Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2024; 21(2):376-395. doi:10.7150/ijms.92131 This issue Cite
Research Paper
Altered Gut Microbiota as a Potential Risk Factor for Coronary Artery Disease in Diabetes: A Two-Sample Bi-Directional Mendelian Randomization Study
Zhaopei Zeng1,2*, Junxiong Qiu1,2*, Yu Chen3*, Diefei Liang4, Feng Wei1,5, Yuan Fu1, Jiarui Zhang1, Xiexiao Wei6, Xinyi Zhang1, Jun Tao1,2# ![]() , Liling Lin2,7#
, Liling Lin2,7# ![]() , Junmeng Zheng1,2#
, Junmeng Zheng1,2# ![]()
1. Department of Cardiovascular Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China.
2. Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China.
3. Department of Surgical Oncology, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China.
4. Department of Endocrinology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China.
5. Department of Cardiothoracic Surgery, Shenshan Medical Center, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Shanwei, China.
6. Department of Cardiology, Chinese PLA General Hospital, Beijing, China.
7. Department of Anesthesiology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China.
* These authors contributed equally to this work and share first authorship.
# These authors share last authorship.
Received 2023-11-11; Accepted 2023-12-8; Published 2024-1-1
Abstract

The current body of research points to a notable correlation between an imbalance in gut microbiota and the development of type 2 diabetes mellitus (T2D) as well as its consequential ailment, coronary artery disease (CAD). The complexities underlying the association, especially in the context of diabetic coronary artery disease (DCAD), are not yet fully understood, and the causal links require further clarification. In this study, a bidirectional Mendelian randomization (MR) methodology was utilized to explore the causal relationships between gut microbiota, T2D, and CAD. By analyzing data from the DIAGRAM, GERA, UKB, FHS, and mibioGen cohorts and examining GWAS databases, we sought to uncover genetic variants linked to T2D, CAD, and variations in gut microbiota and metabolites, aiming to shed light on the potential mechanisms connecting gut microbiota with DCAD. Our investigation uncovered a marked causal link between the presence of Oxalobacter formigenes and an increased incidence of both T2D and CAD. Specifically, a ten-unit genetic predisposition towards T2D was found to be associated with a 6.1% higher probability of an increase in the Oxalobacteraceae family's presence (β = 0.061, 95% CI = 0.002-0.119). In a parallel finding, an augmented presence of Oxalobacter was related to an 8.2% heightened genetic likelihood of CAD (β = 0.082, 95% CI = 0.026-0.137). This evidence indicates a critical pathway by which T2D can potentially raise the risk of CAD via alterations in gut microbiota. Additionally, our analyses reveal a connection between CAD risk and Methanobacteria, thus providing fresh perspectives on the roles of TMAO and carnitine in the etiology of CAD. The data also suggest a direct causal relationship between increased levels of certain metabolites — proline, lysophosphatidylcholine, asparagine, and salicylurate — and the prevalence of both T2D and CAD. Sensitivity assessments reinforce the notion that changes in Oxalobacter formigenes could pose a risk for DCAD. There is also evidence to suggest that DCAD may, in turn, affect the gut microbiota's makeup. Notably, a surge in serum TMAO levels in individuals with CAD, coinciding with a reduced presence of methanogens, has been identified as a potentially significant factor for future examination.
Keywords: coronary artery disease, type 2 diabetes, causality, gut microbiota, metabolites, Mendelian randomization
Introduction
The diverse bacterial population within the human gut, numbering in the billions, plays a critical role in regulating host health and physiological functions [1]. This microbial community is especially significant in the development and progression of various diseases, including cardiovascular maladies, metabolic disorders, neurogenic conditions, and immune system responses, with a particular impact on type 2 diabetes mellitus (T2D) and coronary artery disease (CAD) [2, 3]. The imbalance of gut microbiota, known as dysbiosis, is increasingly acknowledged as a key contributor to metabolic imbalances, leading to persistent low-grade inflammation and oxidative stress, which are characteristic of T2D and its related health issues. Furthermore, the gut microbiota is known to participate actively in critical metabolic processes, contributing to the emergence of CAD by affecting inflammatory pathways and oxidative stress mechanisms [4]. The likelihood of developing cardiovascular conditions is influenced by a confluence of factors, such as existing health conditions, lifestyle choices, and overall health [5, 6]. Current research highlights the gut microbiota's significant role in mediating the risk and progression of CAD, particularly when it emerges as a secondary complication to diabetes [7].
Numerous studies have linked the gut microbiota to the development of T2D and CAD, highlighting the role of gut bacteria in the onset and progression of these conditions. It's well-documented that T2D significantly increases the risk of CAD, to an extent comparable to the risk associated with established heart diseases [8, 9]. T2D-related issues such as hypertension and oxidative stress can lead to metabolic disturbances and impaired lipid metabolism, which in turn can cause both small and large vessel complications. These include a range of cardiovascular conditions that impact the arteries of various organs [10]. Insulin resistance, a hallmark of T2D, is intricately connected to the composition of the gut microbiota [11]. Specific bacterial species, including Butyrivibrio crossotus, Eubacterium siraeum, Streptococcus mutans, and Eggerthella lenta, play significant roles in regulating blood sugar levels by interacting with the gut's microbial ecosystem [12-14]. Interestingly, shifts in the gut microbiome composition have been observed across different ethnic groups, including Asian and European populations, which have been shown to exhibit alterations in their gut microbiota in the context of T2D [15, 16].
Atherosclerotic cardiovascular conditions remain a leading contributor to disability and death among individuals with T2D. There is a growing body of evidence suggesting that the gut microbiota plays a crucial role in the development of atherosclerotic plaques [17, 18]. The progression of atherosclerosis and CAD appears to be intricately linked to how the gut microbiota manages essential metabolic functions, notably affecting purine and lipid metabolism, as well as pathways related to oxidative stress and inflammation [5, 19].
The dynamic interplay between the gut microbiota's composition and diabetic coronary artery disease (DCAD) demands thorough investigation to establish direct causal links [20]. It's increasingly critical to unravel how T2D enhances the susceptibility to CAD. Establishing causality in this domain is crucial not just for maintaining microbial equilibrium in the gut but also for developing strategies to prevent CAD.
Randomized controlled trials (RCTs) stand as the gold standard in epidemiological studies to determine causative relationships. However, their practical application can be restricted by logistical and ethical considerations. An alternative method, Mendelian randomization (MR), circumvents these limitations by employing genetic variants as proxies to draw causal inferences from observational data, thus minimizing confounder effects [21, 22]. Leveraging the capabilities of MR, our research adopted a bidirectional two-sample MR method to substantiate the causal relationships between the gut microbiota and both T2D and CAD. Recent insights suggest that the interaction between gut microbiota and arterial health may play a role in how a lipid-rich diet contributes to atherosclerosis. Our MR examination of metabolites provides insights into their possible causative links with T2D and CAD [23].
