Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Copy Number Variation of PIK3R1...
Mutation Landscape of PIK3R1 in...
Epigenetic Alterations Occurring...
Clinical Drugs Targeting the PAM...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(12):2932-2943. doi:10.7150/ijms.109506 This issue Cite
Review
Cancer-Associated PIK3R1 Genetic Aberrations and Precision Medicine
Huan Xie1,2, Yirong Li1,2, Xinran Li1,2 ![]()
1. Department of Laboratory Medicine, Zhongnan Hospital of Wuhan University, Wuhan, Hubei, China.
2. Hubei Provincial Clinical Research Center for Molecular Diagnostics, Wuhan, China.
Received 2024-12-27; Accepted 2025-5-6; Published 2025-6-12
Abstract

The PIK3R1 gene encodes the class IA PI3K regulatory subunit p85α, which is frequently altered in cancer. PIK3R1 functions as a tumor suppressor by stabilizing and inhibiting the catalytic activity of p110, and it directly binds to and enhances the activity of the PTEN lipid phosphatase. Aberrations in the PIK3R1 gene are associated with poor prognosis in cancer; available data underscore the significant role of PIK3R1 mutations in mediating tumorigenesis by promoting the signaling of the PI3K/AKT/mTOR pathway. Moreover, copy number variations, driver mutations, and epigenetic alterations in PIK3R1 contribute to tumorigenesis and progression through distinct mechanisms. This article reviews the cancer-promoting effects of PIK3R1 gene aberrations across major cancer types and elucidates their underlying mechanisms. It also discusses the targeted therapies for related aberrations, aiming to provide a comprehensive understanding of the dynamic interplay of PIK3R1 in cancer, thereby advancing precision medicine and the development of targeted interventions.
Keywords: PIK3R1, copy number variation, mutation, epigenetic
Introduction
The phosphoinositide 3-kinase (PI3K) signaling pathway plays a crucial role in metabolic control, immune responses, angiogenesis, and cardiovascular homeostasis, and it is among the most frequently dysregulated pathways in cancer [1-3]. All PI3K catalytic subunits have a PI3K core structure consisting of a C2 domain, a helical domain, and a kinase domain [1]. PI3K is categorized into three distinct classes primarily based on the presence of additional protein domains and their interaction with regulatory subunits [4]. Class IA PI3K comprises a p110 catalytic subunit and a regulatory subunit, either p85 or p55. The genes PIK3CA, PIK3CB, and PIK3CD encode the p110α, p110β, and p110δ catalytic subunits, respectively. Meanwhile, the PIK3R1, PIK3R2, and PIK3R3 genes encode the p85α, p85β, and p55γ regulatory subunits, respectively [5].
The p85α protein is predominantly recognized as a regulatory subunit of class IA PI3K. The PIK3R1 gene encodes p85α, which stabilizes and inhibits the catalytic activity of p110α, while the latter catalyzes the conversion of phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol 3,4,5-triphosphate (PIP3) [6]. As a second messenger, PIP3 binds to a variety of target proteins within cells, thereby regulating cell proliferation, differentiation, apoptosis, metabolism, and other physiological processes [7]. Additionally, the p85α regulatory protein binds directly to and enhances the activity of the PTEN lipid phosphatase, which counteracts PI3K signaling by dephosphorylating the PI3K lipid product [8, 9].
This article examines the copy number variation (CNV) of PIK3R1, with particular emphasis on its gene expression levels and the mechanisms through which gene dosage sensitivity influences tumorigenesis. Furthermore, the article explores the cancer-promoting effects of mutations in various domains of PIK3R1. We also discuss the role of epigenetic alterations in PIK3R1 in cancer initiation and progression. Finally, treatment and management guidelines will be provided, addressing three key aspects: PIK3R1 copy number variation, mutations in its domains, and epigenetic changes.
Copy Number Variation of PIK3R1 in Cancer
Within The Cancer Genome Atlas (TCGA) database, PIK3R1 aberration is one of the most prevalent alterations [10]. The TCGA database shows that the loss of PIK3R1 copy number frequently occurs in various cancer types, which is consistent with the tumor suppressor role of p85α (Fig. 1). Cancers exhibiting PIK3R1 copy number loss may promote tumor development through distinct mechanisms that activate downstream signaling pathways. Analysis of data from the TCGA database reveals that PIK3R1 is lowly expressed in most tumors, compared to their corresponding normal tissues, including ovarian, prostate, breast, lung, liver, and kidney cancers [11-13].
Frequency of PIK3R1 copy number variations and mutations. The distribution of PIK3R1 copy number loss, copy number gain, and mutations across various tumor types in the TCGA dataset is illustrated.
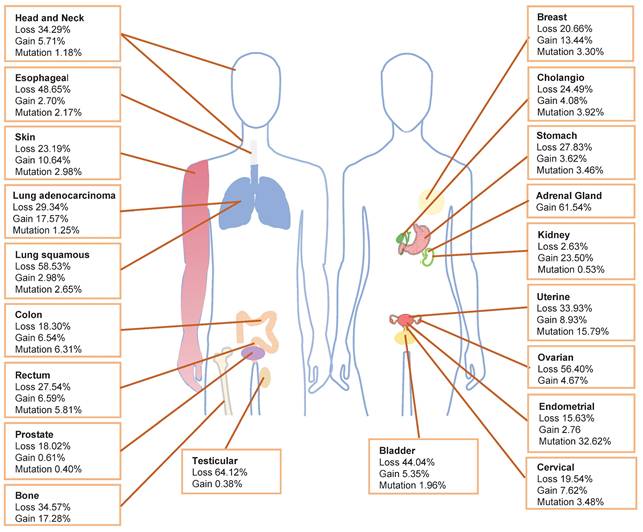
Hemizygous deletion of PIK3R1 is a prevalent occurrence in breast cancer, correlating with markedly reduced PIK3R1 expression in breast tumors [14]. Lower levels of PIK3R1 expression are linked to poorer survival outcomes in breast cancer patients and contribute to tumorigenic transformations in breast cancer models. Furthermore, reduced p85α levels lead to heightened classical AKT signaling, which plays a significant role in these tumorigenic phenotypes [12, 14, 15]. Research indicates that the knockdown of PIK3R1 in breast cells triggers malignant transformation [16]. In the context of prostate cancer, PIK3R1 depletion enhances AKT phosphorylation and promotes the proliferation of prostate cancer cells [17, 18]. Similarly, in renal cancer cells, the depletion of PIK3R1 promotes AKT phosphorylation, proliferation, migration, epithelial-to-mesenchymal transition, and the emergence of stem cell-like properties via the AKT/GSK3β/CTNNB1 pathway, potentially contributing to the progression and metastasis of renal cell carcinoma [19]. Among various cancers, copy-number deletion of PIK3R1 is most commonly observed in ovarian cancer, where its deletion activates AKT and induces p110-independent JAK2/STAT3 signaling through phosphorylation changes in the docking protein Gab2. Additional mechanisms that lead to AKT activation include increased p110α kinase activity and decreased PTEN levels [20].
Haploinsufficiency of PIK3R1 activates the PI3K pathway; conversely, homozygous deletion inhibits this pathway [21]. Partial deletion of p85α enhances the p110α-p85α heterodimer's binding affinity to active receptors, thus amplifying PI3K signaling and oncogenic transformation [14, 22]. On the other hand, homozygous deletion of p85α significantly reduces the amount of p110α-p85α dimers, leading to a marked decrease in PI3K activity and a reduction in PI3K-mediated biological processes such as anti-apoptosis [21].
