Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Conflicting evidence and racial...
Pathogenic mechanisms linking...
Elevated Lp(a) and...
Therapeutic strategies for Lp(a)...
Limitations and prospects
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(2):357-370. doi:10.7150/ijms.102301 This issue Cite
Review
Lipoprotein(a) and Atrial Fibrillation: Mechanistic Insights and Therapeutic Approaches
Zixi Zhang1#, Bo Peng1#, Akedanmu Nuranmubieke2, Yangfan Xu2, Yan Liu2, Tao Tu1, Qiuzhen Lin1, Cancan Wang3, Qiming Liu1, Yichao Xiao1 ![]()
1. Department of Cardiology, The Second Xiangya Hospital, Central South University, Changsha 410011, Hunan Province, People's Republic of China.
2. Xiangya School of Medicine, Central South University, Changsha 410011, Hunan Province, People's Republic of China.
3. Department of Endocrinology, The Second Xiangya Hospital, Central South University, Changsha 410011, Hunan Province, People's Republic of China.
# These authors contributed equally to this study.
Received 2024-8-13; Accepted 2024-12-3; Published 2025-1-1
Abstract

Elevated lipoprotein(a) [Lp(a)] levels are increasingly recognized as a significant risk factor for cardiovascular diseases and may also contribute to atrial fibrillation (AF). This review investigated the indirect mechanisms through which Lp(a) may influence AF, including proatherogenic, prothrombotic, and proinflammatory pathways. Traditional lipid-lowering therapies, such as lifestyle modifications and statins, have limited effects on Lp(a) levels. Emerging treatments, such as proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, lipoprotein apheresis, small interfering RNA, antisense oligonucleotides, cholesterol ester transfer protein inhibitors, and interleukin-6 receptor monoclonal antibodies, are promising alternatives. Notably, only PCSK9 inhibitors and lipoprotein apheresis have been shown to reduce both Lp(a) levels and cardiovascular events. Research indicates varying associations between Lp(a) and AF across different populations, underscoring the need for diverse, large-scale studies to elucidate these differences. Ongoing trials aim to provide clearer insights into these relationships. Addressing these gaps is essential for developing targeted therapies to manage elevated Lp(a) and mitigate the risk of AF and associated cardiovascular events.
Keywords: Lipoprotein(a), Atrial fibrillation, Cardiovascular disease, Pathogenic mechanism, Therapies
Introduction
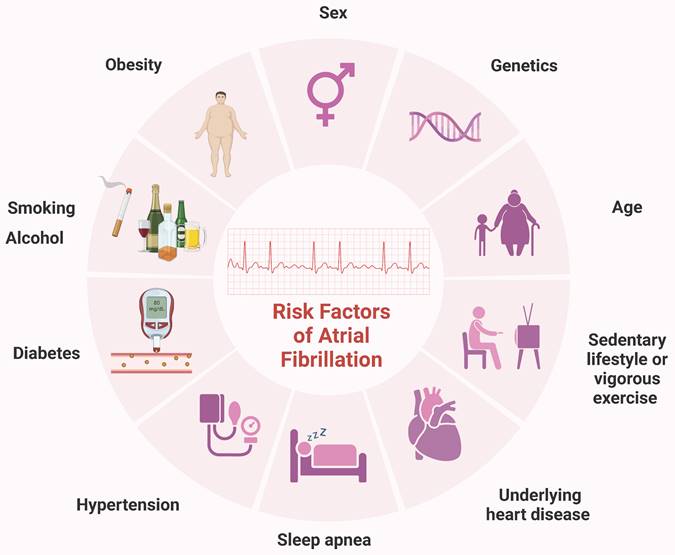
Atrial fibrillation (AF) is the most common sustained arrhythmia in adults and represents a significant global public health challenge. In 2010, over 33 million individuals worldwide were affected by AF, with higher incidence and prevalence rates observed in developed countries [1]. Projections indicate that by 2030, the European Union will have 14-17 million AF patients, with an annual diagnosis of 120000-215000 new cases [2]. AF is influenced by various factors, including sex, age, genetics, hypertension, and obesity [3, 4]. Other potential contributors include diabetes, smoking, obstructive sleep apnea, vigorous exercise, sedentary lifestyles, and alcohol consumption [5, 6] (Figure 1). This complex array of risk factors underscores the necessity for ongoing research to identify additional contributors and to develop novel treatment and prevention strategies.
Common risk factors for AF. Sex, age, genetics, history of underlying heart disease, particularly congestive HF and valvular disease, hypertension, obesity, diabetes, obstructive sleep apnea, smoking, vigorous exercise, sedentary lifestyle, smoking and alcohol have all been linked to the incidence of AF. AF, atrial fibrillation; HF, heart failure.
In this context, the role of lipoprotein(a) [Lp(a)] has garnered significant attention. Lp(a) is a low-density lipoprotein cholesterol (LDL-C)-like lipid particle composed of cholesterol, cholesteryl esters, apolipoprotein B100, apolipoprotein(a) [Apo(a)], and small amounts of triglycerides and carbohydrates. It is synthesized primarily in the liver and is metabolized in the liver or kidneys [7, 8]. Current studies suggest that elevated Lp(a) levels may increase the risk of cardiovascular disease (CVD), identifying Lp(a) as an independent marker of atherosclerosis [9]. Notably, Lp(a) levels are genetically determined and exhibit significant variation among different ethnic groups. While some studies have linked elevated Lp(a) levels to a greater risk of AF, others have suggested that increased Lp(a) may reduce the risk of AF [10-12]. The pathogenic mechanisms of Lp(a) include proatherogenic, prothrombotic, and proinflammatory processes [13, 14]. However, the exact role of Lp(a) in AF risk remains unclear and may be related to these mechanisms. A deeper understanding of the relationship between Lp(a) and AF is crucial for developing novel prevention and treatment strategies.
This article reviews the mechanisms by which Lp(a) may contribute to the development of AF and its role in AF-related comorbidities. We also explore treatment strategies for reducing Lp(a) levels to clarify the connection between Lp(a) and AF, providing new insights for the management and prevention of AF.
Conflicting evidence and racial variability
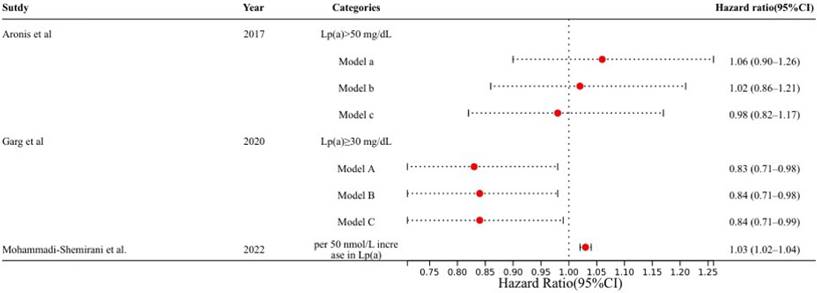
Previous studies have indicated that elevated Lp(a) levels increase the risk of CVD. Research by Satterfield et al. [15] revealed that elevated Lp(a) levels in European pedigrees contributed to AF, suggesting potential clinical implications. However, in 2014, Mora et al. [16] reported no relationship between Lp(a) and AF. This study has several limitations, including a reliance on a single baseline lipoprotein measurement, a lack of continuous electrocardiogram (ECG) monitoring, and the potential exclusion of asymptomatic AF patients. Furthermore, the study population consisted solely of women, which may introduce demographic biases. In 2017, Aronis et al. [12] reported that high Lp(a) levels were not associated with incident AF, but this study excluded individuals with extreme phenotypes of high Lp(a) levels (i.e., > 100 mg/dL). Moreover, the study included only white and black participants, raising concerns about the generalizability of the results to other ethnic groups. A 2020 study by Garg et al. [17] revealed that participants with Lp(a) levels above 30 mg/dL had a lower chance of developing AF. Conversely, Jiang et al. [18] demonstrated that genetically elevated Lp(a) levels increased AF risk. However, their results were only theoretically significant and did not account for multiple hypothesis testing, potentially affecting the robustness of their findings. The study by Mohammadi-Shemirani et al. [19] addressed a common limitation of earlier observational studies—the limited number of participants—by utilizing the UK Biobank, thereby increasing the number of AF cases by up to 20-fold. These findings, which are based on epidemiological and genomic studies, suggest that Lp(a) might be a causal mediator of AF (Figure 2). Higher Lp(a) levels in the UK Biobank were linked with an increased risk of AF incidence. This study also revealed that Lp(a) has a specific residual effect on AF, independent of its association with ischemic heart disease or aortic valve stenosis.