Materials and Methods
Study Design
Our research aimed to explore the genetic underpinnings of gut microbiota profiles and their influence on the incidence of T2D and CAD. By implementing a bidirectional two-sample Mendelian Randomization (MR) model, we assessed combined datasets from extensive genome-wide association studies (GWAS), with this process depicted in Figure 1 and elaborated upon in Supplementary Table S1. Furthermore, we conducted a one-way two-sample MR analysis to probe into the interactions between specific metabolites and the occurrence of T2D and CAD, along with their impact on the composition of the gut microbiota.
Framework for Bidirectional MR Analysis. This diagram details the methodological structure of our bidirectional Mendelian Randomization (MR) investigation, examining the cause-and-effect dynamics between gut microbiota and diseases such as type 2 diabetes (T2D) and coronary artery disease (CAD). Genetic data was primarily extracted from populations of European ancestry. The principal analysis method was inverse variance weighting (IVW), supplemented by sensitivity tests to ensure the reliability of the MR findings. After applying Bonferroni corrections, we identified significant causal links between three gut microbiota characteristics and T2D, and seven with CAD (P < 0.025, adjusted for two hypotheses). Notably, after adjustment for multiple testing (P < 2.36 × 10^-4, adjusted for 211 outcomes), no significant causal effect was observed between T2D/CAD and gut microbiota, although indicative causal links were noted.
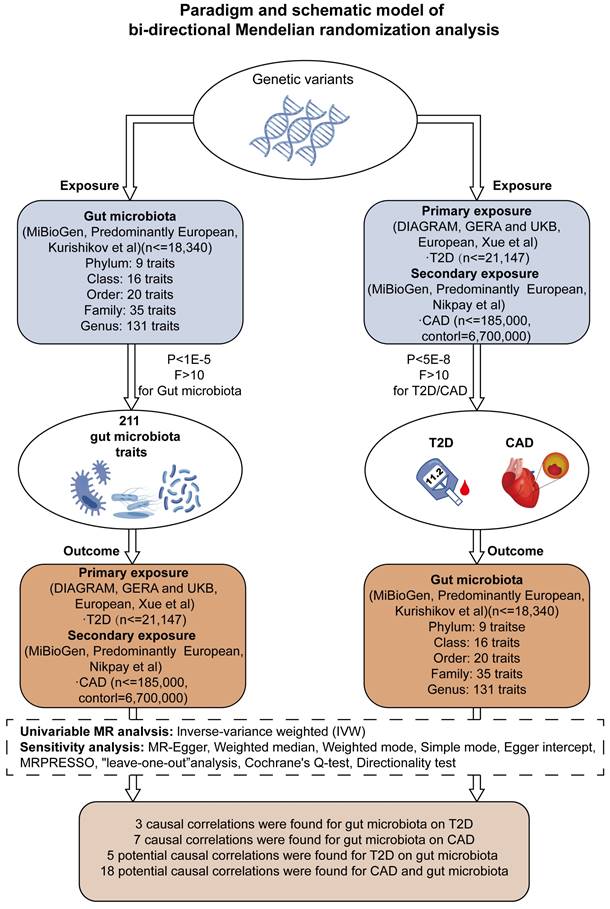
Ethical Considerations and Methodological Conformance
This study incorporates data derived from GWAS databases that have undergone rigorous ethical scrutiny and received clearance for research utilization. The methodology adheres to the protocols established by Burgess and colleagues, and is in compliance with the recommendations outlined in the STROBE-MR guidelines for reporting observational research with Mendelian Randomization frameworks [24, 25].
Data Acquisition and Genetic Marker Selection for T2D Analysis
For our investigation into T2D, we extracted data from a genome-wide association study (GWAS) by Xue et al. [26], which utilized samples from the DIAGRAM, GERA, and UKB cohorts. This pivotal study provided deeper insights into the genetic underpinnings of T2D and pinpointed potential gene loci for more in-depth functional studies. The findings from Xue et al. emphasized the significant impact of rare genetic variations on the risk associated with T2D. Our selection of genetic markers was based on a significance cut-off of 5×10-8, and we incorporated a linkage disequilibrium (LD) filter with an r2 value above 0.01 within a 5000 kb range. We calculated F-statistics for individual SNPs to confirm the strength of the genetic instruments, ensuring that each had an F-value well above 10, which is indicative of their reliability for use in MR analysis.
Data Compilation for Coronary Artery Disease Investigation
For the assessment of CAD, we sourced information from an extensive GWAS meta-analysis undertaken by Nikpay et al. [27]. This meta-analysis incorporated data from 48 distinct studies, totaling a cohort of 141,217 participants and close to 8.6 million SNPs. Instrumental variables selection for CAD mirrored the parameters set in the T2D analysis to maintain uniformity in our methodological approach.
Genomic Insights into Gut Microbiota
For our analysis of gut microbiota, we utilized data from the mibioGen initiative [28], noted for being the most comprehensive GWAS collection to date. This repository includes data from 24 cohort studies, primarily involving individuals of European ancestry. It provides GWAS results for 211 different bacterial groups, spanning 9 phyla, 16 classes, 20 orders, 35 families, and 131 genera. The selection of instrumental variables for this aspect of the study was determined with a P-value threshold of less than 1×10-5, considering the relatively small pool of loci detected. We adopted the same linkage disequilibrium clumping strategy as in our analyses of T2D and CAD to ensure the genetic markers' validity [29].
Compilation and Refinement of Metabolomic Data
We obtained our metabolomic data from a genome-wide association study by Rhee et al. [30], which analyzed blood metabolite profiles from 2,076 individuals of European descent participating in the Framingham Heart Study. This study focused on the relationship between gut microbiota and various host metabolites, taking into account numerous confounding factors such as age, gender, systolic blood pressure, antihypertensive drug use, body mass index (BMI), smoking status in diabetics, prevalence of cardiovascular diseases, and kidney function. These factors were adjusted to evaluate the correlations with 217 distinct metabolite concentrations in the dataset. For the subgroup analysis of metabolites, we set a P-value threshold of less than 1 × 10-5, consistent with the thresholds established in our prior analyses [31].
Methodology for Statistical Analysis and Deduction of Causality
We utilized the inverse-variance weighted (IVW) method to assess causal links between 211 microbiome characteristics and both T2D and CAD. This assessment was conducted within the framework of a two-sample bidirectional MR, leveraging paired GWAS summary statistics. To address the concerns of multiple hypothesis testing and the possibility of horizontal pleiotropy - the scenario where genetic variants might affect disease outcomes via multiple pathways - our analysis incorporated supplementary MR methodologies, including MR-PRESSO, the weighted median approach, and MR Egger. We rigorously tested for the presence of multi-trait pleiotropy using the MR-PRESSO global tests and Cochrane's Q-statistics [32].