Studies conducted in genetically engineered mouse models demonstrate that single-copy deletion of PIK3R1 activates AKT and promotes tumorigenesis [14, 22, 23]. Similar findings were reported in a mouse model of hepatocellular carcinoma characterized by liver-specific PIK3R1 deficiency, which resulted in enhanced tumor development [13]. In animal models of cancer, liver-specific knockout of PIK3R1 has also been shown to increase PI3K pathway activation, thereby facilitating tumorigenesis [13]. Additionally, experiments mimicking human tumors revealed that knockdown of p85α resulted in p110α conjugation to p85β, increased MAP4 interactions, enhanced integration with endosomal membranes, and augmented interactions with activated receptors, culminating in intensified agonist-stimulated PI3K/AKT signaling [24]. Furthermore, low PIK3R1 expression correlates with poor prognosis across multiple cancer types [11]. The reduction of PIK3R1 is particularly associated with dismal prognoses in breast and lung cancers, potentially due to its critical role in stabilizing PTEN [3, 12, 25]. Thus, PIK3R1 deletion activates downstream AKT signaling and facilitates tumorigenesis through various mechanisms across different cancer types, which is associated with unfavorable prognoses for patients.
Notably, PIK3R1 copy-number gain remains under-discussed in the literature. Amplification of PIK3R1 is present in 0.04% of cases recorded in the AACR GENIE database [26]. Additionally, in the TCGA database, breast invasive ductal carcinoma, lung adenocarcinoma, clear cell renal cell carcinoma, adenocarcinoma of unknown primary origin, and adrenal cortex carcinoma have the greatest prevalence of PIK3R1 copy number ampilfication (Fig. 1).
Mutation Landscape of PIK3R1 in Cancer
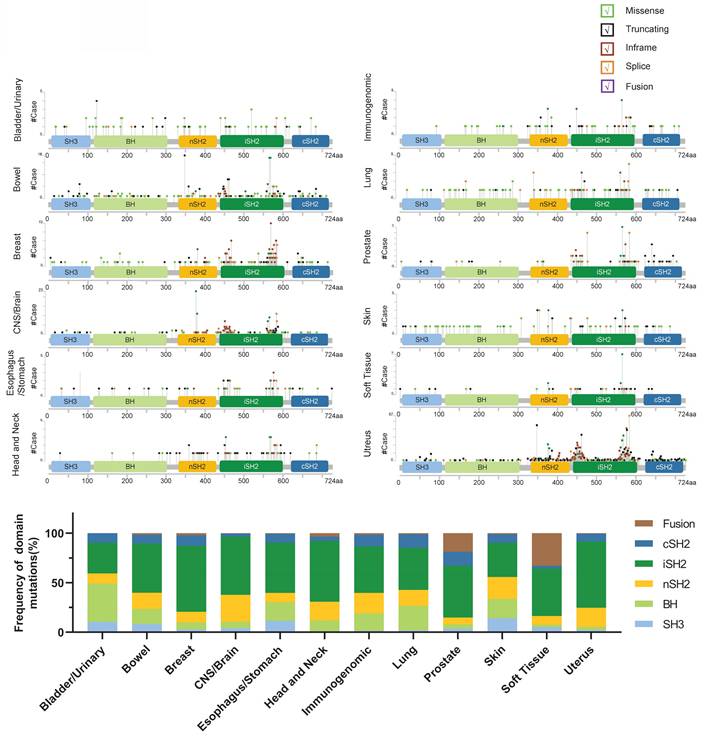
PIK3R1 is located on human chromosome 5 and comprises five protein domains: the Src homology 3 (SH3) domain, the breakpoint cluster region homology (BH) domain, the N-terminal SH2 (nSH2) domain, the middle SH2 (iSH2) domain, and the C-terminal SH2 (cSH2) domain. Notably, cancer-associated mutations have been identified in all five domains [27]. Hotspot mutations in PIK3R1 are predominantly found within the iSH2 and SH2 domains (Fig. 2), supporting a significant role for p110α in stabilizing and inhibiting p85α-p110α heterodimers through these domains [28].
Schematic representation of the PIK3R1 protein and its mutation frequency. The PIK3R1 gene encodes the p85 regulatory subunit, which comprises 724 amino acids and five structural domains: the SH3 domain, the BH domain, the nSH2 domain, the iSH2 domain, and the cSH2 domain. (Upper) The mutation frequency of PIK3R1 across different tissues is shown, based on data from combined studies in the cBioPortal database. The graphical representation depicts the protein domain structure and specific mutation sites. The length of the lines connecting mutations to the protein reflects the number of samples exhibiting those mutations, and the color of the spheres indicates the type of mutation. (Lower) The bar chart shows the mutation frequencies across the SH3, BH, nSH2, iSH2, and cSH2 domains of the PIK3R1 gene, as derived from combined studies of various tissues in the cBioPortal database.
1. SH3 and BH domains
The SH3 and BH domains at the N-terminus of p85 can form homodimers and bind to PTEN [8, 29, 30]. When p85α is devoid of p110α, it can homodimerize via two intermolecular interactions (SH3: proline-rich region; BH: BH) to selectively bind unphosphorylated activated PTEN [31]. PTEN, a tumor suppressor protein, is often lost or mutated in up to 30% of human cancers [32-34]. Acting as a phosphatase, PTEN dephosphorylates the D3 position of PIP3, counteracting the activation of the oncogenic PI3K/AKT/mTOR (PAM) signaling network [35]. The p85α-PTEN interaction is associated with enhanced PTEN stability by inhibiting ubiquitination [30]. Furthermore, homodimers may offer a combinatorial binding site for PTEN and potentially promote the recruitment of other molecules to stabilize PTEN [30]. Mutations in the SH3-BH domain diminish homodimerization and PTEN binding, leading to increased PTEN ubiquitination and reduced total PTEN protein levels. The p85α homodimer has been shown to compete with the E3 ubiquitin-protein ligase WWP2 for binding to the PTEN phosphatase domain, thus protecting PTEN from WWP2-mediated degradation. Disruption of p85α homodimerization increases WWP2-mediated PTEN degradation and enhances its ubiquitination [31]. Notably, cellular expression of p85α lacking the SH3-BH domain significantly elevates the amplitude and duration of AKT phosphorylation in response to growth factor stimulation.
Additionally, the BH domain of the p85α protein exhibits sequence homology with the GAP domain of other proteins and displays GAP activity against several Rab GTPases, particularly Rab5 and Rab4. These GTPases play a crucial role in receptor trafficking and degradation, thus deactivating upstream receptor signaling and the PI3K/AKT pathway [3, 36]. A study revealed that a RabGAP-deficient p85α mutant with a single-point mutation (R274A) has carcinogenic properties [37]. Mutations in the p85α BH domain may contribute to tumorigenesis in human cancers, either by modulating Rab-mediated receptor degradation or by diminishing the positive regulation of PTEN activity [8]. Consequently, mutations in the SH3-BH domain compromise PTEN stability by impairing homodimerization and PTEN binding, resulting in reduced negative feedback to the PI3K/AKT pathway and leading to persistent activation of downstream signals.
2. nSH2 domain
Driver mutations in the p85α nSH2 domain exert a precancerous effect by regulating the PI3Kα pathway through upstream signaling proteins. The p85α nSH2 domain establishes inhibitory contacts with p110α, competing for binding to the phosphorylated tyrosine-containing (pY) consensus sequences (pYXXM) motif in receptor tyrosine kinases (RTKs) [28, 38]. The nSH2 domain interacts reversibly with the C2, helix, and kinase domains of p110α, with these contacts being disrupted upon binding to the pYXXM motif [39-41]. Alkaline residues surrounding the phosphopeptide binding site in the nSH2 domain form inhibitory contacts with the acidic patch in the p110α helix domain. Binding of phosphoproteins to the nSH2 domain may disrupt these inhibitory contacts and activate the p85α/p110α dimer [39]. The release of p85α's inhibitory effect on the p110α catalytic subunit occurs when the nSH2 domain binds to phosphorylated RTKs or adaptor proteins following upstream stimulation [42]. Phosphopeptide binding to nSH2 directly influences the activity of p110α, establishing the nSH2 domain as a direct regulator of p110α [28].
