 | 34% | ↑ Severity |
| Class Actinobacteria | 4 | 1.15 (1.08--1.22) | <0.00001 | 42% | ↑ Severity |
| Order MollicutesRF9 | 4 | 1.14 (1.08--1.20) | <0.00001 | 0% | ↑ Severity |
| Order Selenomonadales | 6 | 1.07 (1.04--1.09) | <0.00001 | 6% | ↑ Severity |
| Family Streptococcaceae | 4 | 0.96 (0.93--0.99) | 0.005 | 0% | ↓ Severity |
| Family Bacteroidaceae | 3 | 1.06 (1.03--1.09) | <0.0001 | 0% | ↑ Severity |
| Genus Tyzzerella3 | 5 | 0.92 (0.88--0.96) | <0.0001 | 24% | ↓ Severity |
| Genus Oxalobacter | 8 | 0.84 (0.78--0.91) | <0.001 | 0% | ↓ Severity |
| Genus Parasutterella | 4 | 0.89 (0.85--0.94) | <0.0001 | 0% | ↓ Severity |
| Genus RuminococcaceaeUCG014 | 3 | 0.87 (0.81--0.93) | <0.0001 | 0% | ↓ Severity |
| Genus RuminococcaceaeUCG011 | 3 | 0.91 (0.86--0.95) | 0.0002 | 0% | ↓ Severity |
| Genus Subdoligranulum | 3 | 0.80 (0.76--0.85) | <0.00001 | 0% | ↓ Severity |
| Genus Alloprevotella | 4 | 0.89 (0.85--0.94) | 0.005 | 83% | ↑ Severity |
| Genus RikenellaceaeRC9 | 3 | 1.08 (1.04--1.13) | 0.0001 | 0% | ↑ Severity |
| Genus Bacteroides | 3 | 1.06 (1.03--1.09) | <0.0001 | 0% | ↑ Severity |
| Genus Bifidobacterium | 4 | 0.89 (0.85--0.94) | 0.001 | 99% | ↑ Severity |
Forest plot of influence of different gut microbiota on the severity of pneumonia. A) Relationship between phylum actinobacteria.id and pneumonia; B) Relationship between class Negativicutes and pneumonia; C) Relationship between class Actinobacteria and pneumonia; D) Relationship between order MollicutesRF9 and pneumonia; E) Relationship between order Selenomonadales and pneumonia; F) Relationship between family Streptococcaceae and pneumonia; G) Relationship between genus Bacteroides and pneumonia; H) Relationship between genus Tyzzerella3 and pneumonia; I) Relationship between genus Oxalobacter and pneumonia; J) Relationship between genus parasutterella and pneumonia; K) Relationship between genus Ruminococcaceae UCG014 and pneumonia; L) Relationship between RuminococcaceaeUCG011 and pneumonia; M) Relationship between genus Subdoligranulum and pneumonia; N) Relationship between genus Alloprevotella and pneumonia; O) Relationship between genus RikenellaceaeRC9 and pneumonia; P) Relationship between genus Bacteroides and pneumonia; Q) Relationship between genus Bifidobacterium and pneumonia.