Causal relationships inferred from the gut microbiota's impact on T2D and CAD were quantified using beta coefficients, complete with 95% confidence intervals. We implemented the Bonferroni method for correcting multiple comparisons, considering causal effects as significant at P-values less than 0.025 for two specific outcomes and less than 2.36 × 10-4 for the broader 211 outcomes. P-values falling between 0.05 and the Bonferroni threshold were interpreted as suggestive of potential causal links.
The robustness of the MR findings was quantified using the mRnd1 online tool. All harmonized data pertinent to our study are accessible in Supplementary Material Data 1, while Supplementary Material Data 2 elaborates on the comprehensive outcomes of the bidirectional MR analysis, encompassing the gut microbiota, T2D, CAD, and related metabolites. Our MR analyses were conducted in the R statistical framework (version 4.2.2), using the TwoSampleMR (version 0.5.6) and MRPRESSO (version 1.0) packages. The TwoSampleMR package was instrumental in integrating exposure and outcome information, based on a thorough compilation of SNP data, including allele information, effect magnitudes, allele frequencies, and standard error metrics.
Results
SNP Selection for T2D and CAD Analysis
In our study, we rigorously filtered SNPs, excluding those within a 5000-kilobase pair range showing linkage disequilibrium (LD) with an r2 value exceeding 0.01, and also removed any duplicates. This stringent selection process identified 1,745 SNPs linked to T2D and 2,801 SNPs associated with CAD, each meeting a significance threshold of P < 1×10-5. Following this, our bidirectional two-sample MR analysis provided substantial evidence indicating an elevated risk of CAD in the context of T2D, as elaborated in Supplementary Table S2.
Our MR analysis identified a total of 81 causal links, including those with potential associations where P < 0.05. This included five gut microbiota traits connected to T2D and ten to CAD, along with 16 metabolite traits associated with each condition. These findings were confirmed using MRPRESSO and leave-one-out analysis techniques, effectively ruling out instances of pleiotropy or heterogeneity. The reliability of these associations was further underscored by the F-statistics for the SNPs used in the MR analysis (see Tables 1-2, and Supplementary Tables S3-S4). A scatter plot in our report illustrates the trends and directionality of effects across different MR methodologies (see Figure 2).
Bidirectional MR Results of Type 2 diabetes and gut microbiota
| Level | Exposure | Outcome | Method | NSNP | Beta(95%CI) | P | Directional pleiotropy | Cochrane's Q-statistic (P) | Steiger P | |
|---|---|---|---|---|---|---|---|---|---|---|
| Egger intercept (P) | MRPRESSO RSSobs (P) | |||||||||
| T2D on Gut microbiota | ||||||||||
| Genus | T2D | Butyrivibrio | MR Egger | 124 | -0.108(-0.278,0.061) | 0.212 | 0.001 (0.870) | 151.361 (0.115) | 143.251 (0.102) | 6.51E-211 |
| Weighted median | 124 | -0.067(-0.195,0.061) | 0.305 | |||||||
| IVW | 124 | -0.096(-0.169,-0.022) | 0.011 | |||||||
| Genus | T2D | Catenibacterium | MR Egger | 114 | 0.046(-0.127,0.22) | 0.603 | 0.004 (0.530) | 126.627 (0.378) | 121.413 (0.277) | 1.51E-202 |
| Weighted median | 114 | 0.044(-0.096,0.184) | 0.537 | |||||||
| IVW | 114 | 0.096(0.02,0.172) | 0.013 | |||||||
| Genus | T2D | Olsenella | MR Egger | 124 | 0.011(-0.14,0.162) | 0.886 | 0.005 (0.363) | 135.497 (0.416) | 122.308 (0.501) | 5.77E-220 |
| Weighted median | 124 | 0.058(-0.072,0.188) | 0.379 | |||||||
| IVW | 124 | 0.074(0.008,0.14) | 0.027 | |||||||
| Family | T2D | Oxalobacteraceae | MR Egger | 125 | 0.124(-0.011,0.258) | 0.073 | -0.005 (0.307) | 154.722 (0.106) | 136.319 (0.212) | 1.47E-212 |
| Weighted median | 125 | 0.065(-0.037,0.167) | 0.215 | |||||||
| IVW | 125 | 0.061(0.002,0.119) | 0.043 | |||||||
| Genus | T2D | Erysipelotrichaceae UCG003 | MR Egger | 14 | 0.203(-0.418,0.823) | 0.534 | -0.004 (0.842) | 23.752 (0.097) | 20.351 (0.087) | 2.09E-15 |
| Weighted median | 14 | 0.173(0.011,0.334) | 0.036 | |||||||
| IVW | 14 | 0.14(0.004,0.276) | 0.043 | |||||||
| Gut microbiota on T2D | ||||||||||
| Genus | Lachnoclostridium | T2D | MR Egger | 8 | 0.524(0.044,1.005) | 0.076 | -0.019(0.230) | 6.420(0.706) | 4.971(0.664) | 1.16E-23 |
| Weighted median | 8 | 0.179(0.03,0.328) | 0.019 | |||||||
| IVW | 8 | 0.206(0.095,0.316) | 0.000 | |||||||
| Genus | Streptococcus | T2D | MR Egger | 11 | 0.118(-0.239,0.474) | 0.533 | 0.002(0.874) | 19.848(0.147) | 13.161(0.215) | 4.19E-37 |
| Weighted median | 11 | 0.116(-0.013,0.245) | 0.077 | |||||||
| IVW | 11 | 0.146(0.046,0.246) | 0.004 | |||||||
| Genus | Actinomyces | T2D | MR Egger | 5 | 0.289(-0.185,0.763) | 0.318 | -0.016(0.514) | 3.149(0.837) | 2.163(0.706) | 4.13E-18 |
| Weighted median | 5 | 0.113(-0.008,0.234) | 0.067 | |||||||
| IVW | 5 | 0.114(0.023,0.205) | 0.014 | |||||||
| Family | Streptococcaceae | T2D | MR Egger | 13 | 0.122(-0.218,0.462) | 0.497 | -0.002(0.867) | 18.677(0.269) | 13.621(0.326) | 1.25E-44 |
| Weighted median | 13 | 0.087(-0.029,0.203) | 0.143 | |||||||
| IVW | 13 | 0.093(0.006,0.18) | 0.035 | |||||||
| Genus | unknown genus id.2041 | T2D | MR Egger | 6 | 0.204(-0.072,0.48) | 0.222 | -0.010(0.472) | 10.065(0.311) | 6.910(0.227) | 8.97E-19 |
| Weighted median | 6 | 0.058(-0.056,0.172) | 0.319 | |||||||
| IVW | 6 | 0.099(0.006,0.192) | 0.037 | |||||||
MR, mendelian randomization; T2D, Type 2 diabetes; IVW, inverse variance weighted; NSNPs, number of single nucleotide polymorphisms; beta, mendelian randomization effect estimate
MR Association Scatterplot for Gut Microbiota and Cardiometabolic Disorders. The scatterplot features in panels A1-B5 illustrate the relationship between various gut microbiota traits and T2D. Panels C1-D17 display associations with CAD, revealing the range of genetic correlations investigated in this study.