Research has identified six nSH2 variants that attenuate these inhibitory contacts by directly affecting p110 binding (G376R, K379E, and L380del), or by disrupting the folding of the nSH2 domain (W333R, G353R, LR372del) [43]. Furthermore, these driver mutations in the nSH2 domain of p85α activate various RTK signaling proteins, such as EGFR, HER2, HER3, c-Met, and IGF-1R, in a p110-independent manner, thereby diminishing the inhibitory effect of p85α on p110α and facilitating its catalytic activity [43]. Mutations that disrupt the nSH2-helix interface were also found to weaken the interaction between the C2-iSH2 domains and the remainder of the adaptor and catalytic subunits, suggesting that the nSH2 domain not only inhibits enzyme activity via the C-leaf interaction with the kinase domain but also plays a crucial scaffold role in stabilizing the enzyme and preventing the inter-domain movements necessary for membrane binding [40, 44]. Moreover, p85α mutants lacking the p110α binding region (e.g., R162*, L380fs, R348*, and the dominant negative mutant p85Δ) failed to interact with p110α [45]. Notably, PIK3R1R348* and PIK3R1L370fs localize to the nucleus, serving as scaffolds for multiple JNK pathway components, and promote malignant phenotypes through ERK and JNK signaling pathways both in vitro and in vivo [46]. Collectively, mutations in the nSH2 domain of p85α not only reduce the inhibitory interaction between p85α and p110α but also enhance p110α activity by inducing the activation of multiple RTKs.
3. iSH2 domain
Somatic mutations in PIK3R1 have been identified in glioblastoma and endometrial cancer, predominantly manifesting as substitutions or indels within the iSH2 domain of p85α, which interacts with p110α [28, 30]. These mutations lead to the uncoupling of p85α from p110, allowing p85α to retain its p110-stabilizing activity while abolishing its inhibitory effect on p110α. This alteration promotes cell survival, activates AKT, supports anchorage-independent cell growth, and facilitates tumorigenesis in a p110α-dependent manner [45]. Furthermore, the iSH2 domain of p85α also engages catalytic subunits through the ABD domain. The ABD domain of class IA PI3K irreversibly binds to the coiled-coil domain of iSH2 across all class IA regulatory subunits [39, 47, 48]. Because the ABD domain remains tightly associated with the iSH2 domain, its detachment may coincide with the disruption of the C2-iSH2 interface, with complete disruption of both interfaces only occurring upon removal of the nSH2 domain [48]. These activating mutations within the iSH2 domain induce the constitutive activation of class IA PI3K kinases and contribute to tumorigenesis by disrupting the boundary between the C2 and iSH2 domains, diminishing the p110α-p85α interaction, and relieving the inhibition of PI3K activity in vivo [47, 49]. Additionally, the oncogenic mutation from Glu545 to Lys545 (E545K) disrupts the charge-charge interaction with the nSH2 domain of p85 [39]. Consequently, driver mutations in the iSH2 domain diminish the inhibitory effect of p85α on p110 kinase activity by disrupting the inhibitory interactions between the iSH2 and p110 C2 domains and targeting the inhibitory interactions between the nSH2 domain and the p110 helix domain.
4. cSH2 domains
In addition to binding to p110, both the nSH2 and cSH2 domains interact with phosphorylated RTKs or pYXXM in adaptor proteins [28]. The cSH2 domain of p85α serves as a negative regulatory element in PI3K signaling, with its deletion resulting in increased signaling activity [50]. Notably, mutations in the cSH2 domain, including the oncogenic truncation mutation (E601*) and the mutation affecting the phosphorylated peptide binding site (R649W) in SHORT syndrome, both lead to diminished sensitivity to PDGFR pY, underscoring the essential role of cSH2 in mediating effective PI3K signaling downstream of activated membrane receptors [51, 52]. Furthermore, the impact of truncated mutants lacking the cSH2 domain on PI3K signaling may mimic a decrease in p85α levels, potentially facilitating engineered cellular responses by increasing available binding sites on activated RTKs, which may promote the formation of signaling-competent p110-p85α heterodimers [14]. An oncogenic variant termed p65-PI3K, which lacks the cSH2 domain and possesses the capacity for constitutive activation and cell transformation, has been isolated from irradiation-induced mouse thymic lymphoma. Additionally, a C-terminal truncated version of p85α, known as p76α, has been identified in human lymphoma cell lines, lacking substantial portions of the cSH2 domain due to frameshift mutations [53-55]. While the cSH2 domain of p85α does not establish an inhibitory interface with the catalytic subunit, its presence may be essential for the complete inhibition of catalytic subunit activity [51, 56]. Deletion of the cSH2 domain can augment the signaling activity of the nSH2 mutant of p85α, and mutations in the cSH2 domain can abolish its negative regulatory influence on PI3K activity through the nSH2 domain of p85α [50, 55]. In summary, mutations in the cSH2 domain can diminish the negative regulatory function of p85α on PI3K activity, thereby modulating the catalytic activity of PI3K.
5. PI3K mutations in HPV-associated tumors
Human papillomavirus (HPV) is a well-established oncogenic virus. Studies have demonstrated that infection with high-risk HPV types is a major risk factor for the development of cervical, vaginal, and vulvar cancers [57]. Real-world data indicate that 48% of HPV-associated gynecological tumors harbor actionable mutations, with PI3K mutations being among the most prevalent oncogenic events [57]. Additionally, approximately 25% of oropharyngeal squamous cell carcinoma (OPSCC) cases worldwide are also attributed to HPV infection [58]. Compared to HPV-negative OPSCC patients, those with HPV-positive OPSCC demonstrate significantly improved overall survival and heightened sensitivity to chemotherapy, radiotherapy, and combined chemoradiotherapy [59-61]. Furthermore, HPV-positive OPSCC exhibits a distinct mutational landscape compared to its HPV-negative counterpart [62]. Among HPV-positive OPSCC cases, the class I subgroup of the PI3K family is most frequently associated with mutational dysregulation. Notably, the PI3K/PTEN/AKT/mTOR pathway has been identified as a critical oncogenic driver in HPV-positive cohorts [58]. Epidemiological studies indicate that 13% to 25% of HPV-positive OPSCC patients experience local or distant recurrence (LDR), which is associated with significantly reduced survival rates [59]. HPV-positive OPSCC patients who develop LDR exhibit a higher mutation burden compared to those without LDR, with mutation frequencies comparable to those observed in HPV-negative OPSCC patients with LDR [59]. Interestingly, HPV-negative OPSCC patients without LDR exhibit the highest overall mutation burden, suggesting that recurrence may be driven more by specific oncogenic mutations rather than total mutation load [59]. In HPV-positive OPSCC, PIK3R1 mutations occur at a higher frequency in LDR patients than in non-LDR patients [59]. Evidence suggests that aberrant PI3K signaling may contribute to cancer progression and poor prognosis by altering PIK3R1 and destabilizing the PIK3CA/PIK3R1 complex [63].
Epigenetic Alterations Occurring in the PIK3R1 Gene
Accumulating evidence indicates that aberrant epigenetic regulation of gene function is closely linked to the development of cancer [64]. Cell transformation, tumor progression, and metastasis are orchestrated by a complex network of interactions involving genomic and epigenomic mutations, particularly those affecting oncogenes and tumor suppressor genes, along with environmental factors that contribute to malignancy and tumorigenesis [65, 66].
1. DNA methylation
Higher levels of promoter methylation of PIK3R1 have been observed across various cancer types, and this methylation is positively correlated with gene expression levels in multiple probes within the promoter region [11]. Notably, hypomethylation of the CpG locus in the PIK3R1 promoter is associated with reduced gene expression and correlates with decreased overall survival and relapse-free survival in pancreatic cancer patients [67]. Furthermore, the potential of PIK3R1 methylation as a biomarker has been reported in esophageal cancer [68]. In breast cancer, the downregulation and hypermethylation of PIK3R1 correlate with poor patient outcomes, suggesting its utility as a diagnostic and prognostic biomarker for breast cancer [69]. Consequently, the methylation level of PIK3R1 is positively correlated with expression levels and is closely related to clinical data from cancer patients, establishing it as a promising cancer biomarker.