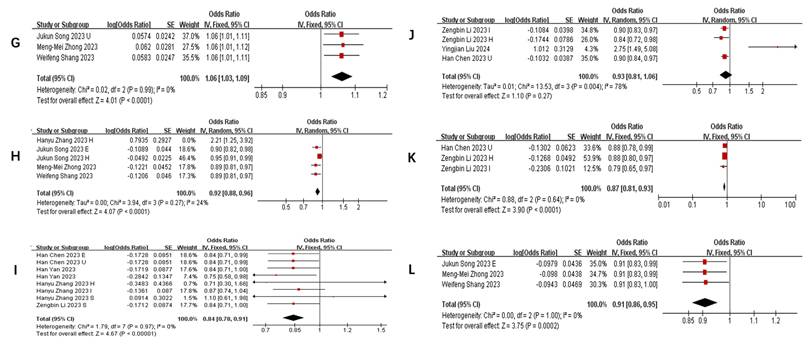
Gut microbiota dynamics across COVID-19 severity stages
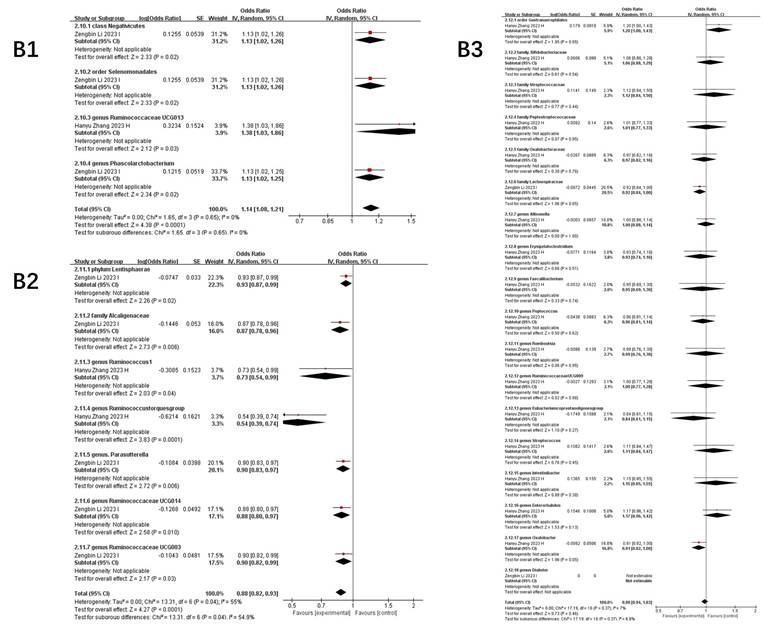
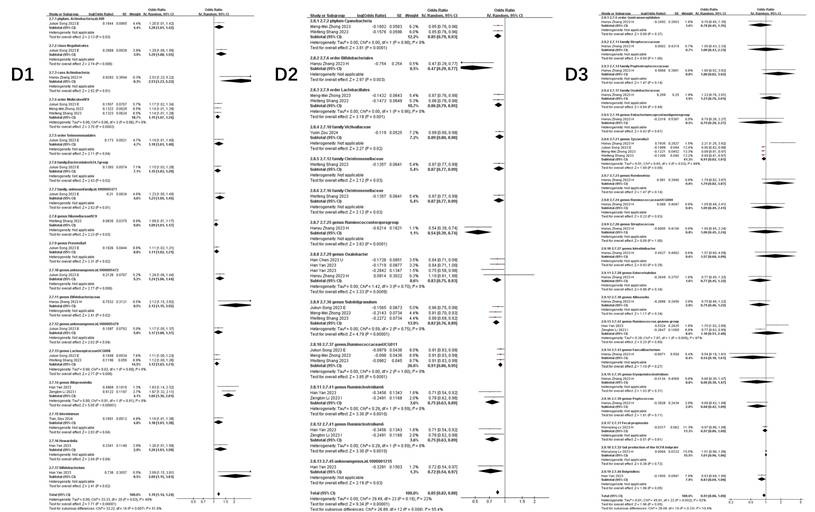
Figure 4 illustrates taxa significantly associated (P<0.05) with COVID-19 susceptibility, infection, hospitalization, and severe disease. The primary analysis of this study focused on the association between the gut microbiome and COVID-19 risk. Secondary, exploratory analyses of other outcomes (e.g., BP, BLA) are presented in Supplementary Table S3.
Forest plot of summary of gut microbiota of different COVID-19 severities. A) COVID-19 susceptibility; B) COVID-19 infection; C) COVID-19 hospitalization; D) COVID-19 severe.


COVID-19 susceptibility
Key microbial signatures identified through Mendelian randomization analysis (Figure 4 A):
Risk-enhancing taxa: genus Dorea (1.05 [1.01-1.09]), class Actinobacteria (1.16 [1.06-1.26]), class Negativicutes (1.06 [1.03-1.09]), phylum Lentisphaerae (1.02 [1.00, 1.04]), genus Alloprevotella (1.09 [1.02, 1.16]), order Selenomonadales (1.05 [1.03, 1.08]), family Bacteroidaceae (1.07 [1.03, 1.10]), genus Coprococcus (1.16 [1.03, 1.30]), and others (Figure 4 A1).
Protective taxa: genus Oxalobacter (0.84 [0.71-0.99]), genus Ruminococcaceae UCG014 (0.88 [0.78-0.99]), genus Parasutterella (0.90 [0.84, 0.97]), class Gammaproteobacteria (0.94 [0.91, 0.97]), and family Streptococcaceae (0.96 [0.93, 0.99]), among others (Figure 4 A2).