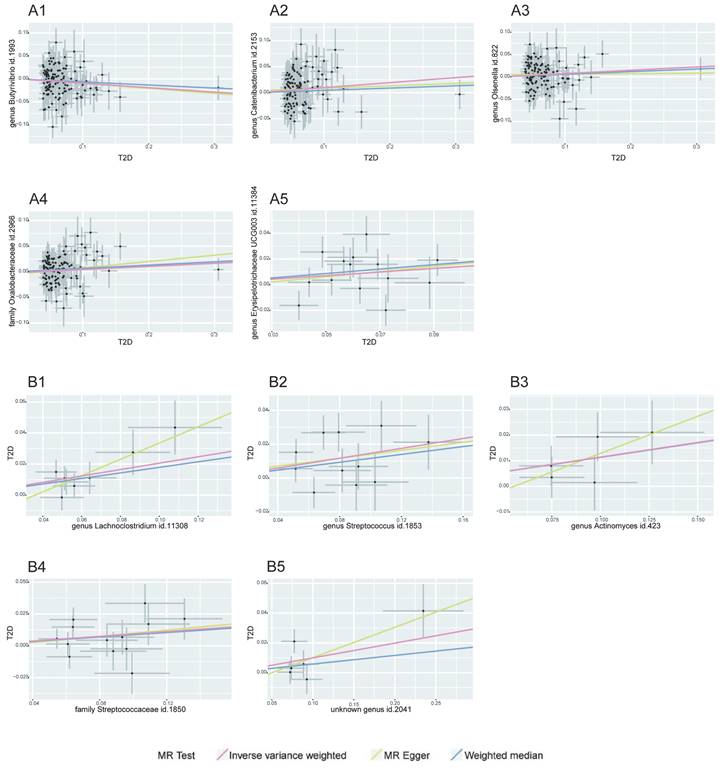
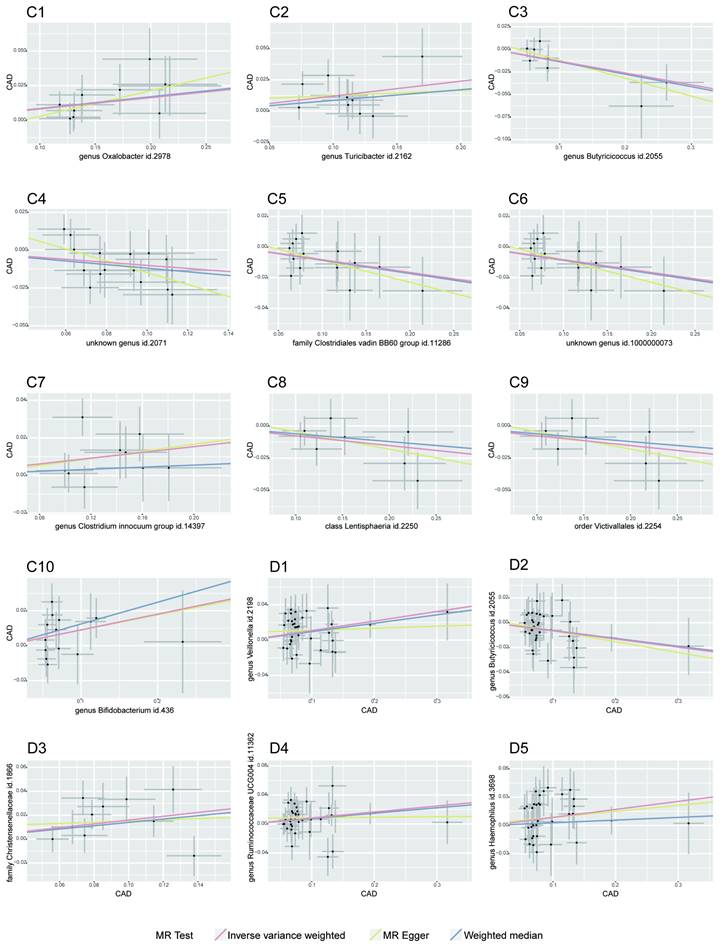
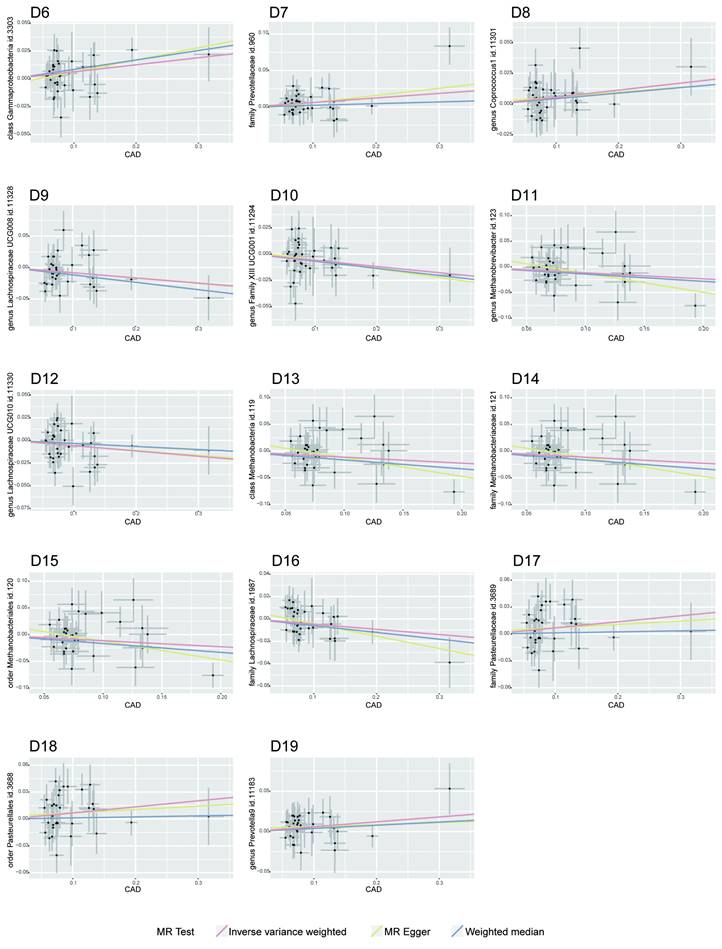
Bidirectional MR Results of Coronary artery disease and gut microbiota
| Level | Exposure | Outcome | Method | NSNP | Beta(95%CI) | P | Directional pleiotropy | Cochrane's Q-statistic (P) | Steiger P | |
|---|---|---|---|---|---|---|---|---|---|---|
| Egger intercept (P) | MRPRESSO RSSobs (P) | |||||||||
| Gut Microbiota on CAD | ||||||||||
| Genus | Oxalobacter | CAD | MR Egger | 11 | 0.184(-0.075,0.444) | 0.197 | -0.016(0.447) | 12.740(0.496) | 4.155(0.940) | 1.30E-36 |
| Weighted median | 11 | 0.085(0.013,0.156) | 0.020 | |||||||
| IVW | 11 | 0.082(0.026,0.137) | 0.004 | |||||||
| Genus | Turicibacter | CAD | MR Egger | 10 | 0.042 (-0.143, 0.226) | 0.827 | 0.008(0.676) | 14.681(0.478) | 7.201(0.616) | 5.77E-40 |
| Weighted median | 10 | 0.085 (0.029, 0.142) | 0.132 | |||||||
| IVW | 10 | 0.119 (0.076, 0.163) | 0.006 | |||||||
| Genus | Butyricicoccus | CAD | MR Egger | 8 | -0.197(-0.381, -0.014) | 0.080 | 0.007(0.426) | 10.029(0.494) | 4.227(0.753) | 5.00E-24 |
| Weighted median | 8 | -0.138(-0.279,0.003) | 0.056 | |||||||
| IVW | 8 | -0.131(-0.234, -0.028) | 0.012 | |||||||
| Genus | unknown genus id.2071 | CAD | MR Egger | 16 | -0.392(-0.764, -0.02) | 0.058 | 0.024(0.139) | 28.083(0.141) | 13.176(0.589) | 9.07E-51 |
| Weighted median | 16 | -0.119(-0.23, -0.008) | 0.036 | |||||||
| IVW | 16 | -0.101(-0.18, -0.021) | 0.013 | |||||||
| Family | Clostridiales vadin BB60 group | CAD | MR Egger | 15 | -0.144(-0.345,0.057) | 0.184 | 0.006(0.536) | 9.383(0.945) | 7.743(0.902) | 2.85E-50 |
| Weighted median | 15 | -0.086(-0.177,0.004) | 0.062 | |||||||
| IVW | 15 | -0.083(-0.153, -0.013) | 0.021 | |||||||
| Genus | unknown genus id.1000000073 | CAD | MR Egger | 15 | -0.144(-0.345,0.057) | 0.184 | 0.006(0.536) | 9.383(0.940) | 7.743(0.902) | 2.85E-50 |
| Weighted median | 15 | -0.086(-0.175,0.003) | 0.057 | |||||||
| IVW | 15 | -0.083(-0.153, -0.013) | 0.021 | |||||||
| Genus | Clostridium innocuum group | CAD | MR Egger | 9 | 0.094(-0.262,0.45) | 0.620 | -0.002(0.924) | 14.581(0.30)9 | 8.562(0.381) | 1.10E-28 |
| Weighted median | 9 | 0.028(-0.06,0.115) | 0.537 | |||||||
| IVW | 9 | 0.077(0.011,0.142) | 0.022 | |||||||
| Class | Lentisphaeria | CAD | MR Egger | 8 | -0.135(-0.371,0.1) | 0.303 | 0.009(0.625) | 5.244(0.908) | 3.979(0.782) | 3.79E-29 |
| Weighted median | 8 | -0.061(-0.152,0.031) | 0.194 | |||||||
| IVW | 8 | -0.076(-0.144, -0.008) | 0.028 | |||||||
| Order | Victivallales | CAD | MR Egger | 8 | -0.135(-0.371,0.1) | 0.303 | 0.009(0.625) | 5.244(0.897) | 3.979(0.782) | 3.79E-29 |
| Weighted median | 8 | -0.061(-0.145,0.024) | 0.160 | |||||||
| IVW | 8 | -0.076(-0.144, -0.008) | 0.028 | |||||||
| Genus | Bifidobacterium | CAD | MR Egger | 14 | 0.087(-0.147,0.321) | 0.482 | 0.000(0.972) | 18.136(0.464) | 11.777(0.546) | 3.50E-58 |
| Weighted median | 14 | 0.125(0.014,0.235) | 0.027 | |||||||
| IVW | 14 | 0.091(0.008,0.173) | 0.031 | |||||||
| CAD on Gut Microbiota | ||||||||||
| Genus | CAD | Veillonella | MR Egger | 36 | 0.023(-0.13,0.177) | 0.77 | 0.009 (0.243) | 34.134 (0.812) | 29.682 (0.722) | 1.80E-85 |
| Weighted median | 36 | 0.095(-0.001,0.192) | 0.052 | |||||||
| IVW | 36 | 0.108(0.045,0.171) | 0.001 | |||||||