2. Histone modifications
The isonicotinylation of lysine residues on histones diminishes the binding capacity between histones and genomic DNA, resulting in a more open chromatin structure that facilitates the transcription of numerous genes [70, 71]. In isoniazid-treated HepG2 cells, RNA sequencing analysis has revealed an upregulation of the PIK3R1 gene, mediated by isoniazid through histone modifications, leading to elevated levels of p85α and the activation of the hepatocellular carcinoma-associated PAM pathway [70]. There exists a CapG-binding region within the PIK3R1 gene, situated near the transcription start site of PIK3R1 variant 3 (P50) [72]. The transcriptional coactivator CREB-binding protein/p300 is recruited to the PIK3R1 promoter via interactions with CapG, enhancing the transcription of PIK3R1/P50 through histone H3 lysine 27 acetylation (H3K27Ac) [72]. This process triggers the activation of the PI3K/AKT signaling pathway, contributing to paclitaxel resistance in breast cancer cells. Chemoresistance in hepatocellular carcinoma is partially attributed to chemotherapy-induced N-acetyltransferase 10, which significantly upregulates H3K27Ac on the PIK3R1 promoter, thereby activating transcription and driving chemoresistance [73]. Additionally, a super-enhancer characterized by a high level of H3K27Ac and mediator bindings has been identified at the PIK3R1 locus in multiple adult T-cell leukemia/lymphoma samples, but not in normal T cells [74]. This finding suggests an involvement of the PI3K/AKT pathway in the pathogenesis of adult T-cell leukemia/lymphoma [74]. Overall, histone modifications of PIK3R1 may enhance p85α expression, thus activating the PI3K/AKT pathway and potentially contributing to tumor chemoresistance.
3. Non-coding RNA
MicroRNAs (miRNAs) are endogenous non-coding RNAs approximately 21 nucleotides in length, which are expressed in most somatic tissues [75]. They regulate gene expression post-transcriptionally and are integral components of the epigenome. In many cancers, miRNA expression is dysregulated, with PIK3R1 being targeted by multiple miRNAs that exert tumor-suppressive effects [76, 77]. For instance, PIK3R1 has been identified as a direct target of miR-155 in breast cancer and B lymphocytes, where it promotes tumor growth by activating glucose metabolism [78-80]. In ovarian cancer, circPLPP4 targets miR-136, acting as a competitive endogenous RNA to regulate PIK3R1 expression and enhance cisplatin resistance [81]. Additionally, p85α, a crucial target of miR-29 in the p53 pathway, upregulates p53, inducing apoptosis in a p53-dependent manner through a PI3K/AKT/MDM2-mediated mechanism [82]. Furthermore, p85α is a direct target of miR-503; its ectopic expression inhibits tumor cell proliferation and metastasis-related traits both in vitro and in vivo [83, 84]. The modulation of miR-503 through overexpression and knockdown partially influences apoptotic activity and alters cisplatin resistance in ovarian cancer cells, suggesting that miR-503 may serve as a sensitizer for cisplatin therapy in ovarian cancer [85]. In colorectal cancer, miR-455-5p acts as a tumor suppressor by inhibiting the proliferation and migration of colorectal cancer cells while promoting their apoptosis; it may also target and downregulate PIK3R1, increasing sensitivity to 5-fluorouracil [86, 87]. Moreover, miR-21's targeting of PIK3R1 inhibits tumor cell migration and invasion by reducing PI3K/AKT signaling, reversing epithelial-to-mesenchymal transition, and predicting clinical outcomes in breast cancer [15]. miR-495 similarly promotes endometrial cell apoptosis and inhibits proliferation by also targeting PIK3R1 [88]. Overall, PIK3R1 emerges as a target of multiple miRNAs and plays a significant role in the initiation and progression of various cancers.
Circular RNAs (circRNAs) are single-stranded, covalently closed RNA molecules formed by the reverse splicing of mRNA exons [89]. Many circRNAs exhibit aberrant expression across various cancers, with their dysregulation being closely associated with tumor progression [90]. In hepatocellular carcinoma cells, circRHBDD1 acts as a scaffold, enhancing the interaction between YTHDF1 and PIK3R1 mRNA and promoting PIK3R1 mRNA translation in an m6A-dependent manner [80]. CircSEMA4B, a protein-coding circRNA significantly downregulated in ovarian cancer, encodes a novel protein, SEMA4B-211aa, which inhibits PIP3 production by binding to p85, thereby inhibiting AKT phosphorylation and breast cancer progression [91]. CircRNAs can exert either anti-immunotherapeutic or anti-tumor effects by regulating the expression of PIK3R1 or binding to p85α.
Clinical Drugs Targeting the PAM Pathway and Targeted Therapies for Cancers Harboring PIK3R1 Aberrations
1. FDA-Approved Clinical Drugs Targeting the PAM Pathway
Due to the alteration of the PI3K pathway being found in multiple cancers, it is considered one of the most commonly targeted signaling cascades, and as a result, several PI3K/AKT inhibitors have been developed [57, 92]. Several classes of drugs target the PI3K pathway, including pan-PI3K inhibitors, isoform-selective PI3K inhibitors (IS PI3Ki), AKT inhibitors, mTOR inhibitors, and dual PI3K/mTOR inhibitors [93]. Pan-PI3K inhibitors effectively target high-level PIP3 tumors by inhibiting the catalytic activity of all four class I PI3K isoforms: PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ [94]. While numerous pan-PI3K inhibitors are currently in clinical development, copanlisib is the only one to have demonstrated significant efficacy in clinical trials [95]. IS PI3Ki specifically inhibit selected PI3K isoforms (α/β/γ or δ), and of the IS PI3Ki, only six have received Food and Drug Administration (FDA) approval: alpelisib, umbralisib, duvelisib, inavolisib, leniolisib, and idelalisib [94, 96-102]. Alpelisib, a potent drug with targeted efficacy against the PI3Kα isoform, has been approved for use in patients with advanced or metastatic breast cancer with hormone receptor (HR)+/HER2-PIK3CA mutations in combination with fulvestrant [94].
AKT inhibitors are classified into three broad categories: ATP-competitive, allosteric, and covalent allosteric inhibitors, with capivasertib being the first drug to receive FDA approval in combination with the estrogen receptor degrader fulvestrant for breast cancer treatment [103].
mTOR inhibitors were the first PAM-targeted drugs to advance to clinical use [104, 105]. These inhibitors are generally classified into three generations based on their common binding sites on mTOR. Current clinical options—rapamycin, temsirolimus, everolimus, and rapamycin bound to albumin—belong to the first generation [106]. The FDA-approved mTOR inhibitors are everolimus, sirolimus, and temsirolimus. Dual PI3K/mTOR inhibitors target both PI3K and mTOR signaling; however, none are currently FDA-approved for cancer treatment [96]. PI3K/mTOR dual ATP-competitive inhibitors directly act on PI3K and mTOR, more effectively inhibiting the PAM signaling pathway, and reducing resistance and side effects compared to single inhibitors [107-109]. Reports of dual PI3K/mTOR inhibitors (Gedatolisib, Omipalisib, Apitolisib, and others) currently in Phase I or II clinical stages indicate that none have received FDA approval for cancer therapy [96, 110-113].
Normal cells depend on PI3K signaling for survival; consequently, serious adverse effects may present before complete inhibition of target tumor cells [114]. Over 40 PI3K pathway inhibitors are undergoing various clinical development stages [114], yet few have received FDA approval (Table 1). Most PI3K inhibitors provide only modest benefits, thus limiting their application in clinical settings.