Neutral association: Bacteroides (genus: 1.02 [0.94-1.10], P=0.68) (Figure 4 A3).
This stratified pattern suggests taxonomic-specific modulation of host susceptibility, with Actinobacteria and Negativicutes emerging as consistent risk predictors.
COVID-19 infection
Stage-specific microbial dynamics revealed divergent associations (Figure 4B):
Positive correlates: Class Negativicutes (1.13 [1.02, 1.26]), order Selenomonadales (1.13 [1.02, 1.26]) (Figure 4 B1).
Negative correlates: genus Rum.inococcustorquesgroup (0.54[0.39, 0.74]), family Alcaligenaceae (0.87 [0.78, 0.96]), genus. Parasutterella (0.90 [0.83, 0.97]), phylun Lentisphaerae (0.93 [0.87, 0.99]), genus Ruminococcaceae UCG014 (0.88 [0.80, 0.97]), genus Ruminococcus1 (0.73 [0.54, 0.99]), genus Ruminococcaceae UCG003 (0.90 [0.82, 0.99]) (Figure 4 B2).
Non-significant taxa: family Bifidobacteriaceae (1.06 [0.88, 1.29]), genus Oxalobacter (0.87 [0.74, 1.04]), family Streptococcaceae (1.12 [0.84, 1.50]), genus Eubacteriumcoprostanoligenesgroup (0.84 [0.61, 1.15]), genus Oxalobacter (0.87 [0.74, 1.04]), genus Allisonella (1.00 [0.88, 1.14]), genus Erysipelatoclostridium (0.93 [0.74, 1.16]) (Figure 4 B3).
Notably, Parasutterella demonstrated dual protective roles across susceptibility (0.90 [0.84-0.97]) and infection stages (0.90 [0.83-0.97]).
COVID-19 hospitalization
Microbial predictors of clinical deterioration (Figure 4 C):
High-risk indicators: order MollicutesRF9 (1.13 [1.04, 1.22]), Alloprevotella (1.25 [1.07, 1.45]), class Actinobacteria (1.11 [1.03, 1.18]), family Bifidobacteriaceae (1.54 [0.52, 4.56]), class Negativicutes (1.23 [1.11, 1.37]), order Selenomonadales (1.13 [1.03, 1.25]). At the genus level, Dorea (1.16 [1.05, 1.28]), Prevotella9 (1.21 [1.04, 1.41]), and an unidentified genus (id.1000005472) (1.10 [1.03, 1.18]) also demonstrated positive correlations. Additionally, family-level taxa such as an unidentified family (id.1000005471) (1.11 [1.03, 1.21]) and FamilyXIII (1.30 [1.03, 1.64]), as well as order Bacteroidales (1.09 [1.01, 1.18]) and genus Eubacteriumruminantiumgroup (1.07 [1.01, 1.12]), were similarly linked to greater hospitalization severity (Figure 4 C1).
Protective factors: Ruminiclostridium6 (0.81 [0.69, 0.94]), gut production of the SCFA butyrate (0.97 [0.94-0.99]), genus Parasutterella (0.84 [0.72, 0.98]), Marvinbryantia (0.89 [0.81, 0.97]), Tyzzerella3 (0.95 [0.91, 0.99]), Ruminococcaceae UCG014 (0.86 [0.77, 0.95]), genus Olsenella (0.94 [0.91, 0.97]), and Alistipes (0.78 [0.63, 0.96]) (Figure 4 C2).
The inverse correlation between SCFA butyrate and hospitalization severity highlights potential therapeutic targets.
COVID-19 severe
Integrated analysis of nine studies (Figure 4D) revealed significant gut microbiota perturbations associated with COVID-19 severity. Three distinct microbial patterns emerged through Mendelian randomization analyses.
Risk-enhancing taxonomic signatures: Phylum-level dysbiosis was characterized by increased Actinobacteria abundance (1.20 [1.01, 1.42]), particularly within the class Negativicutes (1.29 [1.08, 1.55]). Order-level alterations demonstrated consistent elevation of Selenomonadales (1.19 [1.01, 1.40]) and Mollicutes RF9 (1.15 [1.07, 1.24]). Genus-level analysis identified multiple risk biomarkers, including Alloprevotella (1.66 [1.36, 2.01]) and Bifidobacterium (2.09 [1.15, 3.81]), the latter showing paradoxical associations despite its conventional probiotic role. Notably, an uncharacterized genus (id.1000005472) exhibited robust correlation with disease severity (1.24 [1.06, 1.44]), warranting taxonomic clarification (Figure 4 D1).