| Genus | CAD | Butyricicoccus | MR Egger | 36 | -0.088(-0.199,0.024) | 0.134 | 0.002 (0.678) | 29.168 (0.934) | 23.637 (0.928) | 8.15E-91 |
| Weighted median | 36 | -0.063(-0.131,0.005) | 0.069 | |||||||
| IVW | 36 | -0.066(-0.112, -0.019) | 0.005 | |||||||
| Family | CAD | Christensenellaceae | MR Egger | 10 | 0.05(-0.367,0.468) | 0.819 | 0.01 (0.609) | 17.859 (0.182) | 11.086 (0.27) | 4.40E-14 |
| Weighted median | 10 | 0.14(-0.005,0.285) | 0.059 | |||||||
| IVW | 10 | 0.159(0.046,0.272) | 0.006 | |||||||
| Genus | CAD | Ruminococcaceae UCG004 | MR Egger | 36 | 0.007(-0.142,0.156) | 0.928 | 0.008 (0.281) | 34.257 (0.784) | 30.473 (0.686) | 5.60E-87 |
| Weighted median | 36 | 0.074(-0.023,0.171) | 0.134 | |||||||
| IVW | 36 | 0.083(0.021,0.145) | 0.009 | |||||||
| Genus | CAD | Haemophilus | MR Egger | 36 | 0.064(-0.095,0.222) | 0.438 | 0.002 (0.774) | 42.824 (0.453) | 35.787 (0.431) | 7.65E-84 |
| Weighted median | 36 | 0.028(-0.071,0.128) | 0.575 | |||||||
| IVW | 36 | 0.085(0.02,0.149) | 0.010 | |||||||
| Class | CAD | Gammaproteobacteria | MR Egger | 36 | 0.109(-0.008,0.225) | 0.076 | -0.005 (0.408) | 38.906 (0.609) | 28.717 (0.764) | 3.77E-86 |
| Weighted median | 36 | 0.085(0.016,0.154) | 0.016 | |||||||
| IVW | 36 | 0.063(0.015,0.111) | 0.010 | |||||||
| Family | CAD | Prevotellaceae | MR Egger | 36 | 0.098(-0.024,0.219) | 0.124 | -0.004 (0.533) | 31.991 (0.865) | 26.465 (0.85) | 9.13E-91 |
| Weighted median | 36 | 0.023(-0.055,0.101) | 0.563 | |||||||
| IVW | 36 | 0.062(0.012,0.112) | 0.015 | |||||||
| Genus | CAD | Coprococcus1 | MR Egger | 36 | 0.039(-0.074,0.153) | 0.5 | 0.002 (0.74) | 33.98 (0.803) | 26.142 (0.86) | 1.19E-89 |
| Weighted median | 36 | 0.045(-0.024,0.114) | 0.203 | |||||||
| IVW | 36 | 0.057(0.01,0.104) | 0.017 | |||||||
| Genus | CAD | Lachnospiraceae UCG008 | MR Egger | 35 | -0.08(-0.256,0.096) | 0.38 | 0(0.973) | 42.248 (0.401) | 33.612 (0.486) | 1.43E-83 |
| Weighted median | 35 | -0.117(-0.223, -0.011) | 0.03 | |||||||
| IVW | 35 | -0.083(-0.156, -0.01) | 0.025 | |||||||
| Genus | CAD | Family XIII UCG001 | MR Egger | 36 | -0.082(-0.211,0.046) | 0.217 | 0.002 (0.699) | 33.55 (0.819) | 30.732 (0.674) | 1.43E-89 |
| Weighted median | 36 | -0.068(-0.144,0.009) | 0.082 | |||||||
| IVW | 36 | -0.059(-0.113, -0.006) | 0.03 | |||||||
| Genus | CAD | Methanobrevibacter | MR Egger | 34 | -0.37(-0.667, -0.073) | 0.02 | 0.024 (0.083) | 37.82 (0.596) | 32.384 (0.498) | 6.55E-78 |
| Weighted median | 34 | -0.141(-0.304,0.021) | 0.088 | |||||||
| IVW | 34 | -0.117(-0.224, -0.01) | 0.032 | |||||||
| Genus | CAD | Lachnospiraceae UCG010 | MR Egger | 36 | -0.053(-0.188,0.082) | 0.447 | -0.001 (0.934) | 40.419 (0.522) | 38.02 (0.333) | 3.35E-85 |
| Weighted median | 36 | -0.033(-0.115,0.048) | 0.42 | |||||||
| IVW | 36 | -0.058(-0.113, -0.003) | 0.038 | |||||||
| Class | CAD | Methanobacteria | MR Egger | 34 | -0.352(-0.647, -0.057) | 0.026 | 0.023 (0.099) | 40.407 (0.493) | 35.488 (0.352) | 1.44E-76 |
| Weighted median | 34 | -0.166(-0.337,0.005) | 0.057 | |||||||
| IVW | 34 | -0.114(-0.223, -0.004) | 0.042 | |||||||
| Family | CAD | Methanobacteriaceae | MR Egger | 34 | -0.352(-0.647, -0.057) | 0.026 | 0.023 (0.099) | 40.407 (0.494) | 35.488 (0.352) | 1.44E-76 |
| Weighted median | 34 | -0.166(-0.332,0) | 0.05 | |||||||
| IVW | 34 | -0.114(-0.223, -0.004) | 0.042 | |||||||
| Order | CAD | Methanobacteriales | MR Egger | 34 | -0.352(-0.647, -0.057) | 0.026 | 0.023 (0.099) | 40.407 (0.451) | 35.488 (0.352) | 1.44E-76 |