Adjusted models examining the associations between Lp(a) levels and AF in recent studies. Model a: Adjusted for age, sex, and ethnic groupings. Model b: Model a was extended by adding smoking status, systolic and diastolic blood pressure, hypertension therapy, heart rate, height, BMI, ECG left ventricular hypertrophy, PR interval, prevalent HF, CAD, and diabetes mellitus. Model c: Model b was further extended by adding LDL cholesterol, HDL cholesterol, triglycerides, lipid-lowering medication, and a log-transformed N-terminal pro-B-type natriuretic peptide. Model A: Adjusted for age, race, sex, education, income, and location. Model B: Model A was extended by adding height, BMI, smoking status, diabetes mellitus status, SBP, DBP, antihypertensive medication, physical activity, and alcohol intake. Model C further extended Model B by adding total cholesterol, HDL cholesterol, triglycerides, and lipid-lowering treatment. AF, atrial fibrillation; BMI, body mass index; CAD, coronary artery disease; DBP, diastolic blood pressure; ECG, electrocardiogram; HDL, high-density lipoprotein; HF, heart failure; LDL, low-density lipoprotein; Lp(a), lipoprotein(a); SBP, systolic blood pressure.
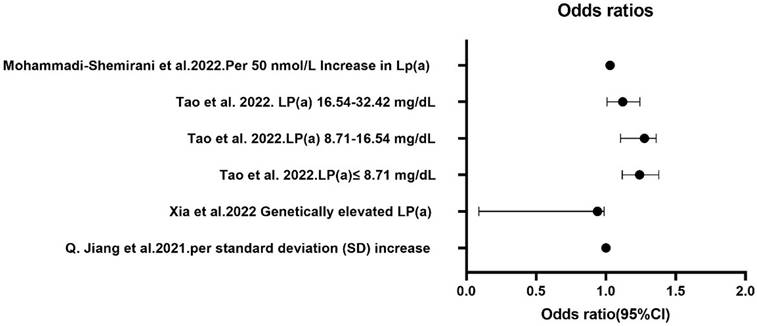
A common limitation of the above studies is that they included only white and black populations, leaving it unclear whether these findings apply to other ethnicities, such as Han Chinese. Lp(a) levels vary significantly across different races, and whether these findings can be generalized to other populations remains uncertain. This area requires further exploration. Additionally, the specific subcomponents of Lp(a) that affect AF and their mechanisms of action remain unanswered questions. Xia et al. [11] proposed that hereditary Lp(a) levels were not linked with AF among the Han Chinese population. However, a study by Tao et al. [20] revealed that low circulating Lp(a) levels were associated with AF, particularly in Han Chinese women, suggesting that Lp(a) might help stratify AF risk in females. The varying results from these studies may be due to differences in Lp(a) levels between races (Figure 3).
Comparison of Lp(a) levels and AF risk in European and Asian populations. The results from several independent studies investigating the relationship between Lp(a) levels and the risk of AF. The findings illustrate notable disparities in AF risk associated with Lp(a) levels between European and Asian populations. AF, atrial fibrillation; Lp(a), lipoprotein(a).
While evidence suggests a positive correlation between high Lp(a) levels and AF, the relationship is complex and influenced by racial and demographic factors. Future research should focus on identifying specific targets of Lp(a) for treating and preventing AF. Large-scale randomized controlled trials (RCTs) are needed to confirm whether these findings apply to East Asian populations and to clarify the mechanisms by which Lp(a) influences AF risk.
Pathogenic mechanisms linking Lp(a) to AF
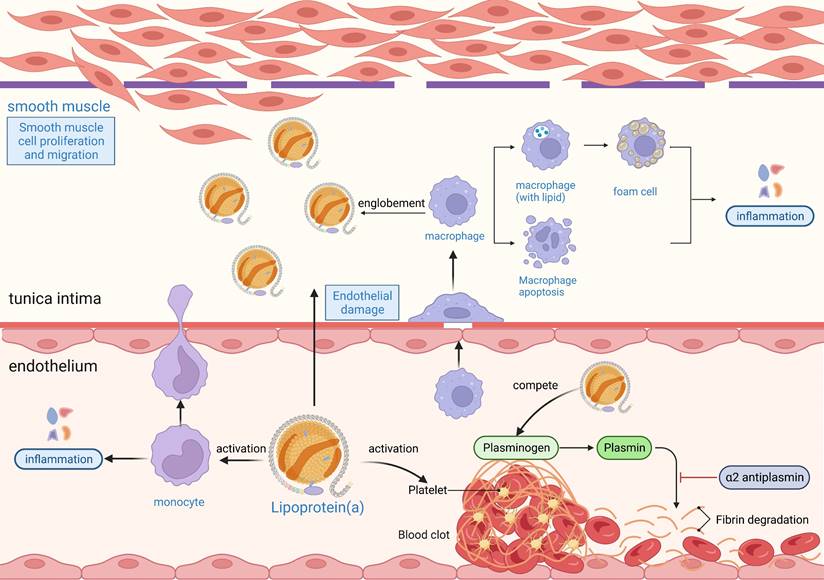
Understanding the pathogenic mechanisms linking Lp(a) to AF is critical for identifying potential therapeutic targets. The relationship between Lp(a) function and AF pathogenesis can be explored from three main perspectives: proatherogenic, prothrombotic, and proinflammatory mechanisms (Figure 4).
The proatherogenic, prothrombotic, and proinflammatory mechanisms of Lp(a). Lp(a) enters the endothelium and is readily oxidized, damaging endothelial cells and smooth muscle cells. The damaged endothelium secretes cytokines or growth factors to induce inflammatory cells such as macrophages and monocytes to enter the endothelium to phagocytose lipids and form foam cells. The damaged endothelium secretes growth factors that activate smooth muscle cells to proliferate and migrate, producing an extracellular matrix that increases the thickness and stiffness of the intima. The foam cells in the intima become necrotic and disintegrate to form atheromatous plaques. The inflammatory response occurs throughout the process of atherosclerosis, and OxPLs in Lp(a) are potent proinflammatory mediators. Lp(a) promotes thrombosis by exacerbating endothelial damage and activating platelet-associated responses, and high concentrations of Lp(a) may inhibit fibrinolytic responses by inhibiting fibrinogen activation. Lp(a), lipoprotein(a); OxPL, oxidized phospholipid.
Proatherogenic mechanisms
Lp(a) plays a significant role in the development of atherosclerosis. Studies indicate that Lp(a) bound to oxidized phospholipids (OxPLs) is more readily taken up by macrophages, increasing their atherogenic potential [21]. Elevated levels of Lp(a) can directly damage the vascular wall and exacerbate atherosclerosis through inflammatory responses. Moreover, the OxPL component of Lp(a) activates endothelial cells and promotes monocyte migration across the endothelium, worsening atherosclerosis [22, 23]. Atherosclerosis can mediate the occurrence of AF. The results from an observational and Mendelian randomization study demonstrated that for each 50 nmol/L (23 mg/dL) genetically predicted increase in Lp(a), the risk of AF increased by 3% [95% confidence interval (CI): 1.02-1.05]. Mendelian randomization via two independent genome-wide association studies (GWASs) provided evidence of a positive causal relationship between Lp(a) levels and AF [19]. Additionally, a meta-analysis of Mendelian randomization data demonstrated that elevated Lp(a) levels were associated with an increased risk of AF [odds ratio (OR): 1.024, 95% CI: 1.007-1.042, I² = 87.72%; P < 0.001], with notable variations across different ethnic groups. In European populations, higher Lp(a) levels were linked to a significantly increased risk of AF (OR: 1.023, 95% CI: 1.007-1.040; P < 0.001). However, in Chinese populations, elevated Lp(a) levels are associated with a comparatively lower risk of AF (OR: 0.940, 95% CI: 0.893-0.990) [24]. These findings suggest that Lp(a) is a potential causal mediator of AF development, with effects that are partly independent of atherosclerotic cardiovascular disease (ASCVD). Atherosclerosis affects atrial structure and electrical function through various mechanisms, including inflammation, myocardial ischemia, atrial infarction, myocardial cell death, atrial fibrosis, and coronary artery narrowing, all of which can contribute to AF [25]. Among these factors, endothelial dysfunction is considered a key element in the development of AF in patients with atherosclerosis. During AF episodes, rapid atrial pacing can decrease shear stress on blood vessels, reduce nitric oxide production by the endothelium, and increase oxidative stress markers, potentially creating a vicious cycle [26]. Given the multifaceted role of Lp(a) in promoting atherosclerosis and its downstream impact on AF development, targeting endothelial dysfunction has emerged as a promising therapeutic strategy. Research has shown that statins can improve endothelial dysfunction in both peripheral and coronary arteries, reduce the risk of AF, and lower the recurrence rate of AF after catheter ablation or dual-chamber pacemaker implantation [27-29]. These findings underscore the importance of comprehensive management approaches to address both the atherosclerotic burden and its arrhythmogenic complications.
The role of Lp(a) in promoting atherosclerosis and influencing AF is complex and multifaceted. Lp(a) can impact atrial structure and function both directly and indirectly. Future research should further explore the specific mechanisms by which Lp(a) contributes to atherosclerosis and its associated AF risk and assess whether reducing Lp(a) levels could effectively decrease the incidence and progression of AF.