Protective microbial consortia: Commensal taxa demonstrating inverse correlations with disease severity included butyrate-producing Oxalobacter (0.84 [0.76, 0.92]) and mucin-degrading Subdoligranulum (0.82 [0.76, 0.89]). The order Lactobacillales (0.86 [0.79, 0.95]) showed particular promise for microbial intervention, potentially through competitive exclusion mechanisms. Cyanobacteria at the phylum level (0.85 [0.79, 0.93]) suggested light-dependent metabolic pathways might influence disease progression (Figure 4 D2).
Neutral microbial associations: Multiple taxa including Faecalibacterium (0.54 [0.18, 1.61]) and Romboutsia (1.79 [0.82, 3.87]) demonstrated non-significant associations (all P>0.05), with confidence intervals spanning protective to risk-enhancing ranges. Gut metabolites showed similar neutrality, with butyrate (1.01 [0.96, 1.06]) and fecal propionate (0.97 [0.86, 1.09]) production levels exhibiting no disease-modifying effects (Figure 4 D3).
Collectively, this analysis delineates a dynamic reciprocity between gut microbiota composition and COVID-19 severity, wherein specific taxa (e.g., Actinobacteria, Negativicutes) demonstrate disease-aggravating effects, while others (Oxalobacter, Parasutterella) confer protection through immunomodulatory pathways, alongside commensal taxa exhibiting neutral disease associations.
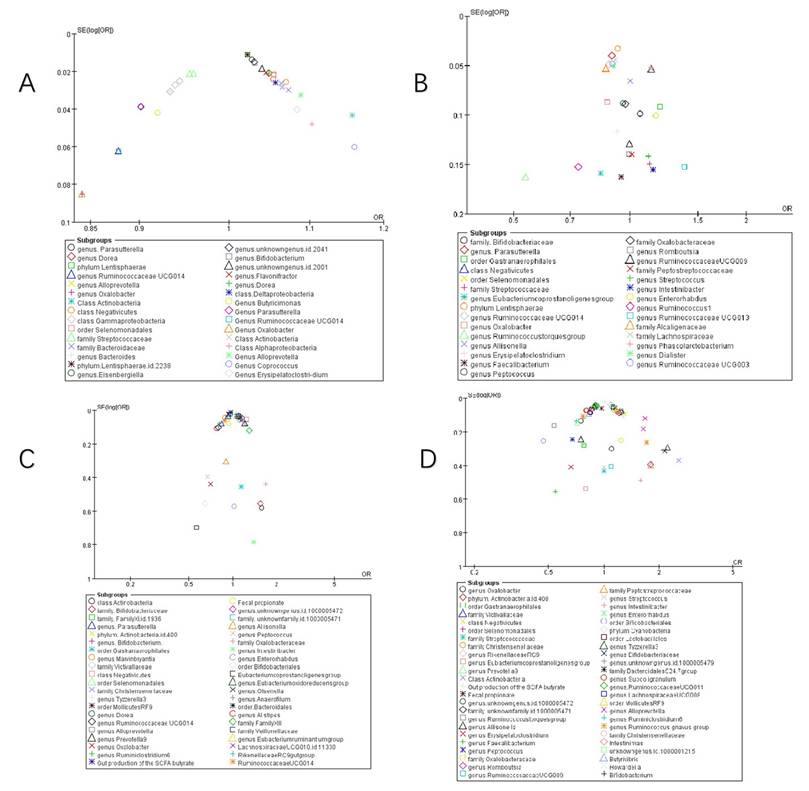
Publication bias
Funnel plots assessed publication bias in pneumonia studies. COVID-19 analyses demonstrated acceptable bias ranges for suspected cases (Figure 5 A), hospitalizations (Figure 5 C), and severe outcomes (Figure 5 D), evidenced by symmetrical distribution of studies near the funnel apex. Conversely, COVID-19 infection studies (Figure 5 B) exhibited significant publication bias, potentially due to limited included reports. Egger's regression modeled the logarithm of effect size against standard error, accounting for sample size influences on estimation precision. Results indicated: no significant publication bias for hospitalizations (t = 1.296, df = 42, p = 0.202; β = -0.019, 95% CI: -0.061 to 0.023), severe cases (t = 0.517, df = 65, p = 0.607; β = -0.037, 95% CI: -0.105 to 0.029), infections (t = 0.585, df = 22, p = 0.565; β = -0.089, 95% CI: -0.180 to 0.003), or susceptibility (t = -0.859, df = 39, p = 0.396; β = 0.039, 95% CI: 0.003 to 0.075). Minimal funnel asymmetry and nonsignificant intercept terms support robust pooled estimates.