| Weighted median | 34 | -0.166(-0.331, -0.001) | 0.048 | |||||||
| IVW | 34 | -0.114(-0.223, -0.004) | 0.042 | |||||||
| Family | CAD | Lachnospiraceae | MR Egger | 36 | -0.11(-0.219, -0.001) | 0.055 | 0.007 (0.217) | 25.906 (0.969) | 21.433 (0.965) | 3.95E-95 |
| Weighted median | 36 | -0.06(-0.125,0.004) | 0.066 | |||||||
| IVW | 36 | -0.046(-0.091, -0.002) | 0.042 | |||||||
| Family | CAD | Pasteurellaceae | MR Egger | 36 | 0.037(-0.127,0.201) | 0.66 | 0.003 (0.692) | 45.756 (0.349) | 39.428 (0.278) | 1.81E-83 |
| Weighted median | 36 | 0.012(-0.089,0.113) | 0.822 | |||||||
| IVW | 36 | 0.068(0.001,0.134) | 0.047 | |||||||
| Order | CAD | Pasteurellales | MR Egger | 36 | 0.037(-0.127,0.201) | 0.66 | 0.003 (0.692) | 45.756 (0.324) | 39.428 (0.278) | 1.81E-83 |
| Weighted median | 36 | 0.012(-0.083,0.106) | 0.81 | |||||||
| IVW | 36 | 0.068(0.001,0.134) | 0.047 | |||||||
| Genus | CAD | Prevotella9 | MR Egger | 36 | 0.024(-0.123,0.172) | 0.748 | 0.004 (0.604) | 26.219 (0.979) | 20.101 (0.979) | 2.75E-95 |
| Weighted median | 36 | 0.039(-0.048,0.127) | 0.377 | |||||||
| IVW | 36 | 0.06(0,0.121) | 0.05 | |||||||
MR, mendelian randomization; CAD, Coronary artery disease; IVW, inverse variance weighted; NSNP, number of single nucleotide polymorphisms; beta, mendelian randomization effect estimate
In the bidirectional MR framework where T2D was considered as the exposure factor influencing CAD, a significant P-value of less than 0.05 was observed. While this result did not meet the criteria of the Cochran's Q test for heterogeneity, the existence of a P-value below 0.05 in a multiplicative random effects model pointed to a potential causal relationship between T2D and CAD, as noted in Supplementary Table S2.
Impact of Gut Microbiota on T2D and CAD
In our investigation, we discerned nine distinct microbial taxa, spanning various taxonomic levels, that exhibit a positive causal relationship with both T2D and CAD. Regarding T2D, a genetic predisposition towards a greater abundance of the genera Lachnoclostridium, Streptococcus, Actinomyces, and the Streptococcaceae family was linked to a higher risk of the disease. Notably, a marked increase in Lachnoclostridium (β = 0.206, 95% CI = 0.095-0.316, P = 0.0002) was observed, indicating a significant rise in T2D risk (refer to Table 1). For CAD, elevated levels of Oxalobacter, Turicibacter, the Clostridium innocuum group, and Bifidobacterium were found to have a causative association with an increased risk, with Turicibacter showing a notable effect (β = 0.119, 95% CI = 0.076-0.163, P = 0.006), implying a considerable risk escalation for CAD (as shown in Table 2).
On the other hand, we identified that certain gut microbiota characteristics exhibit an inverse correlation with CAD risk. Specifically, the Lentisphaeria class, Victivallales order, Clostridiales vadin BB60 family, and Butyricicoccus genus demonstrated a protective effect, as evidenced by beta coefficients ranging from -0.234 to -0.008, suggesting they may mitigate CAD progression.
While our data analysis didn't reveal any significant negative causal effects of gut microbiota on T2D, it did indicate that certain microbes are associated with a reduced CAD risk, pointing towards their potential protective influence against the condition, as detailed in Table 2.