Prothrombotic mechanisms
Lp(a) is closely associated with an increased risk of thrombosis in patients with AF. One of the key components of Lp(a) is Apo(a), which shares structural similarities with plasminogen but has different physiological functions. Apo(a) inhibits tissue plasminogen activator, preventing the conversion of plasminogen to plasmin [30], thereby reducing fibrinolysis and impairing the body's ability to break down clots, promoting thrombus formation. Moreover, Lp(a) enhances coagulation by increasing platelet reactivity, upregulating tissue factor expression, and inhibiting the tissue factor pathway [31, 32]. Although the interaction between Lp(a) and platelets has been studied, the exact receptor involved remains unclear. Current evidence on this interaction is conflicting, with some studies suggesting that Lp(a) may have both activating and inhibitory effects depending on the activation pathway. For example, Lp(a) reportedly enhances platelet activation via thrombin-related activating hexapeptides, although it does not directly interact with thrombin or adenosine diphosphate [33]. Conversely, other studies have indicated that Lp(a) inhibits platelet activation induced by collagen or thrombin [34]. Despite these discrepancies, there is broad consensus that Lp(a) significantly impairs fibrinolysis by competing with plasminogen and tissue plasminogen activator for binding sites on the platelet surface, thereby obstructing thrombus resolution [35]. This prothrombotic activity contributes to a hypercoagulable state and is associated with systemic inflammation and atrial fibrosis, both of which are linked to adverse outcomes in AF patients [36]. Elevated Lp(a) levels are particularly concerning because they correlate with the progression of atrial fibrosis and the disruption of atrial structure and electrical activity, increasing the risk of clot formation [37]. Additionally, Lp(a) may exacerbate inflammatory responses in the atrial endocardium [38], further increasing thrombotic risk.
There is increasing evidence that AF is associated with a prothrombotic state characterized by various hematological abnormalities, including elevated Lp(a) levels [39, 40]. Igarashi et al. [41] reported a correlation between thrombi in the left atrium of elderly patients with chronic AF and elevated Lp(a) levels, although this study had a small sample size. Another study on nonvalvular AF revealed that high Lp(a) levels were linked to thrombotic events in individuals with low CHA2DS2-VASc scores [42]. A cross-sectional investigation revealed that elevated Lp(a) levels in patients with nonvalvular AF increased the likelihood of ischemic stroke and thrombosis [43]. These findings collectively suggest a significant association between elevated Lp(a) levels and increased thrombotic risk in patients with AF.
Given the role of Lp(a) in promoting coagulation and inhibiting fibrinolysis in AF patients, targeting Lp(a) with specific treatments could be clinically significant [44]. Recent studies have shown that proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors can significantly lower Lp(a) levels [45, 46], potentially offering benefits in thrombus prevention for AF patients. Further research is needed to evaluate the effectiveness of these therapeutic strategies in reducing thrombotic events in AF patients and to determine how these strategies can be integrated into comprehensive AF management plans.
Proinflammatory mechanisms
Recent evidence highlights the proinflammatory effects of Lp(a), which are driven primarily by its OxPL content [47]. OxPLs associated with Lp(a) are potent proinflammatory mediators that contribute to cardiovascular inflammation and play significant roles in endothelial dysfunction, vascular inflammation, and oxidative stress [48]. Current research indicates that AF mechanisms are intricately linked to atrial electrical and structural remodeling, processes heavily influenced by inflammation [49-51]. Inflammatory mediators modulate ion channels and calcium homeostasis, disrupt cardiac action potential duration, and promote aberrant calcium handling in cardiomyocytes. Such dysregulation fosters atrial ectopic activity and reentry circuits, which are critical in AF initiation and perpetuation. Concurrently, inflammation activates fibroblasts, driving excessive collagen deposition and atrial fibrosis while also inducing cardiomyocyte apoptosis, further contributing to atrial structural remodeling [52].
Inflammation is closely associated with both the prevalence and progression of AF. Mediators of the inflammatory response can alter the electrophysiological and structural substrates of the atria, thereby increasing susceptibility to AF. A cross-sectional study demonstrated that elevated levels of interleukin-6 (IL-6) were associated with the development of both paroxysmal and chronic AF [53]. Additionally, a study by Jia et al. [54] revealed that AF patients with higher IL-6 levels had a significantly increased risk of stroke [hazard ratio (HR): 3.81, 95% CI: 1.11-13.05; P = 0.033) and all-cause mortality (HR: 3.11, 95% CI: 1.25-7.72; P = 0.015). Furthermore, risk factors for AF, such as obesity, diabetes, hypertension, and metabolic syndrome, are also associated with inflammatory states [55]. These findings indicate that inflammation plays a crucial role in the onset and progression of AF. Notably, OxPLs carried by Lp(a) can amplify these inflammatory pathways by activating Toll-like receptors and downstream nuclear factor-kappa B signaling, both of which have been implicated in atrial fibrosis and oxidative stress [56]. This dual proinflammatory and pro-oxidative action creates a pathological environment conducive to AF. Oxidative stress, exacerbated by OxPLs, further initiates the remodeling process. Excess reactive oxygen species (ROS) promote the oxidation of calcium-handling proteins, such as ryanodine receptors, impairing their function and contributing to intracellular calcium overload—a hallmark of AF [57]. ROS also facilitate the activation of matrix metalloproteinases, enzymes that degrade extracellular matrix components, disrupting atrial structural integrity and promoting fibrosis [58].
Given the overlap between OxPL-mediated inflammation, oxidative stress, and AF pathogenesis, targeting Lp(a) and its associated OxPLs represents a promising therapeutic strategy. Anti-inflammatory approaches, such as interleukin-1 blockers and Toll-like receptor inhibitors, may mitigate atrial remodeling and slow fibrosis progression, thereby preserving atrial structure and function. Preclinical studies have shown that strategies that reduce ROS levels can improve calcium homeostasis and prevent electrical remodeling, highlighting the potential of antioxidative therapies in AF management [59-61].
Future investigations should focus on the direct modulation of Lp(a)-OxPL pathways in AF through clinical trials, exploring agents such as PCSK9 inhibitors and antisense oligonucleotides to reduce Lp(a) levels. Additionally, therapies targeting oxidative stress and inflammation, potentially in combination, could offer synergistic benefits by addressing both structural and electrical remodeling, thereby reducing the AF burden and improving patient outcomes.
Elevated Lp(a) and cardiovascular risk
Association of elevated Lp(a) with AF and coronary artery disease
Coronary artery disease (CAD) is characterized by narrowing or blockage of coronary arteries due to atheromatous plaques, leading to myocardial ischemia, hypoxia, or necrosis. CAD shares several common risk factors with AF, including smoking, obesity, hypertension, dyslipidemia, age, and race [51, 62, 63]. Patients with CAD are more prone to developing AF, which can exacerbate the severity of CAD and pose greater challenges in clinical management [25] (Figure 5). Identifying common factors between these diseases can enhance prevention and treatment strategies.
Atherosclerosis, thrombosis, and stroke in patients with AF. AF increases the risk of thrombosis in the left atrium due to weak atrial contractility, stagnant blood flow, and a potential prethrombotic state. Thromboembolism, where a clot formed in the heart moves to other parts of the body, is a dangerous complication of AF, with ischemic stroke being the most serious outcome. Furthermore, AF and CAD are synergistic conditions. AF can exacerbate CAD through thrombosis, inadequate blood supply, and the promotion of atherosclerosis. AF, atrial fibrillation; CAD, coronary artery disease.
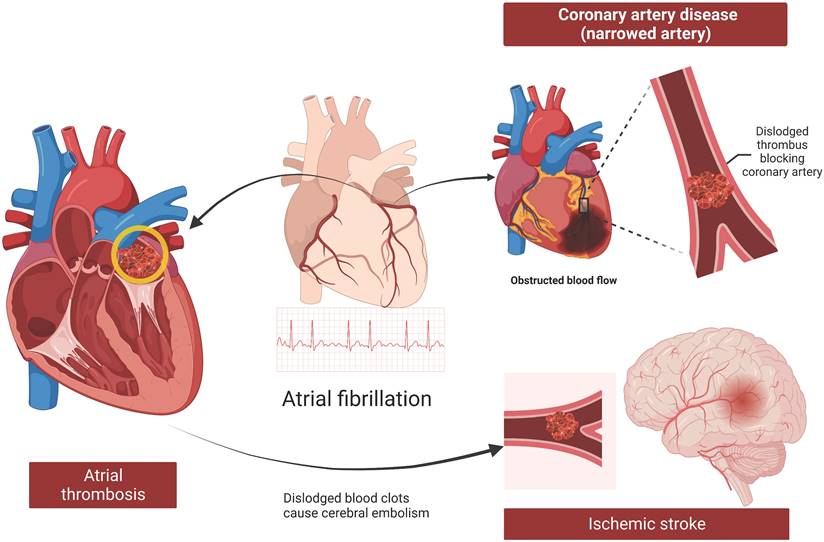
Elevated Lp(a) levels are associated with an increased risk of CAD [64]. Independent of CAD, Mendelian randomization studies have shown a strong association between elevated Lp(a) levels and the development of AF [18, 19]. A study by Li et al. [65] revealed that patients with both CVD and AF were significantly older and had higher Lp(a) levels than those with CAD without AF. Multivariate logistic regression analysis revealed that increased Lp(a) levels and age were independent risk factors for AF in patients with CAD. Therefore, high Lp(a) levels may increase the incidence of AF in patients with CAD, but large-scale RCTs are needed to further validate this association.