Funnel plot of summary of gut microbiota of different COVID-19 severities. A) COVID-19 susceptibility; B) COVID-19 infection; C) COVID-19 hospitalization; D) COVID-19 severe.
Discussion
The current body of research, encompassing data from approximately 32, 748, 274 participants, has demonstrated a significant association between COVID-19 (new coronary pneumonia) and intestinal microbiota. An increasing number of scientific studies have progressively elucidated the role of gut microbiota in the development and progression of COVID-19 [28-30]. This study is the first to employ Mendelian randomization (MR) to systematically summarize these findings. The MR approach assumes that the observed associations are independent of traditional confounding factors, thereby providing a robust framework for causal inference.
Our analysis incorporated data from 11 studies, involving over 32, 748, 274 participants and 1, 487 single nucleotide polymorphisms (SNPs), to investigate the causal relationship between intestinal microbiota, its metabolites, and COVID-19. Among these studies, two focused on COVID-19 infection, four on COVID-19 susceptibility, nine on COVID-19 severe, six on COVID-19 hospitalization, and others on related conditions, including bacterial pneumonia (BP), pneumococcal pneumonia (PP), and bronchopneumonia or lung abscess (Table S3). The analysis revealed that certain microbial taxa, such as order Bifidobacteriales, genus Ruminococcustorquesgroup, genus Ruminiclostridium6, genus Oxalobacter, genus Ruminococcaceae UCG014, genus Olsenella, genus Tyzzerella3, genus Subdoligranulum, family Christensenellaceae, phylum Cyanobacteria, order Lactobacillales, class Gammaproteobacteria, genus Anaerofilum, genus Parasutterella, and family Streptococcaceae, were negatively correlated with an increased risk of pneumonia. Conversely, other taxa, such as class Actinobacteria, genus Prevotella 9, genus Alloprevotella, genus Lachnospiraceae UCG008, genus Rikenellaceae RC9, order Mollicutes RF9, genus Bacteroides, and family Bacteroidaceae, were positively correlated with an increased risk of pneumonia. Interestingly, the order Gastroaerophilales exhibited an inconspicuous association with pneumonia. These findings suggest that the causal relationship between gut microbiota and pneumonia, including COVID-19, may become clearer as more MR studies are conducted.
The gut microbiota critically regulates spatiotemporal dynamics of mucosal immune homeostasis, demonstrating pathogen-specific immunomodulatory patterns in bacterial, viral, and fungal pneumonias [31]. This microbial regulation directly influences host immune responses according to pathogen class. Notably, COVID-19-induced gut dysbiosis correlates with neuropsychiatric sequelae [32-35] and exacerbates disease severity in obese NASH patients, where Peptococcus abundance associates with pro-inflammatory signatures in pulmonary and hepatic tissues [36]. Although Streptococcus enrichment occurs in COVID-19 cohorts [37] - contrasting with our findings (Table S3) - emerging evidence implicates elevated streptococcal loads in upregulating viral entry receptors, potentially facilitating infection [38]. Similarly, anaerobic genera such as Prevotella and Veillonella may propagate under hypoxia, potentially contributing to secondary pulmonary infections [39].
Consistent with our data, multiple studies report reduced α-diversity in COVID-19 fecal microbiomes [40-43], diminished SCFA-producing taxa (e.g., Ruminococcaceae), and decreased Ruminococcus abundance [42, 44]. Gut-derived SCFAs, particularly butyrate, translocate hematogenously to the pulmonary compartment where they enhance alveolar macrophage bactericidal capacity while suppressing pro-inflammatory cytokine cascades [31]. Clinically, severe COVID-19 exhibits marked depletion of butyrogenic bacteria including Faecalibacterium [34]. Therapeutic Bifidobacterium supplementation partially restores microbial diversity, fortifies intestinal barrier integrity, and demonstrates efficacy against mycoplasma pneumonia [35]. A paradoxical positive association (odds ratio [OR] > 2.0) was observed for the Bifidobacterium genus, contradicting prior expectations [35]. This finding may be attributable to genetic pleiotropy, wherein genetic variants underlying the instrumental variables could directly influence COVID-19 severity through alternative biological mechanisms, independent of their effects on microbial abundance. Furthermore, microbial impacts are likely highly context-dependent, modulated by specific host immune status and environmental context.