Effect of T2D and CAD on Gut Microbiota Dynamics
Our study explored the causal impact of T2D and CAD on the composition of gut microbiota, assessing causal links across 210 microbiotas for T2D and 211 for CAD. Four gut microbiotas exhibited positive causal links with T2D as a genetic factor, including the genera Catenibacterium, Olsenella, and Erysipelotrichaceae UCG-003, as well as the Oxalobacteraceae family. A genetic inclination towards T2D correlated with a heightened presence of these groups (Catenibacterium β = 0.096, 95% CI = 0.020-0.172, P = 0.013; Olsenella β = 0.074, 95% CI = 0.008-0.140, P = 0.027; Erysipelotrichaceae UCG-003 β = 0.140, 95% CI = 0.004-0.276, P = 0.043; Oxalobacteraceae β = 0.061, 95% CI = 0.002-0.119, P = 0.043), as indicated in Table 1. For CAD, an augmentation in several gut microbiota genera and families was noted, implying a possible connection post-Bonferroni adjustment (refer to Table 2).
In contrast, the Butyrivibrio genus showed a decrease in abundance with T2D, hinting at a possible protective role. Regarding CAD, a diminution in the abundance of certain gut microbiotas, such as Butyricicoccus and Methanobacteriaceae, was evident. Notably, the Methanobacteria genus displayed a significant reduction in abundance, suggesting a substantial protective influence against CAD.
To validate these conclusions, we conducted various sensitivity analyses, including MR-PRESSO, Cochrane's Q-test, and MR-Egger intercept tests. These procedures did not reveal any signs of heterogeneity or horizontal pleiotropy, thereby confirming the reliability of the identified causal relationships. Additionally, the F-values of the SNPs showing statistical significance consistently exceeded the threshold of 10, adding further credibility to our findings (as detailed in Supplementary Table S5).
Metabolomic Influences on T2D and CAD
In conducting a MR study, coupled with Bonferroni adjustments for dual hypotheses (setting the significance threshold at P < 0.025), we identified a subset of 22 metabolites from a total of 217, which were genetically associated with a reduced risk of T2D. This selection encompassed a diverse array of metabolite classes, including but not limited to sphingomyelin (specifically SM14_0), selected amino acids, lysophosphatidylcholine (notably LPC18_2), triacylglycerol (specifically TAG58_8), certain adenosine derivatives, salicylurate, and glycerol. These metabolites demonstrated beta effect sizes in the range of -0.072 to -0.010, indicating their inverse relationship with T2D risk. In contrast, an increase in specific metabolites such as taurocholate, phosphatidylcholine (particularly PC36_1), and suberic acid was found to be genetically correlated with an elevated risk of T2D, with beta effect sizes ranging from 0.011 to 0.067.
Metabolite-Gut Microbiota Interactions and CAD
In an analysis utilizing unidirectional MR, refined through Bonferroni adjustments (threshold set at P < 2.36 × 10-4), we were able to pinpoint four metabolites exhibiting causative links with both T2D and CAD. This assessment uncovered a negative causal association between proline levels and the presence of Eubacterium xylanophilum (yielding a beta coefficient of -0.038, within a 95% confidence interval of -0.058 to -0.019, and a P-value of 1.18×10-4). Furthermore, LPC18_2 demonstrated a causal relationship with alterations in four distinct gut microbiota taxa. Significantly, an inverse correlation was observed between asparagine and the genus Desulfovibrio (beta coefficient of -0.059, 95% CI between -0.090 and -0.028, P = 1.80×10-4), while the Bacteroidales S24-7 group showed a positive correlation. Additionally, salicylurate was identified as having causative connections with both the Christensenellaceae family and the genus Coprococcus1, as detailed in Supplementary Data 2.
Metabolite Associations with CAD
Utilizing a directional two-sample MR approach, followed by a Bonferroni correction accommodating dual hypotheses (establishing a significance threshold at P < 0.025), our analysis discerned associations of 16 metabolites with CAD. Within this group, seven metabolites, notably LPC18_2 and asparagine, were found to be genetically correlated with an increased predisposition to CAD. This correlation was quantified with beta effects spanning from 0.008 to 0.057. In contrast, a set of nine metabolites, which included amino acids like lysine and proline, exhibited a negative genetic association with CAD risk. The beta effect values for these metabolites varied from -0.067 to -0.007, as depicted in Figure 3.
Forest Plots of MR-Derived Causal Estimates. Displayed here are the results from inverse variance-weighted MR analyses, examining the causal effects of different metabolites on T2D and CAD. Beta coefficients, along with 95% confidence intervals (CI), are shown, illustrating the variation in disease risk associated with each 10-unit increase in metabolite concentration. Analyzed metabolites include sphingomyelin (SM), lysophosphatidylcholine (LPC), triacylglycerol (TAG), phosphatidylcholine (PC), and cholesterol ester (CE).
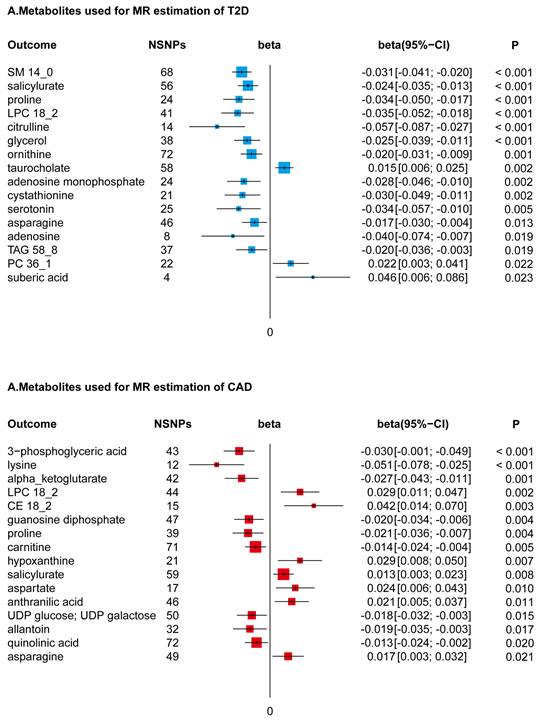
Discussion
In this investigation, we explored the reciprocal genetic relationships between the composition of the gut microbiota and the incidence of T2D and CAD. Our findings identified causal links of five gut microbiota characteristics with T2D, and ten with CAD. Conversely, our results suggest potential causal relationships of T2D with five gut microbiota types, and CAD with eighteen types. Additionally, we noted that certain metabolites, particularly those related to energy and lipids, exhibit causal connections with both T2D and CAD [33, 34].
The study identified five gut microbiota changes associated with T2D and ten with CAD. Of these, three microbiota types were causally linked to T2D, and seven to CAD. A notable causal association was observed between the increase in Oxalobacteraceae family abundance and T2D. In a surprising finding, a rise in the genus Oxalobacter was positively associated with an increased risk of CAD [35, 36]. Noteworthy was the discovery that both Turicibacter and the Clostridium innocuum group shared the same single nucleotide polymorphism (SNP), rs4869133, suggesting its significance in the heightened risk of CAD linked to gut microbiota. Furthermore, the Clostridiales vadin BB60 family, an unknown genus with the identifier id.1000000073, the Lentisphaeria class, and the Victivallales order all displayed identical SNPs in our final MR analysis. This genetic congruence might be attributed to the categorization of the unknown genus id.1000000073 under the Clostridiales vadin BB60 family, and a shared lineage between the Victivallales order and the Lentisphaeria class, indicating a limited range of genetic markers within these groups, as detailed in Table 3.