FDA-approved inhibitors of the PAM pathway
| Drug name | Target | Indication | Launch year |
|---|---|---|---|
| Umbralisib | PI3Kδ; CK1ε | Adults with relapsed or refractory marginal zone and follicular lymphoma. | 2021 |
| Alpelisib | PI3Kα | Alpelisib in combination with fulvestrant for postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer. | 2019 |
| Duvelisib | PI3Kδ; PI3Kγ | Chronic lymphocytic leukemia/small lymphocytic lymphoma and follicular lymphoma. | 2018 |
| Copanlisib | Pan-class PI3K | Adults with relapsed follicular lymphoma who have received at least two prior systemic therapies. | 2017 |
| Idelalisib | PI3Kδ | Relapsed/refractory chronic lymphocytic leukemia, follicular lymphoma, and small lymphocytic lymphoma. | 2014 |
| Inavolisib | PI3Kα | Inavolisib with palbociclib and fulvestrant for endocrine-resistant, PIK3CA-mutated, HR-positive, HER2-negative, advanced breast cancer. | 2024 |
| Leniolisib | PI3Kδ | Adults and children 12 years of age or older with Activated Phosphoinositide-3-kinase-delta Syndrome (APDS). | 2023 |
| Capivasertib | Pan-AKT | Capivasertib with fulvestrant for adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer with one or more PIK3CA/AKT1/PTEN-alterations. | 2023 |
| Everolimus | mTOR | Postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer in combination with exemestane, after the failure of treatment with letrozole or anastrozole. Adult patients with progressive neuroendocrine tumors of pancreatic origin (PNET) with unresectable, locally advanced or metastatic disease. Adult patients with advanced RCC after failure of treatment with sunitinib or sorafenib. Adult patients with renal angiomyolipoma and tuberous sclerosis complex (TSC), not requiring immediate surgery. Adult and pediatric patients, 3 years of age or older, with SEGA associated with TSC who require therapeutic intervention but are not candidates for curative surgical resection. | 2009 |
| Sirolimus | mTOR | The prophylaxis of organ rejection in patients aged ≥13 years receiving renal transplants. | 1999 |
| Temsirolimus | mTOR | Advanced renal cell carcinoma. | 2007 |
Understanding the aberrant expression of cancer pathway genes that play crucial roles in cancer initiation and progression can positively impact clinical outcomes for patients with genetic anomalies. The degree to which these aberrations drive tumor behavior and serve as critical therapeutic targets necessitates further investigation to establish predictive biomarkers of response to PI3K inhibition [115].
2. Targeted therapy for PIK3R1 gene aberrations in pre-clinical studies
Specific molecular aberrations in cancer-associated genes may have functional consequences that influence treatment sensitivity [46, 116, 117]. Selective pharmacological inhibition of Pan-PI3K and p110α effectively block transformations driven by partial p85α deletion in both in vitro and in vivo models, indicating that p85α functions as a tumor suppressor in transformation processes. This suggests that p110α may represent a potential therapeutic target for treating breast cancer patients with PIK3R1 deletions [14]. The blockade of AKT or STAT3 may benefit ovarian cancer patients experiencing copy number losses or reduced expression of PIK3R1. The combination of AKT and STAT3 inhibitors demonstrates synergistic antitumor effects in vitro in 3D spheroid models and shows enhanced efficacy in vivo compared to either inhibitor alone [20].
PIK3R1 mutations across different domains exhibit varying impacts on cancer, necessitating diverse therapeutic strategies for effectiveness [3]. A growing body of evidence suggests that the efficacy of these drugs may partly depend on specific mutations in target proteins and the genetic context surrounding those mutations. A comprehensive understanding of how PIK3R1 mutations affect their interactions and regulatory functions will aid in identifying cancer-associated mutations that dysregulate the PI3K pathway and pinpoint the most effective therapeutic targets for inhibitor therapy [3]. While a small percentage of patients have observed clinical benefits from treatments based on newly identified hotspot mutations, further exploration into the ramifications of various combinations of mutations and genetic alterations on cancer biology and therapeutic sensitivity complicates the landscape [118].
Mutation characteristics in cancer cells provide insights into tumorigenesis and reveal candidates for targeted therapies. Different mutations can result in varying susceptibilities to specific PI3K pathway inhibitors [30]. Inhibitors targeting the AKT or HER families can diminish the carcinogenic potential of driver mutations, with combined use yielding significant synergistic effects [43]. Cells harboring nSH2 domain-driven mutations exhibit similar traits to those expressing HER3 [43]. Notably, HER3-expressing cells activate the PI3K pathway without engaging the MAPK pathway [119, 120]. In 19 endometrial cancer cell lines, HER3 overexpression correlated with responsiveness to the dual epidermal growth factor receptor-1/2 inhibitor lapatinib [121]. Furthermore, patients with PIK3R1R348*, PIK3R1L370fs, or adjacent PIK3R1 mutations may benefit from concurrent treatment with RAS and PI3K pathway inhibitors, conferring unexpected sensitivity to MEK and JNK inhibitors in both in vitro and in vivo settings [46]. Targeting p85α homodimerization or p85α:PTEN interactions may present new therapeutic avenues for mutations in the p85α BH and SH3 domains [31]. Mutations in the iSH2 domain may facilitate the development of pharmacological compounds that inhibit PI3-kinase by stabilizing the regulated C2-iSH2 interface between p85 and p110 [47]. Tumors exhibiting iSH2 mutations are likely to respond favorably to inhibitor therapies targeting PI3K or its downstream effectors, such as AKT [45].
Given the significance of epigenetic alterations in cancer and their potential for reprogramming, molecular regulators of these modifications have emerged as promising targets in cancer therapy [64, 122]. Currently, the most extensively studied epigenetic molecular inhibitors include histone deacetylase inhibitors, histone lysine demethylase inhibitors, and DNA methyltransferase inhibitors, alongside dietary interventions involving epigenetically modified proteins and metabolic molecules as promising treatment strategies [123]. However, responses to monotherapy often fall short of expectations, with resistance to such therapies appearing inevitable [124]. The introduction of new therapies, including the application of miRNAs, multidrug combinations, and immunotherapies, may enhance cancer treatment outcomes while mitigating drug resistance in comparison to monotherapy [64, 125-127]. Various combination strategies have been proposed and tested, necessitating further research to bolster the potential of these epigenetic drugs as innovative treatments.
Conclusion
We summarize evidence indicating that PIK3R1 manifests characteristics consistent with a tumor suppressor gene. In several cancer types, PIK3R1 experiences copy number loss, leading to the activation of downstream signaling molecules that promote cancer development, suggesting that the occurrence of PIK3R1 copy number deletion may be a key indicator linked to poor cancer prognosis. Cancer-associated mutations have been identified across all five protein domains of the PIK3R1 gene. Driver mutations in the BH and SH3 domains can diminish the negative feedback of the PI3K/AKT pathway by reducing homodimerization or binding to PTEN, thereby activating downstream signals. Driver mutations within the iSH2 domain can trigger the downstream AKT signaling pathway by weakening the interaction between p110α and p85α. Meanwhile, driver mutations found in the nSH2 domain may exert catalytic effects by activating a series of upstream RTK signaling proteins to diminish the inhibitory effect of p85α on p110α. However, there is a scarcity of comprehensive literature regarding mutations in the cSH2 domain, necessitating further investigations to elucidate the oncogenic mechanisms underlying driver mutations in this domain. Additionally, epigenetic changes associated with the PIK3R1 gene remain a focal point of research, and PIK3R1 methylation could potentially serve as a cancer biomarker indicative of malignancy prognosis, as well as a target for multiple miRNAs that perform tumor-suppressive functions, thus providing a robust foundation for future development of specific epigenetic drugs.
In conclusion, PIK3R1 plays a crucial role in cancer as a regulatory subunit of PI3K, and the phenotypic variations observed in human cancers stem from a complex interplay of genetic factors, transcriptomic profiles, epigenetics, and proteomics. Copy number variations, mutations, and epigenetic alterations in PIK3R1 contribute to a proliferative phenotype characterized by recurrent mutations across diverse cancers. Developing therapeutic strategies that target the PI3K signaling network holds significant promise for enhancing cancer treatment outcomes. The accumulating body of evidence related to genetic aberrations in PIK3R1 and corresponding cancer markers reinforces the notion that targeting the aberrant PIK3R1 pathway could be advantageous for anticancer therapy.