Association of Lp(a) with AF and ischemic stroke
AF increases the risk of thrombus formation within the left atrium due to reduced atrial contractility, stagnant blood flow, and a prethrombotic state. This predisposes patients to thromboembolism, where a clot formed in the heart can travel to other parts of the body, leading to serious complications such as ischemic stroke (Figure 5). Current evidence suggests that Lp(a) may increase the risk of ischemic stroke [66]. However, the direct correlation between Lp(a) and stroke triggered by AF remains inconclusive.
Research by Aronis et al. [12], which focused on patients without AF, did not determine whether elevated Lp(a) levels are associated with an increased incidence of stroke in those with AF. Despite this limitation, their study revealed that high Lp(a) levels were linked to an increased risk of ischemic stroke. Arora et al. [67] reported that high Lp(a) levels were linked to an increased risk of ischemic stroke. However, their study did not find a sex-specific correlation between Lp(a) levels and stroke risk. Although their research suggested a potential racial disparity in Lp(a)-associated stroke risk, the results were not statistically significant, indicating that further investigation is needed. A retrospective case-control study by Fu et al. [68] in a Han Chinese population reached similar conclusions, noting a correlation between Lp(a) and ischemic stroke but not addressing the role of AF.
While most studies indicate a correlation between Lp(a) and ischemic stroke, few have explored whether AF plays a role in this association. This represents a significant research gap, and addressing it could lead to breakthroughs in prevention and treatment strategies for stroke and AF.
Association of Lp(a) with AF and heart failure
Heart failure (HF) and AF frequently coexist, each exacerbating the clinical course of the other [69]. Elevated Lp(a) levels have been implicated in the development and progression of HF, potentially impacting the incidence of AF in these patients. Studies indicate that Lp(a) may contribute to HF through its proatherogenic, prothrombotic, and proinflammatory properties [70-72]. Elevated Lp(a) levels can lead to atherosclerosis, increasing the likelihood of ischemic events and subsequent HF. Moreover, the prothrombotic nature of Lp(a) can exacerbate HF by promoting microvascular thrombosis, further impairing cardiac function. Inflammatory responses driven by Lp(a) can also worsen HF by promoting myocardial fibrosis and adverse remodeling.
A study by Masson et al. [73] demonstrated a positive relationship between Lp(a) levels and HF. Among individuals with stage A or B HF, higher Lp(a) and OxPL concentrations are independent risk factors for progression to symptomatic HF or cardiovascular death [74]. Given the close relationship between HF and AF, elevated Lp(a) levels may impact the development and progression of both conditions through various mechanisms. Future research should focus on investigating the specific role of Lp(a) in both HF and AF and assessing its potential as a therapeutic target.
Therapeutic strategies for Lp(a) reduction
Among the lipoproteins associated with CVD, Lp(a) plays a crucial role in determining disease risk. Lowering Lp(a) levels can reduce the incidence of CVD and mitigate associated lipid abnormalities. Therefore, identifying effective methods to lower Lp(a) is essential for improving patient outcomes. However, current lipid-lowering drugs have had limited success in reducing Lp(a), and no specific drug for this purpose has been approved. This review summarized existing treatments for lowering Lp(a) and aimed to provide evidence for potential clinical applications (Table 1).
Current therapies for Lp(a).
| Therapies | Mechanism | Impact on Lp(a) | Effect on AF or CVD |
|---|---|---|---|
| Statins | Reducing cholesterol synthesis through competition with endogenous enzymes | Do not reduce Lp(a), and may even increase its level | Reduces the risk of HF and stroke events in patients with AF |
| LA | The blood lipids were removed by extracorporeal circulation blood purification | Lp(a) decreased by an average of 71.1% in patients treated with LA | No studies have examined using LA to lower the incidence of AF by lowering Lp(a) levels |
| PCSK9 inhibitors | Enhancing the number of LDL receptors | PCSK9 inhibitors not only have the clinical effect of decreasing Lp(a) but also reduce the occurrence of cardiovascular events | PCSK9 shows promise in lowering the risk of AF, but it is essential to consider the possible increased risks of developing Alzheimer's disease and asthma |
| Olpasiran | Effectively reduces PCSK9 production | The efficacy of Olpasiran in reducing Lp(a) levels increased with higher concentrations | No studies have examined using Olpasiran to lower the incidence of AF by lowering Lp(a) levels |
| Antisense oligonucleotides | Bind directly to mRNA in hepatocytes, inhibiting the target gene's protein synthesis and thereby reducing Lp(a) | Treated with antisense oligonucleotides experienced an average 80% reduction in Lp(a) levels when given the highest dose | Animal experiments have shown that antisense oligonucleotides reduce the frequency and duration of AF by downregulating SK3 channels, but clinical evidence is lacking |
| CETP inhibitors | Block the transfer of cholesterol esters from nonatherogenic particles to atherogenic lipoprotein fractions | TA-8995 reduced LDL cholesterol levels by 45.3%, increased HDL cholesterol levels by 179.1%, and boosted ApoA-1 levels by 63.4% | CETP inhibitors may reduce the incidence of CVD, including heart disease and stroke |
| IL-6 receptor monoclonal antibody | Inhibiting IL-6-induced Lp(a) mRNA | Tocilizumab can reduce Lp(a) levels in RA patients by approximately 30%-40% | No studies have examined using IL-6 receptor monoclonal antibody to lower the incidence of AF by lowering Lp(a) levels |
AF, atrial fibrillation; CETP, cholesterol ester transfer protein; CVD, cardiovascular disease; HDL, high-density lipoprotein; HF, heart failure; IL-6, interleukin-6; LA, lipoprotein apheresis; LDL, low-density lipoprotein; Lp(a), lipoprotein(a); mRNA, messenger RNA; PCSK9, proprotein convertase subtilisin/kexin type 9; RA, rheumatoid arthritis; SK3, small conductance Ca2+-activated K+ channel.
Lifestyle modifications
Many cardiovascular events are associated with elevated Lp(a) levels, which are primarily genetically determined. Although lifestyle changes are key interventions for CVD, studies have shown that lifestyle modifications, such as dietary control, increased exercise, or smoking cessation, do not significantly alter Lp(a) levels. Research indicates that Lp(a) levels remain unaffected by different diets, whether they are fasted or not [75]. While some studies suggest that high-carbohydrate and high-protein diets can slightly increase Lp(a) levels compared with diets rich in unsaturated fatty acids, these increases are minimal compared with the impact of alcohol consumption [76]. Therefore, specific therapies targeting Lp(a) are needed to manage cardiovascular risks associated with elevated Lp(a) levels more effectively.
Statins
Statins, which are hydroxymethylglutaryl coenzyme A reductase inhibitors, work by reducing cholesterol synthesis through competition with endogenous enzymes [77]. This mechanism effectively lowers the risk of CVD. However, while statins are beneficial for managing overall cholesterol levels, they do not reduce Lp(a) levels and may even increase them [78]. A meta-analysis pooling data from 45044 patients across seven studies revealed that the risk of Lp(a)-associated CVD persisted in patients on statins. The HR was 1.31 (95% CI: 1.08-1.58) before statin use and increased to 1.43 (95% CI: 1.15-1.76) after statin use, indicating an increasing trend in Lp(a) levels following statin therapy [79]. Consequently, statins are not suitable for lowering Lp(a) levels.
Despite their limitations in reducing Lp(a), statins have proven effective in other cardiovascular contexts. Studies have shown that statin use is associated with a decreased risk of incident HF in a duration-dependent manner among patients with AF [80]. In addition, the results from a retrospective cohort study revealed that statin therapy was independently associated with a lower incidence of stroke after adjusting for confounding variables (HR: 0.87, 95% CI: 0.78-0.97; P = 0.01) [81]. These benefits underscore the importance of statins in specific cardiovascular conditions, even though their role in Lp(a) management remains limited.
Lipoprotein apheresis
Patients with elevated Lp(a) levels benefit significantly from lipoprotein apheresis (LA), which has the greatest efficacy in preventing cardiovascular events. In addition to its lipid-lowering effects, LA can reduce the serum levels of proinflammatory and prothrombotic factors, decrease blood viscosity, enhance microvascular myocardial perfusion, and potentially exert beneficial effects on endothelial function [82]. The German Lipoprotein Apheresis Registry summarized data on lipoprotein changes following LA treatment in 1435 patients with CVD enrolled between 2011 and 2016 [83]. On the basis of the results of all patients treated with LA, an average reduction of 71.1% in Lp(a) was achieved. Additionally, LA treatment was associated with a low rate of adverse events (5.9%), mostly puncture-related problems, and resulted in a 90% reduction in cardiovascular events [83]. A retrospective cohort study demonstrated that after a mean duration of 7.1 years of LA treatment, patients' median Lp(a) levels decreased from 95.0 to 31.1 mg/dL (-67.3%, P < 0.0001), mean LDL-C levels decreased from 1.85 to 0.76 mmol/L (-58.9%, P < 0.0001), and the annual rate of major adverse cardiac events decreased from 0.34 to 0.006 (-0.33, P = 0.0002) [84]. These findings indicate that LA not only lowers Lp(a) and LDL-C levels but also concomitantly reduces the incidence of cardiovascular events. Moreover, Pokrovsky et al. [85] reported that eliminating Lp(a) from the bloodstream can reduce inflammatory and thrombotic processes within months and lead to the regression of atherosclerotic plaques within 1.5 years. In both clinical trials and real-world settings, LA treatment for 2-5 years has further demonstrated that a 60-80% reduction in Lp(a) is associated with a proportional decrease in the incidence and risk of cardiovascular events. However, no studies have investigated whether LA can reduce the incidence of AF by lowering Lp(a) levels. Nevertheless, LA remains a highly effective method for specifically reducing Lp(a). If a causal relationship between lowering Lp(a) and a reduced incidence of AF can be established, LA may provide a promising therapeutic approach for the management of AF.