Furthermore, COVID-19 severity inversely correlates with Actinomycetota abundance but positively associates with Bacteroidetes prevalence, alongside pandemic-associated shifts in antimicrobial resistance genes [37]. Critically, Ruminococcus torques group abundance confers reduced infection risk, while Bifidobacteriales enrichment predicts lower severe disease incidence [44] - observations aligning with our dataset. Collectively, these findings position the gut microbiota as a viable target for COVID-19 adjuvant therapies [45].
Given the extensive assessment of gut microbial taxa in this study, potential false-positive associations may arise due to multiple testing. Furthermore, as the primary GWAS summary statistics were derived from European ancestry cohorts, the generalizability of findings to other populations is constrained. Future replication studies in more diverse ethnic populations are required to validate these associations.
While Mendelian randomization analyses cannot establish definitive causality and remain limited by cohort sizes, they provide compelling evidence for gut microbiota-COVID-19 interactions. This synthesis resolves taxonomic controversies and advances evidence-based medicine, suggesting that clinical microbiota modulation may improve COVID-19 prognoses. Further mechanistic investigations are warranted to translate these associations into therapeutic strategies.
Conclusion
In summary, we conducted a comprehensive assessment to estimate the causal relationship between gut microbiota and COVID-19. Our meta-analysis incorporated data from 11 studies, encompassing over 32, 748, 274 participants and 1, 487 single nucleotide polymorphisms (SNPs). This analysis identified potential causal links between 52 types of gut microbiota, two gut microbiota metabolites, and four levels of COVID-19 severity. These findings contribute to a deeper understanding of the role of gut microbiota in the progression of COVID-19 and aim to provide an evidence-based foundation for exploring the interplay between the gut microbiome and the disease.
Supplementary Material
Supplementary tables.
Acknowledgements
Funding
This study was supported by Zhuhai Social Development Field Science and Technology Plan-Key Project (No. 2320004000286, to RYY.), State Key Laboratory of Traditional Chinese Medicine Syndrome Project (No. QZ2023ZZ23, to R.Y.Y.), Science and Technology Planning Project of Guangdong Province ( No.2023B1212060062, to R.Y.Y.), Science and Technology Planning Project of Guangdong Province (No.2023B1212060062, to R.Y.Y.), Joint Research Project of the Chinese Society of Traditional Chinese Medicine (No. 2023DEPLHGG-06, to R.Y.Y.).
Data availability statement
The data that supports the findings of this study are available in the supplementary material of this article.
Author contributions
QL contributed to conceptualization, methodology and designed the study. QPD and YYL contributed to literature search, data curation, formal analysis and made major contribution to write the first version of manuscript. QL and RYY contributed to supervision of the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Organization WH. WHO coronavirus (COVID-19) dashboard. 2021.
2. Huang J, Zhang L, Liu X, Wei Y, Liu C, Lian X. et al. Global prediction system for COVID-19 pandemic. Science bulletin. 2020;65:1884-7
3. Kucharski AJ, Russell TW, Diamond C, Liu Y, Edmunds J, Funk S. et al. Early dynamics of transmission and control of COVID-19: a mathematical modelling study. The Lancet Infectious diseases. 2020;20:553-8
4. Wu JT, Leung K, Leung GM. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet (London, England). 2020;395:689-97
5. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nature medicine. 2018;24:392-400
6. Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science (New York, NY). 2005;307:1915-20
7. Mancabelli L, Milani C, Fontana F, Lugli GA, Tarracchini C, Viappiani A. et al. Untangling the link between the human gut microbiota composition and the severity of the symptoms of the COVID-19 infection. Environmental microbiology. 2022;24:6453-62
8. Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H. et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology. 2020;159:944-55.e8
9. Cheng X, Zhang Y, Li Y, Wu Q, Wu J, Park SK. et al. Meta-analysis of 16S rRNA microbial data identified alterations of the gut microbiota in COVID-19 patients during the acute and recovery phases. BMC microbiology. 2022;22:274
10. Zhang F, Lau RI, Liu Q, Su Q, Chan FKL, Ng SC. Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nature reviews Gastroenterology & hepatology. 2023;20:323-37
11. Bingqing S, Qiu W, Yue G. Effect of microbiota-gut-liver communication during SARS-CoV-2 infection on liver injury. Chemistry of Life. 2024;44:216-24
12. Moon Y. Gut distress and intervention via communications of SARS-CoV-2 with mucosal exposome. Frontiers in public health. 2023;11:1098774
13. Patra S, Saxena S, Sahu N, Pradhan B, Roychowdhury A. Systematic Network and Meta-analysis on the Antiviral Mechanisms of Probiotics: A Preventive and Treatment Strategy to Mitigate SARS-CoV-2 Infection. Probiotics and antimicrobial proteins. 2021;13:1138-56
14. Woodall CA, McGeoch LJ, Hay AD, Hammond A. Respiratory tract infections and gut microbiome modifications: A systematic review. PloS one. 2022;17:e0262057
15. Li J, Ghosh TS, McCann R, Mallon P, Hill C, Draper L. et al. Robust cross-cohort gut microbiome associations with COVID-19 severity. Gut microbes. 2023;15:2242615
16. Mengyuan W, Hengmin X, Jingxuan W, Kaifeng P, Wenqing L. Mendelian Randomization Analysis of Research on Risk Factors for Gastric Cancer. Cancer Research on Prevention and Treatment. 2023;50:470-6
17. Chen H, Ye B, Su W, Song Y, Sun PL, Zhou X. et al. The causal role of gut microbiota in susceptibility and severity of COVID-19: A bidirectional Mendelian randomization study. Journal of medical virology. 2023;95:e28880
18. Li Z, Zhu G, Lei X, Tang L, Kong G, Shen M. et al. Genetic support of the causal association between gut microbiome and COVID-19: a bidirectional Mendelian randomization study. Frontiers in immunology. 2023;14:1217615
19. Liu Y, Zhu Q, Guo G, Xie Z, Li S, Lai C. et al. Causal associations of genetically predicted gut microbiota and blood metabolites with inflammatory states and risk of infections: a Mendelian randomization analysis. Frontiers in microbiology. 2024;15:1342653
20. Shang W, Zhang S, Qian H, Pan X, Huang S, Wen Z. et al. Association of gut microbiota with COVID-19 susceptibility and severity: A two-sample Mendelian randomization study. Journal of medical virology. 2023;95:e28734
21. Song J, Wu Y, Yin X, Ma H, Zhang J. The causal links between gut microbiota and COVID-19: A Mendelian randomization study. Journal of medical virology. 2023;95:e28784
22. Tian S, Huang W. The causal relationship between gut microbiota and COVID-19: A two-sample Mendelian randomization analysis. Medicine. 2024;103:e36493
23. Zhang H, Zhou Z. Association of gut microbiota and dietary component intake with COVID-19: A mendelian randomization study. Clinical nutrition (Edinburgh, Scotland). 2023;42:1308-13
24. Zhong MM, Xie JH, Feng Y, Zhang SH, Xia JN, Tan L. et al. Causal effects of the gut microbiome on COVID-19 susceptibility and severity: a two-sample Mendelian randomization study. Frontiers in immunology. 2023;14:1173974
25. Zou Y, Pan M, Zhou T, Yan L, Chen Y, Yun J. et al. Critical COVID-19, Victivallaceae abundance, and celiac disease: A mediation Mendelian randomization study. PloS one. 2024;19:e0301998
26. Lv W, He J, Shao J, Chen Y, Xia L, Zhang L. Causal relationships between short-chain fatty acids and L-isoleucine biosynthesis and susceptibility and severity of COVID-19: Evidence from Mendelian randomization. The Journal of infection. 2023;87:e16-e8
27. Yan H, Zhao S, Huang HX, Xie P, Cai XH, Qu YD. et al. Systematic Mendelian randomization study of the effect of gut microbiome and plasma metabolome on severe COVID-19. Frontiers in immunology. 2023;14:1211612
28. Nagata N, Takeuchi T, Masuoka H, Aoki R, Ishikane M, Iwamoto N. et al. Human Gut Microbiota and Its Metabolites Impact Immune Responses in COVID-19 and Its Complications. Gastroenterology. 2023;164:272-88