Abbreviations
PI3K, phosphoinositide 3-kinase; PIP3, phosphatidylinositol 3,4,5-triphosphate; TCGA, The Cancer Genome Atlas; SH3, Src homology 3; BH, breakpoint cluster region homology; nSH2, N-terminal SH2; iSH2, middle SH2; cSH2, C-terminal SH2; PAM, PI3K/AKT/mTOR; pY, phosphorylated tyrosine-containing; pYXXM, phosphorylated tyrosine-containing consensus sequences; RTKs, receptor tyrosine kinases; HPV, Human papillomavirus; OPSCC, oropharyngeal squamous cell carcinoma; LDR, local or distant recurrence; H3K27Ac, histone H3 lysine 27 acetylation; miRNAS, MicroRNAs; circRNAs, Circular RNAs; IS PI3Ki, isoform-selective PI3K inhibitors; FDA, Food and Drug Administration.
Acknowledgements
The research was supported by National Natural Science Foundation of China (82102793), Program of Excellent Doctoral (Postdoctoral) of Zhongnan Hospital of Wuhan University (ZNYB2021019), Program of Excellent Young Scientists Fund of Zhongnan Hospital of Wuhan University (ZNYQ2023012).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329-41
2. Noorolyai S, Shajari N, Baghbani E, Sadreddini S, Baradaran B. The relation between PI3K/AKT signalling pathway and cancer. Gene. 2019;698:120-8
3. Marshall JDS, Whitecross DE, Mellor P, Anderson DH. Impact of p85alpha Alterations in Cancer. Biomolecules. 2019;9:29
4. Jean S, Kiger AA. Classes of phosphoinositide 3-kinases at a glance. J Cell Sci. 2014;127:923-8
5. Cui W, Cai Y, Zhou X. Advances in subunits of PI3K class I in cancer. Pathology. 2014;46:169-76
6. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-7
7. Tsay A, Wang JC. The Role of PIK3R1 in Metabolic Function and Insulin Sensitivity. Int J Mol Sci. 2023;24:12665
8. Chagpar RB, Links PH, Pastor MC, Furber LA, Hawrysh AD, Chamberlain MD. et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2010;107:5471-6
9. Mellor P, Furber LA, Nyarko JN, Anderson DH. Multiple roles for the p85alpha isoform in the regulation and function of PI3K signalling and receptor trafficking. Biochem J. 2012;441:23-37
10. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
11. Liu Y, Wang D, Li Z, Li X, Jin M, Jia N. et al. Pan-cancer analysis on the role of PIK3R1 and PIK3R2 in human tumors. Sci Rep. 2022;12:5924
12. Cizkova M, Vacher S, Meseure D, Trassard M, Susini A, Mlcuchova D. et al. PIK3R1 underexpression is an independent prognostic marker in breast cancer. BMC Cancer. 2013;13:545
13. Taniguchi CM, Winnay J, Kondo T, Bronson RT, Guimaraes AR, Aleman JO. et al. The phosphoinositide 3-kinase regulatory subunit p85alpha can exert tumor suppressor properties through negative regulation of growth factor signaling. Cancer Res. 2010;70:5305-15
14. Thorpe LM, Spangle JM, Ohlson CE, Cheng H, Roberts TM, Cantley LC. et al. PI3K-p110alpha mediates the oncogenic activity induced by loss of the novel tumor suppressor PI3K-p85alpha. Proc Natl Acad Sci U S A. 2017;114:7095-100
15. Yan LX, Liu YH, Xiang JW, Wu QN, Xu LB, Luo XL. et al. PIK3R1 targeting by miR-21 suppresses tumor cell migration and invasion by reducing PI3K/AKT signaling and reversing EMT, and predicts clinical outcome of breast cancer. Int J Oncol. 2016;48:471-84
16. Turturro SB, Najor MS, Yung T, Portt L, Malarkey CS, Abukhdeir AM. et al. Somatic loss of PIK3R1 may sensitize breast cancer to inhibitors of the MAPK pathway. Breast Cancer Res Treat. 2019;177:325-33
17. Munkley J, Livermore KE, McClurg UL, Kalna G, Knight B, McCullagh P. et al. The PI3K regulatory subunit gene PIK3R1 is under direct control of androgens and repressed in prostate cancer cells. Oncoscience. 2015;2:755-64
18. Chakraborty G, Nandakumar S, Hirani R, Nguyen B, Stopsack KH, Kreitzer C. et al. The Impact of PIK3R1 Mutations and Insulin-PI3K-Glycolytic Pathway Regulation in Prostate Cancer. Clin Cancer Res. 2022;28:3603-17
19. Lin Y, Yang Z, Xu A, Dong P, Huang Y, Liu H. et al. PIK3R1 negatively regulates the epithelial-mesenchymal transition and stem-like phenotype of renal cancer cells through the AKT/GSK3beta/CTNNB1 signaling pathway. Sci Rep. 2015;5:8997
20. Li X, Mak VCY, Zhou Y, Wang C, Wong ESY, Sharma R. et al. Deregulated Gab2 phosphorylation mediates aberrant AKT and STAT3 signaling upon PIK3R1 loss in ovarian cancer. Nat Commun. 2019;10:716
21. Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol. 2002;22:965-77
22. Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ. et al. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest. 2002;109:141-9
23. Luo J, Sobkiw CL, Logsdon NM, Watt JM, Signoretti S, O'Connell F. et al. Modulation of epithelial neoplasia and lymphoid hyperplasia in PTEN+/- mice by the p85 regulatory subunits of phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2005;102:10238-43
24. Thapa N, Chen M, Cryns VL, Anderson R. A p85 isoform switch enhances PI3K activation on endosomes by a MAP4- and PI3P-dependent mechanism. Cell Rep. 2024;43:114119
25. Lu Y, Lemon W, Liu PY, Yi Y, Morrison C, Yang P. et al. A gene expression signature predicts survival of patients with stage I non-small cell lung cancer. PLoS Med. 2006;3:e467
26. Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7:818-31
27. Cheung LW, Mills GB. Targeting therapeutic liabilities engendered by PIK3R1 mutations for cancer treatment. Pharmacogenomics. 2016;17:297-307
28. Yu J, Wjasow C, Backer JM. Regulation of the p85/p110alpha phosphatidylinositol 3'-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J Biol Chem. 1998;273:30199-203
29. Harpur AG, Layton MJ, Das P, Bottomley MJ, Panayotou G, Driscoll PC. et al. Intermolecular interactions of the p85alpha regulatory subunit of phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:12323-32
30. Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B. et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170-85
31. Cheung LW, Walkiewicz KW, Besong TM, Guo H, Hawke DH, Arold ST. et al. Regulation of the PI3K pathway through a p85alpha monomer-homodimer equilibrium. Elife. 2015;4:e06866
32. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497-510
33. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527-41
34. Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77-82
35. Alvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of PTEN loss in cancer: It's all about diversity. Semin Cancer Biol. 2019;59:66-79
36. Chamberlain MD, Berry TR, Pastor MC, Anderson DH. The p85alpha subunit of phosphatidylinositol 3'-kinase binds to and stimulates the GTPase activity of Rab proteins. J Biol Chem. 2004;279:48607-14
37. Chamberlain MD, Chan T, Oberg JC, Hawrysh AD, James KM, Saxena A. et al. Disrupted RabGAP function of the p85 subunit of phosphatidylinositol 3-kinase results in cell transformation. J Biol Chem. 2008;283:15861-8
38. Miller MS, Thompson PE, Gabelli SB. Structural Determinants of Isoform Selectivity in PI3K Inhibitors. Biomolecules. 2019;9:82
39. Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y. et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239-42
40. Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH. et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106:16996-7001