PCSK9 inhibitors
It is hypothesized that PCSK9 inhibitors reduce Lp(a) levels by increasing the number of LDL receptors, thereby increasing Lp(a) clearance. PCSK9 inhibitors not only decrease Lp(a) levels but also reduce the incidence of cardiovascular events [86]. The results from the FOURIER-OLE study demonstrated that patients treated with evolocumab experienced a 23% reduction in the risk of cardiovascular death (HR: 0.77; 95% CI: 0.60-0.99; P = 0.04) and a 20% reduction in the risk of myocardial infarction or stroke (HR: 0.80; 95% CI: 0.68-0.93; P = 0.003) compared with those in the placebo group [87]. A secondary analysis of the FOURIER-OLE study indicated that achieving long-term low LDL-C levels with evolocumab, as low as < 20 mg/dL (< 0.5 mmol/L), was associated with a reduced risk of cardiovascular outcomes without significant safety concerns [88]. Furthermore, a meta-analysis demonstrated that PCSK9 inhibitors provide substantial and durable reductions in LDL-C levels and improve cardiovascular outcomes [89]. These findings underscore the significant potential of PCSK9 inhibitors in enhancing cardiovascular health.
Two monoclonal antibodies, evolocumab and alirocumab, both of which are fully human monoclonal antibodies injected subcutaneously, have been developed and are in clinical use as PCSK9 inhibitors [46]. For different baseline plasma PCSK9 levels, evolocumab resulted in a reduction in lipoprotein levels. The effect of alirocumab at the maximum dose, whether used as monotherapy or in combination, achieved a 60% decrease in plasma lipoproteins, which is consistent with the effects of evolocumab [46]. From a safety perspective, the safety profile of PCSK9 inhibitors is positive, with only a few patients experiencing mild injection site reactions. However, there is some evidence that PCSK9 inhibitors do not reduce the risk of vascular wall inflammation, possibly because PCSK9 inhibitors do not significantly decrease Lp(a) in people with persistently increasing Lp(a) [90]. A Mendelian randomized trial suggested that a targeted reduction in the PCSK9 concentration is significant for reducing the occurrence of AF (OR: 0.90, 95% CI: 0.83-0.97) and has a protective effect on patients at risk of AF [91]. These findings suggest that a lower concentration of PCSK9 is associated with a lower risk of AF, and other methods to reduce PCSK9 concentrations may also prevent AF, providing a valid approach for treatment. However, low PCSK9 concentrations increase the risk of developing Alzheimer's disease and asthma [91].
These findings highlight the potential of PCSK9 inhibitors in reducing Lp(a) levels and associated cardiovascular events. However, despite their efficacy, PCSK9 inhibitors may not significantly impact vascular wall inflammation in patients with persistently elevated Lp(a). Furthermore, while a targeted reduction in PCSK9 concentrations shows promise in lowering the risk of AF, it is essential to consider the possible increased risks of developing Alzheimer's disease and asthma. Thus, comprehensive research and clinical evaluations are necessary to balance these benefits and risks, guiding the effective use of PCSK9 inhibitors in managing CVD.
Olpasiran
Olpasiran, a novel small interfering RNA (siRNA) targeting PCSK9, effectively reduces PCSK9 production, leading to decreased Lp(a) synthesis in the liver. In a placebo-controlled trial, patients with ASCVD treated with Olpasiran presented significantly lower Lp(a) levels than did those receiving a placebo. The efficacy of Olpasiran in reducing Lp(a) levels is dose-dependent, with higher doses resulting in greater reductions, while adverse events remain minimal and primarily involve injection site pain [92]. Consequently, Olpasiran shows promise as a therapeutic option for ASCVD patients with the aim of reducing Lp(a). Furthermore, recent results from the OCEAN(a)-DOSE study demonstrated that participants receiving doses of ≥ 75 mg every 12 weeks sustained an approximately 40-50% reduction in Lp(a) levels nearly one year after the last dose, with no significant adverse effects observed [93]. Given that PCSK9 inhibitors have been shown to lower AF incidence by reducing Lp(a), it is plausible that Olpasiran may similarly reduce the risk of AF. However, further evidence is needed to confirm its therapeutic efficacy in reducing AF risk.
Antisense oligonucleotides
Antisense oligonucleotides bind directly to messenger RNA (mRNA) in hepatocytes, inhibiting target gene protein synthesis and thereby reducing Lp(a). Like siRNA therapy, antisense oligonucleotide therapy works by binding to RNA in target cells through base pairing, inhibiting gene function. AKCEA-APO(a)-LRX is an antisense oligonucleotide that decreases Lp(a) in hepatocytes in a dose-dependent manner. Patients treated with AKCEA-APO(a)-LRX experienced an average 80% reduction in Lp(a) levels when given the highest dose [94].
Some studies suggest a relationship between small conductance Ca2+-activated K+ (SK3) channels and the development of AF, indicating that downregulating the expression of these cardiac proteins may prevent AF. In an experiment in which antisense oligonucleotides were used to knock down SK3 channels in rats, the group with downregulated SK3 protein expression presented 78% shorter episodes of AF than did the control group with artificially induced AF. There was a 68% reduction in spontaneous AF episodes, and the duration of spontaneous AF episodes was significantly shorter, with the experimental group experiencing episodes lasting 7.2 seconds compared with 29.7 seconds in the control group [95]. Electrophysiological parameters were measured to verify the protective effect of SK3 channels on AF. Although the experimental group exhibited delayed prolongation of the effective refractory period, SK3 channels downregulation still conferred a protective effect against AF [95]. Given the close association of SK3 channel with AF, this trial indicates significant prospects for antisense oligonucleotides in treating AF. However, this approach lacks clinical evidence, and since rats do not fully represent human electrophysiological activity, more clinical data are needed to demonstrate the effects of antisense oligonucleotides on AF.
Cholesterol ester transfer protein inhibitors
Cholesterol ester transfer protein (CETP) inhibitors block the transfer of cholesterol esters from nonatherogenic high-density lipoprotein (HDL) particles to atherogenic lipoprotein fractions, including LDL. Anacetrapib and evacetrapib are notable CETP inhibitors, with TA-8995 emerging as a new CETP inhibitor with high tolerability. A phase 2 trial revealed that TA-8995 reduced LDL cholesterol levels by 45.3%, increased HDL cholesterol levels by 179.1%, and increased ApoA-1 levels by 63.4%. When used in combination with statins, TA-8995 further reduced LDL levels by an additional 39.8-50.2% [96]. However, the impact on cardiovascular event risk requires further validation through trials.
In a Mendelian randomized evaluation trial, targeted CETP inhibition had an OR of 0.99 (95% CI: 0.96-1.01) for AF, indicating no significant association [91]. Nevertheless, CETP inhibitors may reduce the incidence of CVD, including heart disease and stroke. The combined use of CETP inhibitors and PCSK9 inhibitors, which impact lipoprotein metabolism differently, could provide greater benefits. Both classes lower Lp(a) levels, and PCSK9 inhibitors have been shown to reduce the incidence of AF [91]. Ongoing studies are investigating the combined effects of these inhibitors on AF, potentially providing improved prognoses for patients.
IL-6 receptor monoclonal antibodies
Tocilizumab, a humanized monoclonal antibody, targets IL-6 receptors and is used to treat severe inflammatory conditions such as rheumatoid arthritis (RA). By inhibiting IL-6-induced Lp(a) mRNA and protein expression in human hepatocytes, tocilizumab reduces inflammation and joint damage in patients with RA [48]. Studies have shown that tocilizumab can reduce Lp(a) levels in RA patients by approximately 30-40% [97]. In addition, in patients with COVID-19, treatment with tocilizumab also reduces Lp(a) levels by approximately 30% [98].
IL-6 is commonly used as an indicator of inflammation and is used to gauge the efficacy of anti-inflammatory agents. Inflammation is also a known cause of AF, suggesting that IL-6 receptor monoclonal antibodies are promising for reducing AF occurrence. However, more research is needed to determine whether they reduce the incidence of AF by lowering Lp(a) levels.
Limitations and prospects
AF, as one of the most common and persistent arrhythmias, often presents without typical clinical symptoms, making early screening, diagnosis, and treatment crucial to prevent serious adverse events. The relationship between Lp(a) and AF has shown conflicting results across different ethnic groups. For example, high Lp(a) levels are positively associated with AF in Europeans, whereas studies in Eastern populations indicate a negative association. This discrepancy highlights several limitations in current research, such as ancestral homogeneity and insufficient sample sizes, which impedes a comprehensive understanding of the global role of Lp(a).