29. Ancona G, Alagna L, Alteri C, Palomba E, Tonizzo A, Pastena A. et al. Gut and airway microbiota dysbiosis and their role in COVID-19 and long-COVID. Frontiers in immunology. 2023;14:1080043
30. Dhar D, Mohanty A. Gut microbiota and Covid-19- possible link and implications. Virus research. 2020;285:198018
31. Ye F, Li L, Wang J, Yang H. Advances in gut-lung axis research: clinical perspectives on pneumonia prevention and treatment. Frontiers in immunology. 2025;16:1576141
32. Blackett JW, Sun Y, Purpura L, Margolis KG, Elkind MSV, O'Byrne S. et al. Decreased Gut Microbiome Tryptophan Metabolism and Serotonergic Signaling in Patients With Persistent Mental Health and Gastrointestinal Symptoms After COVID-19. Clinical and translational gastroenterology. 2022;13:e00524
33. Sen P, Molinero-Perez A, O'Riordan KJ, McCafferty CP, O'Halloran KD, Cryan JF. Microbiota and sleep: awakening the gut feeling. Trends in molecular medicine. 2021;27:935-45
34. Fan R, Liu S, Sun N, Yang Y, Deng X, Hu B. et al. Gut microbiota composition is associated with disease severity and host immune responses in COVID-19. Frontiers in cellular and infection microbiology. 2023;13:1274690
35. Ling Z, Liu X, Guo S, Cheng Y, Shao L, Guan D. et al. Role of Probiotics in Mycoplasma pneumoniae Pneumonia in Children: A Short-Term Pilot Project. Frontiers in microbiology. 2018;9:3261
36. Sencio V, Benech N, Robil C, Deruyter L, Heumel S, Machelart A. et al. Alteration of the gut microbiota's composition and metabolic output correlates with COVID-19-like severity in obese NASH hamsters. Gut microbes. 2022;14:2100200
37. Tao W, Zhang G, Wang X, Guo M, Zeng W, Xu Z. et al. Analysis of the intestinal microbiota in COVID-19 patients and its correlation with the inflammatory factor IL-18. Medicine in microecology. 2020;5:100023
38. Peng Y, Zhang D, Chen T, Xia Y, Wu P, Seto WK. et al. Gut microbiome and resistome changes during the first wave of the COVID-19 pandemic in comparison with pre-pandemic travel-related changes. Journal of travel medicine. 2021;28:taab067
39. Gupta A, Karyakarte R, Joshi S, Das R, Jani K, Shouche Y. et al. Nasopharyngeal microbiome reveals the prevalence of opportunistic pathogens in SARS-CoV-2 infected individuals and their association with host types. Microbes and infection. 2022;24:104880
40. Langevin S, Pichon M, Smith E, Morrison J, Bent Z, Green R. et al. Early nasopharyngeal microbial signature associated with severe influenza in children: a retrospective pilot study. The Journal of general virology. 2017;98:2425-37
41. Gaibani P, D'Amico F, Bartoletti M, Lombardo D, Rampelli S, Fornaro G. et al. The Gut Microbiota of Critically Ill Patients With COVID-19. Frontiers in cellular and infection microbiology. 2021;11:670424
42. Gu S, Chen Y, Wu Z, Chen Y, Gao H, Lv L. et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2020;71:2669-78
43. Xu R, Lu R, Zhang T, Wu Q, Cai W, Han X. et al. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Communications biology. 2021;4:240
44. Ren Z, Wang H, Cui G, Lu H, Wang L, Luo H. et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut. 2021;70:1253-65
45. Europe must come together to confront omicron. BMJ (Clinical research ed). 2022; 376: o90.
Author contact
![]() Corresponding author: Hui Rong, Professor, E-mail: 525342938com, Wuhan, Hubei 430065, P. R. China; phone 86-13986096556, fax 86-13986096556. Qing Liu, Associate chief physician, E-mail: 851757626com, 111 Dade Road, Guangzhou, Guangdong 510120, P. R. China; phone 86-13631223512, fax 8602081887233. Rongyuan Yang, Professor, E-mail: yangrongyuancom, phone 8602081887233, fax 8602081887233.
Corresponding author: Hui Rong, Professor, E-mail: 525342938com, Wuhan, Hubei 430065, P. R. China; phone 86-13986096556, fax 86-13986096556. Qing Liu, Associate chief physician, E-mail: 851757626com, 111 Dade Road, Guangzhou, Guangdong 510120, P. R. China; phone 86-13631223512, fax 8602081887233. Rongyuan Yang, Professor, E-mail: yangrongyuancom, phone 8602081887233, fax 8602081887233.