41. Burke JE, Williams RL. Dynamic steps in receptor tyrosine kinase mediated activation of class IA phosphoinositide 3-kinases (PI3K) captured by H/D exchange (HDX-MS). Adv Biol Regul. 2013;53:97-110
42. Cuevas BD, Lu Y, Mao M, Zhang J, LaPushin R, Siminovitch K. et al. Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3-kinase. J Biol Chem. 2001;276:27455-61
43. Li X, Lau AYT, Ng ASN, Aldehaiman A, Zhou Y, Ng PKS. et al. Cancer-associated mutations in the p85alpha N-terminal SH2 domain activate a spectrum of receptor tyrosine kinases. Proc Natl Acad Sci U S A. 2021;118:e2101751118
44. Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proc Natl Acad Sci U S A. 2012;109:15259-64
45. Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W. et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16:463-74
46. Cheung LW, Yu S, Zhang D, Li J, Ng PK, Panupinthu N. et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell. 2014;26:479-94
47. Wu H, Shekar SC, Flinn RJ, El-Sibai M, Jaiswal BS, Sen KI. et al. Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110alpha and are disrupted in oncogenic p85 mutants. Proc Natl Acad Sci U S A. 2009;106:20258-63
48. Jenkins ML, Ranga-Prasad H, Parson MAH, Harris NJ, Rathinaswamy MK, Burke JE. Oncogenic mutations of PIK3CA lead to increased membrane recruitment driven by reorientation of the ABD, p85 and C-terminus. Nat Commun. 2023;14:181
49. Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci U S A. 2010;107:15547-52
50. Ito Y, Vogt PK, Hart JR. Domain analysis reveals striking functional differences between the regulatory subunits of phosphatidylinositol 3-kinase (PI3K), p85alpha and p85beta. Oncotarget. 2017;8:55863-76
51. Dornan GL, Stariha JTB, Rathinaswamy MK, Powell CJ, Boulanger MJ, Burke JE. Defining How Oncogenic and Developmental Mutations of PIK3R1 Alter the Regulation of Class IA Phosphoinositide 3-Kinases. Structure. 2020;28:145-56 e5
52. Solheim MH, Winnay JN, Batista TM, Molven A, Njolstad PR, Kahn CR. Mice Carrying a Dominant-Negative Human PI3K Mutation Are Protected From Obesity and Hepatic Steatosis but Not Diabetes. Diabetes. 2018;67:1297-309
53. Jucker M, Sudel K, Horn S, Sickel M, Wegner W, Fiedler W. et al. Expression of a mutated form of the p85alpha regulatory subunit of phosphatidylinositol 3-kinase in a Hodgkin's lymphoma-derived cell line (CO). Leukemia. 2002;16:894-901
54. Jimenez C, Jones DR, Rodriguez-Viciana P, Gonzalez-Garcia A, Leonardo E, Wennstrom S. et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J. 1998;17:743-53
55. Hofmann BT, Jucker M. Activation of PI3K/Akt signaling by n-terminal SH2 domain mutants of the p85alpha regulatory subunit of PI3K is enhanced by deletion of its c-terminal SH2 domain. Cell Signal. 2012;24:1950-4
56. Jani V, Sonavane U, Sawant S. Structural insights into the activation mechanism of phosphoinositide 3-kinase alpha. Comput Biol Chem. 2024;108:107994
57. Kraus FBT, Sultova E, Heinrich K, Jung A, Westphalen CB, Tauber CV. et al. Genetics and beyond: Precision Medicine Real-World Data for Patients with Cervical, Vaginal or Vulvar Cancer in a Tertiary Cancer Center. Int J Mol Sci. 2024;25:2345
58. Brennan S, Baird AM, O'Regan E, Sheils O. The Role of Human Papilloma Virus in Dictating Outcomes in Head and Neck Squamous Cell Carcinoma. Front Mol Biosci. 2021;8:677900
59. Reder H, Wagner S, Wuerdemann N, Langer C, Sandmann S, Braeuninger A. et al. Mutation patterns in recurrent and/or metastatic oropharyngeal squamous cell carcinomas in relation to human papillomavirus status. Cancer Med. 2021;10:1347-56
60. Liu C, Mann D, Sinha UK, Kokot NC. The molecular mechanisms of increased radiosensitivity of HPV-positive oropharyngeal squamous cell carcinoma (OPSCC): an extensive review. J Otolaryngol Head Neck Surg. 2018;47:59
61. Economopoulou P, de Bree R, Kotsantis I, Psyrri A. Diagnostic Tumor Markers in Head and Neck Squamous Cell Carcinoma (HNSCC) in the Clinical Setting. Front Oncol. 2019;9:827
62. Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L. et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709-20
63. Reder H, Wagner S, Gamerdinger U, Sandmann S, Wuerdemann N, Braeuninger A. et al. Genetic alterations in human papillomavirus-associated oropharyngeal squamous cell carcinoma of patients with treatment failure. Oral Oncol. 2019;93:59-65
64. Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics. 2019;14:1164-76
65. Werner HM, Mills GB, Ram PT. Cancer Systems Biology: a peek into the future of patient care? Nat Rev Clin Oncol. 2014;11:167-76
66. Park JW, Han JW. Targeting epigenetics for cancer therapy. Arch Pharm Res. 2019;42:159-70
67. Faleiro I, Roberto VP, Demirkol Canli S, Fraunhoffer NA, Iovanna J, Gure AO. et al. DNA Methylation of PI3K/AKT Pathway-Related Genes Predicts Outcome in Patients with Pancreatic Cancer: A Comprehensive Bioinformatics-Based Study. Cancers (Basel). 2021;13:6354
68. Peng X, Xue H, Lu L, Shi P, Wang J, Wang J. Accumulated promoter methylation as a potential biomarker for esophageal cancer. Oncotarget. 2017;8:679-91
69. Qi L, Zhou B, Chen J, Hu W, Bai R, Ye C. et al. Significant prognostic values of differentially expressed-aberrantly methylated hub genes in breast cancer. J Cancer. 2019;10:6618-34
70. Jiang Y, Li Y, Liu C, Zhang L, Lv D, Weng Y. et al. Isonicotinylation is a histone mark induced by the anti-tuberculosis first-line drug isoniazid. Nat Commun. 2021;12:5548
71. Li Y, Jiang Y, Yan H, Qin Z, Peng Y, Lv D. et al. Global isonicotinylome analysis identified SMAD3 isonicotinylation promotes liver cancer cell epithelial-mesenchymal transition and invasion. iScience. 2024;27:110775
72. Chi Y, Xue J, Huang S, Xiu B, Su Y, Wang W. et al. CapG promotes resistance to paclitaxel in breast cancer through transactivation of PIK3R1/P50. Theranostics. 2019;9:6840-55
73. Wang Y, Su K, Wang C, Deng T, Liu X, Sun S. et al. Chemotherapy-induced acetylation of ACLY by NAT10 promotes its nuclear accumulation and acetyl-CoA production to drive chemoresistance in hepatocellular carcinoma. Cell Death Dis. 2024;15:545
74. Wong RWJ, Ngoc PCT, Leong WZ, Yam AWY, Zhang T, Asamitsu K. et al. Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood. 2017;130:2326-38
75. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597-610
76. Ferragut Cardoso AP, Banerjee M, Nail AN, Lykoudi A, States JC. miRNA dysregulation is an emerging modulator of genomic instability. Semin Cancer Biol. 2021;76:120-31
77. Liu X, Chen X, Yu X, Tao Y, Bode AM, Dong Z. et al. Regulation of microRNAs by epigenetics and their interplay involved in cancer. J Exp Clin Cancer Res. 2013;32:96
78. Kim S, Lee E, Jung J, Lee JW, Kim HJ, Kim J. et al. microRNA-155 positively regulates glucose metabolism via PIK3R1-FOXO3a-cMYC axis in breast cancer. Oncogene. 2018;37:2982-91
79. Huang X, Shen Y, Liu M, Bi C, Jiang C, Iqbal J. et al. Quantitative proteomics reveals that miR-155 regulates the PI3K-AKT pathway in diffuse large B-cell lymphoma. Am J Pathol. 2012;181:26-33
80. Cai J, Chen Z, Zhang Y, Wang J, Zhang Z, Wu J. et al. CircRHBDD1 augments metabolic rewiring and restricts immunotherapy efficacy via m(6)A modification in hepatocellular carcinoma. Mol Ther Oncolytics. 2022;24:755-71