Most current studies, primarily those utilizing Mendelian randomization and cohort analyses, are limited by a lack of ethnic diversity. To elucidate the ethnic variations in disease incidence and the impact of allele distribution, future research must include larger and more diverse populations. Furthermore, the direct connection between specific components of Lp(a) and AF remains unclear, with current findings suggesting only indirect effects through proatherogenic, prothrombogenic, and proinflammatory mechanisms. Addressing these gaps could lead to new strategies for preventing and treating AF by targeting Lp(a).
A study by Mohammadi-Shemirani et al. [19] using UK biobank data revealed that elevated Lp(a) levels are linked to new-onset AF, independent of ASCVD. These findings point to potential new treatments for patients with high Lp(a) levels. However, effective lipid-lowering therapies specifically targeting elevated Lp(a) levels are limited, and no approved treatments are currently available. Therefore, developing effective Lp(a)-lowering strategies is critically needed.
Emerging Lp(a)-lowering therapies, such as PCSK9 inhibitors, siRNAs, and antisense oligonucleotides, have shown promise for significantly lowering Lp(a) levels. Drugs such as Olpasiran, PCSK9 inhibitors, and TA-8995 have demonstrated minimal severe side effects in clinical trials, whereas the antisense oligonucleotide mipomersen has been associated with hepatic side effects, limiting its applicability. Additionally, nonlipid therapies addressing elevated Lp(a)-induced problems, such as antiplatelet agents, have demonstrated effectiveness in modifying coagulation and platelet aggregation [99], suggesting alternative treatment options.
Despite numerous treatment options and clinical trials showing significant reductions in Lp(a) levels, only PCSK9 inhibitors and LA have demonstrated clear improvements in clinical outcomes and reduced cardiovascular events. This finding underscores the uncertainty surrounding the optimal clinical treatment regimen for patients with elevated Lp(a). Although many drugs can lower Lp(a), their ability to reduce CVD incidence remains unclear. Reducing cardiovascular events in patients with elevated Lp(a) is essential for improving patient prognosis and quality of life. Statins and antiplatelet agents, despite not directly lowering Lp(a), may reduce cardiovascular risk through their functions. This finding requires validation in larger trials.
An important consideration is whether lowering Lp(a) in patients with elevated levels is correlated with a reduced risk of cardiovascular events. Current evidence suggests that only PCSK9 inhibitors and LA reduce both Lp(a) levels and CVD risk. These findings indicate the potential benefit of combining Lp(a)-lowering drugs with cardiovascular medications to improve patient outcomes. The ongoing HORIZON trial (trial node: NCT04023552) is expected to provide valuable insights into these relationships, significantly advancing cardiovascular event prevention.
Conclusion
Elevated Lp(a) levels are associated with an increased risk of AF through mechanisms such as proatherogenic, prothrombotic, and proinflammatory effects. While current lipid-lowering therapies, such as statins, are ineffective at reducing Lp(a), emerging treatments, including PCSK9 inhibitors, siRNAs, and antisense oligonucleotides, show promise. LA also significantly decreases Lp(a) levels, but further study is needed to determine its impact on AF. Future research should focus on diverse populations to better understand the role of Lp(a) in AF and validate the effectiveness of new therapies.
Abbreviations
AF: atrial fibrillation
Apo(a): apolipoprotein(a)
ASCVD: atherosclerotic cardiovascular disease
BMI: body mass index
CAD: coronary artery disease
CETP: cholesterol ester transfer protein
CI: confidence interval
CVD: cardiovascular disease
DBP: diastolic blood pressure
ECG: electrocardiogram
GWAS: genome-wide association studies
HDL-C: high-density lipoprotein cholesterol
HF: heart failure
HR: hazard ratio
IL-6: interleukin-6
LA: lipoprotein apheresis
LDL-C: low-density lipoprotein cholesterol
Lp(a): lipoprotein(a)
mRNA: messenger RNA
OR: odds ratio
OxPL: oxidized phospholipid
PCSK9: proprotein convertase subtilisin/kexin type 9
RA: rheumatoid arthritis
RCTs: randomized controlled trials
ROS: reactive oxygen species
SBP: systolic blood pressure
siRNA: small interfering RNA
SK3: small conductance Ca2+-activated K+ channel
Acknowledgements
Figure 4 and Figure 5 were created with BioRender (https:// www.biorender.com/).
Funding
This work was supported by the National Natural Science Foundation of China [No. 82070356, 81770337, 82470333], the Key Project of Hunan Provincial Science and Technology Innovation [No. 2020SK1013, 2024JK2119], the Hunan Provincial Natural Science Foundation of China [No. 2021JJ30033, 2023JJ30791], and the Clinical Medical Technology Innovation Guidance Project of Hunan Science and Technology Agency [No. 2021SK53519].
Author contributions
Under the guidance of Professor Yichao Xiao. Cancan Wang and Qiuzhen Lin collected relevant information. Akedanmu Nuranmubieke, Yangfan Xu, and Yan Liu performed the image visualization. Zixi Zhang wrote the manuscript, which was revised by Bo Peng, Tao Tu, and Qiming Liu. All the authors contributed to the article and approved the final manuscript.
Consent to participate
All the authors participated in the study and made significant intellectual contributions to the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Naser N, Kulic M, Dilic M, Dzubur A, Durak A, Pepic E. et al. The Cumulative Incidence of Stroke, Myocardial infarction, Heart Failure and Sudden Cardiac Death in Patients with Atrial Fibrillation. Med Arch. 2017;71:316-9
2. Wańczura P, Aebisher D, Kłosowicz M, Myśliwiec A, Dynarowicz K, Bartusik-Aebisher D. Application of Photodynamic Therapy in Cardiology. Int J Mol Sci. 2024;25:3206
3. Lin CY, Hu YF, Lin YJ, Chen SA. Can Genetic Risk Scoring Predict Atrial Fibrillation Ablation Outcomes? Korean Circ J. 2019;49:350-2
4. Hindricks G, Potpara T, Dagres N, Arbelo E, Bax JJ, Blomström-Lundqvist C. et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur Heart J. 2021;42:373-498
5. Yao Y, Liu F, Wang Y, Liu Z. Lipid levels and risk of new-onset atrial fibrillation: A systematic review and dose-response meta-analysis. Clin Cardiol. 2020;43:935-43
6. Shamloo AS, Dagres N, Arya A, Hindricks G. Atrial fibrillation: A review of modifiable risk factors and preventive strategies. Rom J Intern Med. 2019;57:99-109
7. Pearson K, Rodriguez F. Lipoprotein(a) and Cardiovascular Disease Prevention across Diverse Populations. Cardiol Ther. 2020;9:275-92
8. Brandt EJ, Brandt DJ, Desai NR, Spatz ES, Nasir K, Mani A. Association of vitamins, minerals, and lead with lipoprotein(a) in a cross-sectional cohort of US adults. Int J Vitam Nutr Res. 2023;93:99-110
9. Lin L, Deng K-Q, Chen Z, Lei F, Qin J-J, Huang X. et al. Lipoprotein(a) distribution and its association with carotid arteriopathy in the Chinese population. Atherosclerosis. 2023;372:1-9
10. Tao Y, Wang Y, Yin Y, Zhang K, Gong Y, Ying H. et al. Associations of lipids and lipid-modifying drug target genes with atrial fibrillation risk based on genomic data. Lipids Health Dis. 2024;23:175
11. Xia J, Guo C, Liu K, Xie Y, Cao H, Peng W. et al. Association of Lipoprotein (a) variants with risk of cardiovascular disease: a Mendelian randomization study. Lipids Health Dis. 2021;20:57
12. Aronis KN, Zhao D, Hoogeveen RC, Alonso A, Ballantyne CM, Guallar E. et al. Associations of Lipoprotein(a) Levels With Incident Atrial Fibrillation and Ischemic Stroke: The ARIC (Atherosclerosis Risk in Communities) Study. J Am Heart Assoc. 2017;6:e007372
13. Yuen T, Mancini GBJ, Hegele RA, Pearson GJ. Consideration and Application of Lipoprotein(a) in the Risk Assessment of Atherosclerotic Cardiovascular Disease Risk in Adults. CJC Open. 2024;6:597-606