81. Li H, Lin R, Zhang Y, Zhu Y, Huang S, Lan J. et al. N6-methyladenosine-modified circPLPP4 sustains cisplatin resistance in ovarian cancer cells via PIK3R1 upregulation. Mol Cancer. 2024;23:5
82. Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol. 2009;16:23-9
83. Yang Y, Liu L, Zhang Y, Guan H, Wu J, Zhu X. et al. MiR-503 targets PI3K p85 and IKK-beta and suppresses progression of non-small cell lung cancer. Int J Cancer. 2014;135:1531-42
84. Yan W, Wu Q, Yao W, Li Y, Liu Y, Yuan J. et al. MiR-503 modulates epithelial-mesenchymal transition in silica-induced pulmonary fibrosis by targeting PI3K p85 and is sponged by lncRNA MALAT1. Sci Rep. 2017;7:11313
85. Wu D, Lu P, Mi X, Miao J. Downregulation of miR-503 contributes to the development of drug resistance in ovarian cancer by targeting PI3K p85. Arch Gynecol Obstet. 2018;297:699-707
86. Lou T, Zhang L, Jin Z, Miao C, Wang J, Ke K. miR-455-5p enhances 5-fluorouracil sensitivity in colorectal cancer cells by targeting PIK3R1 and DEPDC1. Open Med (Wars). 2022;17:847-56
87. Wang J, Lu Y, Zeng Y, Zhang L, Ke K, Guo Y. Expression profile and biological function of miR-455-5p in colorectal carcinoma. Oncol Lett. 2019;17:2131-40
88. Tan A, Luo R, Ruan P. miR-495 promotes apoptosis and inhibits proliferation in endometrial cells via targeting PIK3R1. Pathol Res Pract. 2019;215:594-9
89. Liu X, Zhang Y, Zhou S, Dain L, Mei L, Zhu G. Circular RNA: An emerging frontier in RNA therapeutic targets, RNA therapeutics, and mRNA vaccines. J Control Release. 2022;348:84-94
90. He Z, Zhu Q. Circular RNAs: Emerging roles and new insights in human cancers. Biomed Pharmacother. 2023;165:115217
91. Wang X, Jian W, Luo Q, Fang L. CircSEMA4B inhibits the progression of breast cancer by encoding a novel protein SEMA4B-211aa and regulating AKT phosphorylation. Cell Death Dis. 2022;13:794
92. Hanker AB, Kaklamani V, Arteaga CL. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019;9:482-91
93. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140-56
94. Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH. et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22:138
95. Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA. et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110alpha and p110delta activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12:2319-30
96. Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int J Mol Sci. 2021;22:3464
97. Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS. et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2019;380:1929-40
98. Dhillon S, Keam SJ. Umbralisib: First Approval. Drugs. 2021;81:857-66
99. Blair HA. Duvelisib: First Global Approval. Drugs. 2018;78:1847-53
100. Turner NC, Im SA, Saura C, Juric D, Loibl S, Kalinsky K. et al. Inavolisib-Based Therapy in PIK3CA-Mutated Advanced Breast Cancer. N Engl J Med. 2024;391:1584-96
101. Duggan S, Al-Salama ZT. Leniolisib: First Approval. Drugs. 2023;83:943-8
102. Markham A. Idelalisib: first global approval. Drugs. 2014;74:1701-7
103. Pervanidis KA, D'Angelo GD, Weisner J, Brandherm S, Rauh D. Akt Inhibitor Advancements: From Capivasertib Approval to Covalent-Allosteric Promises. J Med Chem. 2024;67:6052-63
104. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A. et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271-81
105. Coren CV. Burn injuries in children. Pediatr Ann. 1987;16:328-32 35, 38-9
106. Oleksak P, Nepovimova E, Chrienova Z, Musilek K, Patocka J, Kuca K. Contemporary mTOR inhibitor scaffolds to diseases breakdown: A patent review (2015-2021). Eur J Med Chem. 2022;238:114498
107. Liu Y, Wan WZ, Li Y, Zhou GL, Liu XG. Recent development of ATP-competitive small molecule phosphatidylinostitol-3-kinase inhibitors as anticancer agents. Oncotarget. 2017;8:7181-200
108. Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Okada M. et al. Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem. 2006;14:6847-58
109. Lv X, Ma X, Hu Y. Furthering the design and the discovery of small molecule ATP-competitive mTOR inhibitors as an effective cancer treatment. Expert Opin Drug Discov. 2013;8:991-1012
110. Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Evers BM. PKI-587 and sorafenib targeting PI3K/AKT/mTOR and Ras/Raf/MAPK pathways synergistically inhibit HCC cell proliferation. J Surg Res. 2012;176:542-8
111. Sutherlin DP, Bao L, Berry M, Castanedo G, Chuckowree I, Dotson J. et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011;54:7579-87
112. Lukey PT, Harrison SA, Yang S, Man Y, Holman BF, Rashidnasab A. et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur Respir J. 2019;53:1801992
113. Luo L, Sun X, Yang Y, Xia L, Wang S, Fu Y. et al. A Novel Dual PI3K/mTOR Inhibitor, XIN-10, for the Treatment of Cancer. Int J Mol Sci. 2023;24:14821
114. Ellis H, Ma CX. PI3K Inhibitors in Breast Cancer Therapy. Curr Oncol Rep. 2019;21:110
115. Salvesen HB, Werner HM, Krakstad C. PI3K pathway in gynecologic malignancies. Am Soc Clin Oncol Educ Book. 2013
116. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129-39
117. Lutzky J, Bauer J, Bastian BC. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008;21:492-3
118. Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P. et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov. 2018;8:174-83
119. Kirouac DC, Du J, Lahdenranta J, Onsum MD, Nielsen UB, Schoeberl B. et al. HER2+ Cancer Cell Dependence on PI3K vs. MAPK Signaling Axes Is Determined by Expression of EGFR, ERBB3 and CDKN1B. PLoS Comput Biol. 2016;12:e1004827
120. Huang Y, Burns DJ, Rich BE, MacNeil IA, Dandapat A, Soltani SM. et al. Development of a test that measures real-time HER2 signaling function in live breast cancer cell lines and primary cells. BMC Cancer. 2017;17:199
121. Konecny GE, Venkatesan N, Yang G, Dering J, Ginther C, Finn R. et al. Activity of lapatinib a novel HER2 and EGFR dual kinase inhibitor in human endometrial cancer cells. Br J Cancer. 2008;98:1076-84
122. Yang Q, Jiang W, Hou P. Emerging role of PI3K/AKT in tumor-related epigenetic regulation. Semin Cancer Biol. 2019;59:112-24
123. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13:877-919
124. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-26
125. Garofalo M, Croce CM. MicroRNAs as therapeutic targets in chemoresistance. Drug Resist Updat. 2013;16:47-59
126. Glasgow MD, Chougule MB. Recent Developments in Active Tumor Targeted Multifunctional Nanoparticles for Combination Chemotherapy in Cancer Treatment and Imaging. J Biomed Nanotechnol. 2015;11:1859-98
127. Crea F, Nobili S, Paolicchi E, Perrone G, Napoli C, Landini I. et al. Epigenetics and chemoresistance in colorectal cancer: an opportunity for treatment tailoring and novel therapeutic strategies. Drug Resist Updat. 2011;14:280-96
Author contact
![]() Corresponding author: Xinran Li: Department of Laboratory Medicine, Zhongnan Hospital of Wuhan University, No.169 Donghu Road, Wuhan, 430071, China; E-mail: xinran2827edu.cn.
Corresponding author: Xinran Li: Department of Laboratory Medicine, Zhongnan Hospital of Wuhan University, No.169 Donghu Road, Wuhan, 430071, China; E-mail: xinran2827edu.cn.