14. Vinci P, Di Girolamo FG, Panizon E, Tosoni LM, Cerrato C, Pellicori F. et al. Lipoprotein(a) as a Risk Factor for Cardiovascular Diseases: Pathophysiology and Treatment Perspectives. Int J Environ Res Public Health. 2023;20:6721
15. Satterfield BA, Dikilitas O, Safarova MS, Clarke SL, Tcheandjieu C, Zhu X. et al. Associations of Genetically Predicted Lp(a) (Lipoprotein [a]) Levels With Cardiovascular Traits in Individuals of European and African Ancestry. Circ Genom Precis Med. 2021;14:e003354
16. Mora S, Akinkuolie AO, Sandhu RK, Conen D, Albert CM. Paradoxical association of lipoprotein measures with incident atrial fibrillation. Circ Arrhythm Electrophysiol. 2014;7:612-9
17. Garg PK, Guan W, Karger AB, Steffen BT, O'Neal W, Heckbert SR. et al. Lp(a) (Lipoprotein [a]) and Risk for Incident Atrial Fibrillation: Multi-Ethnic Study of Atherosclerosis. Circ Arrhythm Electrophysiol. 2020;13:e008401
18. Jiang Q, Qin D, Yang L, Lin Y, Zhai L, Zhang Y. et al. Causal effects of plasma lipids on the risk of atrial fibrillation: A multivariable mendelian randomization study. Nutr Metab Cardiovasc Dis. 2021;31:1569-78
19. Mohammadi-Shemirani P, Chong M, Narula S, Perrot N, Conen D, Roberts JD. et al. Elevated Lipoprotein(a) and Risk of Atrial Fibrillation: An Observational and Mendelian Randomization Study. J Am Coll Cardiol. 2022;79:1579-90
20. Tao J, Yang X, Qiu Q, Gao F, Chen W, Hu L. et al. Low lipoprotein(a) concentration is associated with atrial fibrillation: a large retrospective cohort study. Lipids Health Dis. 2022;21:119
21. Ugovšek S, Šebeštjen M. Lipoprotein(a)-The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules. 2021;12:26
22. Schnitzler JG, Hoogeveen RM, Ali L, Prange KHM, Waissi F, van Weeghel M. et al. Atherogenic Lipoprotein(a) Increases Vascular Glycolysis, Thereby Facilitating Inflammation and Leukocyte Extravasation. Circ Res. 2020;126:1346-59
23. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD. et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation. 2016;134:611-24
24. Singh S, Baars DP, Desai R, Singh D, Pinto-Sietsma S-J. Association Between Lipoprotein (a) and Risk of Atrial Fibrillation: A Systematic Review and Meta-analysis of Mendelian Randomization Studies. Curr Probl Cardiol. 2024;49:102024
25. Liang F, Wang Y. Coronary heart disease and atrial fibrillation: a vicious cycle. Am J Physiol Heart Circ Physiol. 2021;320:H1-H12
26. Corban MT, Toya T, Ahmad A, Lerman LO, Lee H-C, Lerman A. Atrial Fibrillation and Endothelial Dysfunction: A Potential Link? Mayo Clin Proc. 2021;96:1609-21
27. Gillis AM, Morck M, Exner DV, Soo A, Rose MS, Sheldon RS. et al. Beneficial effects of statin therapy for prevention of atrial fibrillation following DDDR pacemaker implantation. Eur Heart J. 2008;29:1873-80
28. Peña JM, MacFadyen J, Glynn RJ, Ridker PM. High-sensitivity C-reactive protein, statin therapy, and risks of atrial fibrillation: an exploratory analysis of the JUPITER trial. Eur Heart J. 2012;33:531-7
29. Liu C, Shen M, Tan WLW, Chen IY, Liu Y, Yu X. et al. Statins improve endothelial function via suppression of epigenetic-driven EndMT. Nat Cardiovasc Res. 2023;2:467-85
30. Romagnuolo R, Marcovina SM, Boffa MB, Koschinsky ML. Inhibition of plasminogen activation by apo(a): role of carboxyl-terminal lysines and identification of inhibitory domains in apo(a). J Lipid Res. 2014;55:625-34
31. Boffa MB. Beyond fibrinolysis: The confounding role of Lp(a) in thrombosis. Atherosclerosis. 2022;349:72-81
32. Caplice NM, Panetta C, Peterson TE, Kleppe LS, Mueske CS, Kostner GM. et al. Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood. 2001;98:2980-7
33. Rand ML, Sangrar W, Hancock MA, Taylor DM, Marcovina SM, Packham MA. et al. Apolipoprotein(a) enhances platelet responses to the thrombin receptor-activating peptide SFLLRN. Arterioscler Thromb Vasc Biol. 1998;18:1393-9
34. Tsironis LD, Mitsios JV, Milionis HJ, Elisaf M, Tselepis AD. Effect of lipoprotein (a) on platelet activation induced by platelet-activating factor: role of apolipoprotein (a) and endogenous PAF-acetylhydrolase. Cardiovasc Res. 2004;63:130-8
35. Ezratty A, Simon DI, Loscalzo J. Lipoprotein(a) binds to human platelets and attenuates plasminogen binding and activation. Biochemistry. 1993;32:4628-33
36. Spronk HMH, De Jong AM, Verheule S, De Boer HC, Maass AH, Lau DH. et al. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur Heart J. 2017;38:38-50
37. Chehab O, Abdollahi A, Whelton SP, Wu CO, Ambale-Venkatesh B, Post WS. et al. Association of Lipoprotein(a) Levels With Myocardial Fibrosis in the Multi-Ethnic Study of Atherosclerosis. J Am Coll Cardiol. 2023;82:2280-91
38. Dzobo KE, Kraaijenhof JM, Stroes ESG, Nurmohamed NS, Kroon J. Lipoprotein(a): An underestimated inflammatory mastermind. Atherosclerosis. 2022;349:101-9
39. Watson T, Shantsila E, Lip GYH. Mechanisms of thrombogenesis in atrial fibrillation: Virchow's triad revisited. Lancet. 2009;373:155-66
40. Ding WY, Protty MB, Davies IG, Lip GYH. Relationship between lipoproteins, thrombosis, and atrial fibrillation. Cardiovasc Res. 2022;118:716-31
41. Igarashi Y, Yamaura M, Ito M, Inuzuka H, Ojima K, Aizawa Y. Elevated serum lipoprotein(a) is a risk factor for left atrial thrombus in patients with chronic atrial fibrillation: a transesophageal echocardiographic study. Am Heart J. 1998;136:965-71
42. Yan S, Li Q, Xia Z, Yan S, Wei Y, Hong K. et al. Risk factors of thromboembolism in nonvalvular atrial fibrillation patients with low CHA2DS2-VASc score. Medicine (Baltimore). 2019;98:e14549
43. Song J, Zhang X, Wei M, Bo Y, Zhou X, Tang B. Association between lipoprotein(a) and thromboembolism in patients with non-valvular atrial fibrillation: a cross-sectional study. Lipids Health Dis. 2022;21:78
44. Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57:745-57
45. Stătescu C, Anghel L, Benchea L-C, Tudurachi B-S, Leonte A, Zăvoi A. et al. A Systematic Review on the Risk Modulators of Myocardial Infarction in the "Young"-Implications of Lipoprotein (a). Int J Mol Sci. 2023 24
46. O'Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I. et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation. 2019;139:1483-92
47. Berliner JA, Leitinger N, Tsimikas S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res. 2009;50(Suppl):S207-S12
48. Reyes-Soffer G, Westerterp M. Beyond Lipoprotein(a) plasma measurements: Lipoprotein(a) and inflammation. Pharmacol Res. 2021;169:105689
49. Harada M, Nattel S. Implications of Inflammation and Fibrosis in Atrial Fibrillation Pathophysiology. Card Electrophysiol Clin. 2021;13:25-35
50. Boffa MB, Koschinsky ML. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat Rev Cardiol. 2019;16:305-18
51. Zhou X, Dudley SC. Evidence for Inflammation as a Driver of Atrial Fibrillation. Front Cardiovasc Med. 2020;7:62
52. Hu Y-F, Chen Y-J, Lin Y-J, Chen S-A. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. 2015;12:230-43
53. Bai W, Liu Z-Q, He P-Y, Muhuyati. The role of IL-6, IL-10, TNF-α and PD-1 expression on CD4 T cells in atrial fibrillation. Heliyon. 2023;9:e18818
54. Jia X, Cheng X, Wu N, Xiang Y, Wu L, Xu B. et al. Prognostic value of interleukin-6 in atrial fibrillation: A cohort study and meta-analysis. Anatol J Cardiol. 2021;25:872-9
55. da Silva RMFL. Influence of Inflammation and Atherosclerosis in Atrial Fibrillation. Curr Atheroscler Rep. 2017;19:2
56. Liu C, Liu R, Fu H, Li J, Wang X, Cheng L. et al. Pioglitazone attenuates atrial remodeling and vulnerability to atrial fibrillation in alloxan-induced diabetic rabbits. Cardiovasc Ther. 2017 35
57. Peng X, Li L, Zhang M, Zhao Q, Wu K, Bai R. et al. Sodium-Glucose Cotransporter 2 Inhibitors Potentially Prevent Atrial Fibrillation by Ameliorating Ion Handling and Mitochondrial Dysfunction. Front Physiol. 2020;11:912
58. Ghafouri-Fard S, Askari A, Shoorei H, Seify M, Koohestanidehaghi Y, Hussen BM. et al. Antioxidant therapy against TGF-β/SMAD pathway involved in organ fibrosis. J Cell Mol Med. 2024;28:e18052
59. Xiao J, Zhang H, Liang D, Liu Y, Liu Y, Zhao H. et al. Taxol, a microtubule stabilizer, prevents atrial fibrillation in in vitro atrial fibrillation models using rabbit hearts. Med Sci Monit. 2010;16:BR353-BR60
60. Liu T, Li T, Xu D, Wang Y, Zhou Y, Wan J. et al. Small-conductance calcium-activated potassium channels in the heart: expression, regulation and pathological implications. Philos Trans R Soc Lond B Biol Sci. 2023;378:20220171
61. Tai B-Y, Lu M-K, Yang H-Y, Tsai C-S, Lin C-Y. Ziprasidone Induces Rabbit Atrium Arrhythmogenesis via Modification of Oxidative Stress and Sodium/Calcium Homeostasis. Biomedicines. 2022;10:976
62. Gensini GF, Comeglio M, Colella A. Classical risk factors and emerging elements in the risk profile for coronary artery disease. Eur Heart J. 1998 19 Suppl A: A53-A61
63. Benjamin EJ, Levy D, Vaziri SM, D'Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA. 1994;271:840-4
64. Patel AP, Wang M, Pirruccello JP, Ellinor PT, Ng K, Kathiresan S. et al. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights From a Large National Biobank. Arterioscler Thromb Vasc Biol. 2021;41:465-74
65. Li Y, Chen L, Shao Y, Zhang M, Zhi L, Lu Y. The effect of apolipoprotein E gene polymorphism and Lp(a) levels on coronary artery disease with atrial fibrillation. J Int Med Res. 2022;50:3000605221109387
66. Li J-J, Ma C-S, Zhao D, Yan X-W. Lipoprotein(a) and Cardiovascular Disease in Chinese Population: A Beijing Heart Society Expert Scientific Statement. JACC Asia. 2022;2:653-65
67. Arora P, Kalra R, Callas PW, Alexander KS, Zakai NA, Wadley V. et al. Lipoprotein(a) and Risk of Ischemic Stroke in the REGARDS Study. Arterioscler Thromb Vasc Biol. 2019;39:810-8
68. Fu H, Zhang D, Zhu R, Cui L, Qiu L, Lin S. et al. Association between lipoprotein(a) concentration and the risk of stroke in the Chinese Han population: a retrospective case-control study. Ann Transl Med. 2020;8:212
69. Zhang Z, Xiao Y, Dai Y, Lin Q, Liu Q. Device therapy for patients with atrial fibrillation and heart failure with preserved ejection fraction. Heart Fail Rev. 2024;29:417-30
70. Li Z, Liu J, Shen J, Chen Y, He L, Li M. et al. Association of lipoprotein (a) and 1 year prognosis in patients with heart failure with reduced ejection fraction. ESC Heart Fail. 2022;9:2399-406
71. Singh S, Baars DP, Aggarwal K, Desai R, Singh D, Pinto-Sietsma S-J. Association between lipoprotein (a) and risk of heart failure: A systematic review and meta-analysis of Mendelian randomization studies. Curr Probl Cardiol. 2024;49:102439
72. Kamstrup PR, Nordestgaard BG. Elevated Lipoprotein(a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC Heart Fail. 2016;4:78-87
73. Masson W, Barbagelata L, Lavalle-Cobo A, Corral P, Nogueira JP. Lipoprotein(a) and heart failure: a systematic review. Heart Fail Rev. 2023;28:1307-14
74. Januzzi JL, van Kimmenade RRJ, Liu Y, Hu X, Browne A, Plutzky J. et al. Lipoprotein(a), Oxidized Phospholipids, and Progression to Symptomatic Heart Failure: The CASABLANCA Study. J Am Heart Assoc. 2024;13:e034774
75. Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2020;41:2275-84
76. Langsted A, Kamstrup PR, Nordestgaard BG. Lipoprotein(a): fasting and nonfasting levels, inflammation, and cardiovascular risk. Atherosclerosis. 2014;234:95-101
77. Fu B, Wang J, Wang L, Wang Q, Guo Z, Xu M. et al. Integrated proteomic and metabolomic profile analyses of cardiac valves revealed molecular mechanisms and targets in calcific aortic valve disease. Front Cardiovasc Med. 2022;9:944521
78. Haring B, von Ballmoos MCW, Appel LJ, Sacks FM. Healthy dietary interventions and lipoprotein (a) plasma levels: results from the Omni Heart Trial. PLoS One. 2014;9:e114859
79. Willeit P, Ridker PM, Nestel PJ, Simes J, Tonkin AM, Pedersen TR. et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet. 2018;392:1311-20
80. Huang J-Y, Chan Y-H, Tse Y-K, Yu S-Y, Li H-L, Chen C. et al. Statin Therapy Is Associated With a Lower Risk of Heart Failure in Patients With Atrial Fibrillation: A Population-Based Study. J Am Heart Assoc. 2023;12:e032378
81. Shweikialrefaee B, Ko DT, Fang J, Pang A, Austin PC, Dorian P. et al. Statin Use and Stroke Rate in Older Adults With Atrial Fibrillation: A Population-Based Cohort Study. J Am Heart Assoc. 2023;12:e028381
82. Gianos E, Duell PB, Toth PP, Moriarty PM, Thompson GR, Brinton EA. et al. Lipoprotein Apheresis: Utility, Outcomes, and Implementation in Clinical Practice: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2024;44:12
83. Schettler VJJ, Neumann CL, Peter C, Zimmermann T, Julius U, Roeseler E. et al. The German Lipoprotein Apheresis Registry (GLAR) - almost 5 years on. Clin Res Cardiol Suppl. 2017;12:44-9
84. Schumann F, Kassner U, Spira D, Zimmermann FF, Bobbert T, Steinhagen-Thiessen E. et al. Long-term lipoprotein apheresis reduces cardiovascular events in high-risk patients with isolated lipoprotein(a) elevation. J Clin Lipidol. 2024;18:e738-e745
85. Pokrovsky SN, Afanasieva OI, Ezhov MV. Therapeutic Apheresis for Management of Lp(a) Hyperlipoproteinemia. Curr Atheroscler Rep. 2020;22:68
86. Rikhi R, Hammoud A, Ashburn N, Snavely AC, Michos ED, Chevli P. et al. Relationship of low-density lipoprotein-cholesterol and lipoprotein(a) to cardiovascular risk: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2022;363:102-8
87. O'Donoghue ML, Giugliano RP, Wiviott SD, Atar D, Keech A, Kuder JF. et al. Long-Term Evolocumab in Patients With Established Atherosclerotic Cardiovascular Disease. Circulation. 2022;146:1109-19
88. Gaba P, O'Donoghue ML, Park J-G, Wiviott SD, Atar D, Kuder JF. et al. Association Between Achieved Low-Density Lipoprotein Cholesterol Levels and Long-Term Cardiovascular and Safety Outcomes: An Analysis of FOURIER-OLE. Circulation. 2023;147:1192-203
89. Steffens D, Bramlage P, Scheeff C, Kasner M, Hassanein A, Friebel J. et al. PCSK9 inhibitors and cardiovascular outcomes. Expert Opin Biol Ther. 2020;20:35-47
90. Stiekema LCA, Stroes ESG, Verweij SL, Kassahun H, Chen L, Wasserman SM. et al. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur Heart J. 2019;40:2775-81
91. Schmidt AF, Hunt NB, Gordillo-Marañón M, Charoen P, Drenos F, Kivimaki M. et al. Cholesteryl ester transfer protein (CETP) as a drug target for cardiovascular disease. Nat Commun. 2021;12:5640
92. O'Donoghue ML, Rosenson RS, Gencer B, López JAG, Lepor NE, Baum SJ. et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. The New England journal of medicine. 2022;387:1855-64
93. O'Donoghue ML, G López JA, Knusel B, Gencer B, Wang H, Wu Y. et al. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am Heart J. 2022;251:61-9
94. Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E. et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. The New England journal of medicine. 2020;382:244-55
95. Saljic A, Soattin L, Trachsel DS, Boddum K, Jespersen T. In vivo knockdown of SK3 channels using antisense oligonucleotides protects against atrial fibrillation in rats. J Mol Cell Cardiol. 2020;147:18-26
96. Hovingh GK, Kastelein JJ, van Deventer SJ, Round P, Ford J, Saleheen D. et al. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2015;386:452-60
97. Koutsogianni AD, Liamis G, Liberopoulos E, Adamidis PS, Florentin M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels-Potent Clinical Implications. Pharmaceuticals (Basel). 2023;16:750
98. Bianconi V, Mannarino MR, Ramondino F, Fusaro J, Giglioni F, Braca M. et al. Lipoprotein(a) Does Not Predict Thrombotic Events and In-Hospital Outcomes in Patients with COVID-19. J Clin Med. 2023;12:3543
99. Bhatia HS, Wilkinson MJ. Lipoprotein(a): Evidence for Role as a Causal Risk Factor in Cardiovascular Disease and Emerging Therapies. J Clin Med. 2022;11:6040
Author contact
![]() Corresponding author: Yichao Xiao, Ph.D. Department of Cardiology, The Second Xiangya Hospital, Central South University Address: 139 Renmin Road, Furong District, Changsha, 410011, Hunan Province, People's Republic of China.
Corresponding author: Yichao Xiao, Ph.D. Department of Cardiology, The Second Xiangya Hospital, Central South University Address: 139 Renmin Road, Furong District, Changsha, 410011, Hunan Province, People's Republic of China.