Particulars of SNPs used in MR analyses of gut microbiota
| Exposure traits | SNPs | EA | OA | Beta | Se | samplesize | P-value | R2 | F-statistic |
|---|---|---|---|---|---|---|---|---|---|
| Type 2 diabetes (P<1×10-12) | rs2296173 | G | A | 0.065 | 0.0087 | 62892 | 7.65773E-14 | 0.001 | 55.820 |
| rs340874 | C | T | 0.0626 | 0.0073 | 62892 | 8.40621E-18 | 0.001 | 73.536 | |
| rs2972144 | G | A | 0.0913 | 0.0075 | 62892 | 2.55094E-34 | 0.002 | 148.190 | |
| rs243019 | C | T | 0.0566 | 0.0071 | 62892 | 2.28981E-15 | 0.001 | 63.550 | |
| rs780094 | C | T | 0.0692 | 0.0074 | 62892 | 5.15941E-21 | 0.001 | 87.448 | |
| rs17334919 | T | C | -0.1398 | 0.0128 | 62892 | 6.68652E-28 | 0.002 | 119.287 | |
| rs13389219 | T | C | -0.0722 | 0.0074 | 62892 | 2.1062E-22 | 0.002 | 95.194 | |
| rs6808574 | C | T | 0.0552 | 0.0076 | 62892 | 4.38531E-13 | 0.001 | 52.753 | |
| rs11708067 | G | A | -0.0965 | 0.0086 | 62892 | 5.93335E-29 | 0.002 | 125.909 | |
| rs6795735 | T | C | -0.0558 | 0.0073 | 62892 | 1.63005E-14 | 0.001 | 58.428 | |
| rs7651090 | G | A | 0.1204 | 0.0076 | 62892 | 3.8539E-57 | 0.004 | 250.972 | |
| rs1899951 | T | C | -0.1118 | 0.0109 | 62892 | 1.63682E-24 | 0.002 | 105.204 | |
| rs1496653 | G | A | -0.0769 | 0.0088 | 62892 | 2.57217E-18 | 0.001 | 76.364 | |
| rs1801214 | T | C | 0.0903 | 0.0074 | 62892 | 5.51569E-34 | 0.002 | 148.906 | |
| rs459193 | G | A | 0.0711 | 0.0083 | 62892 | 8.80846E-18 | 0.001 | 73.381 | |
| rs7729395 | T | C | 0.1373 | 0.016 | 62892 | 1.10103E-17 | 0.001 | 73.638 | |
| rs7756992 | G | A | 0.1297 | 0.0078 | 62892 | 5.99929E-62 | 0.004 | 276.497 | |
| rs1063355 | G | T | 0.0709 | 0.0079 | 62892 | 3.71535E-19 | 0.001 | 80.545 | |
| rs17168486 | T | C | 0.0742 | 0.0094 | 62892 | 2.17721E-15 | 0.001 | 62.309 | |
| rs2191348 | T | G | 0.0652 | 0.0073 | 62892 | 3.44429E-19 | 0.001 | 79.772 | |
| rs13234269 | A | T | -0.0583 | 0.0078 | 62892 | 6.9775E-14 | 0.001 | 55.866 | |
| rs849135 | A | G | -0.0999 | 0.0072 | 62892 | 1.04112E-43 | 0.003 | 192.516 | |
| rs3802177 | A | G | -0.1217 | 0.008 | 62892 | 2.32113E-52 | 0.004 | 231.420 | |
| rs516946 | C | T | 0.0824 | 0.0085 | 62892 | 3.15864E-22 | 0.001 | 93.976 | |
| rs10974438 | C | A | 0.0591 | 0.0075 | 62892 | 3.01301E-15 | 0.001 | 62.094 | |
| rs10811661 | C | T | -0.1569 | 0.0098 | 62892 | 4.13238E-58 | 0.004 | 256.327 | |
| rs2796441 | A | G | -0.0715 | 0.0073 | 62892 | 1.962E-22 | 0.002 | 95.933 | |
| rs1063192 | A | G | 0.0634 | 0.0073 | 62892 | 3.29837E-18 | 0.001 | 75.428 | |
| rs4918796 | C | T | 0.0623 | 0.0086 | 62892 | 4.01328E-13 | 0.001 | 52.478 | |
| rs7923866 | T | C | -0.0972 | 0.0074 | 62892 | 9.33684E-40 | 0.003 | 172.532 | |
| rs11257655 | T | C | 0.0737 | 0.0087 | 62892 | 1.96607E-17 | 0.001 | 71.762 | |
| rs7903146 | T | C | 0.3059 | 0.0077 | 62892 | 1E-200 | 0.024 | 1578.256 | |
| rs10830963 | G | C | 0.0909 | 0.008 | 62892 | 5.84655E-30 | 0.002 | 129.106 | |
| rs1552224 | C | A | -0.1034 | 0.0101 | 62892 | 8.63575E-25 | 0.002 | 104.809 | |
| rs5215 | T | C | -0.0678 | 0.0073 | 62892 | 2.08882E-20 | 0.001 | 86.261 | |
| rs10842994 | T | C | -0.0755 | 0.0091 | 62892 | 1.01508E-16 | 0.001 | 68.835 | |
| rs2261181 | T | C | 0.0985 | 0.0118 | 62892 | 9.1791E-17 | 0.001 | 69.680 | |
| rs825476 | T | C | 0.0524 | 0.0073 | 62892 | 6.80456E-13 | 0.001 | 51.525 | |
| rs61953351 | T | G | -0.07 | 0.0091 | 62892 | 1.97606E-14 | 0.001 | 59.172 | |
| rs1359790 | A | G | -0.0796 | 0.008 | 62892 | 2.79512E-23 | 0.002 | 99.003 | |
| rs7177055 | A | G | 0.0647 | 0.0079 | 62892 | 2.746E-16 | 0.001 | 67.074 | |
| rs7185735 | G | A | 0.1056 | 0.0073 | 62892 | 1.59001E-47 | 0.003 | 209.258 | |
| rs77258096 | A | C | -0.1171 | 0.0134 | 62892 | 1.7832E-18 | 0.001 | 76.367 | |
| rs8068804 | A | G | 0.0587 | 0.0078 | 62892 | 4.41062E-14 | 0.001 | 56.635 | |
| rs9894220 | G | A | -0.0585 | 0.0079 | 62892 | 1.51705E-13 | 0.001 | 54.835 | |
| rs8108269 | G | T | 0.0644 | 0.0079 | 62892 | 3.11387E-16 | 0.001 | 66.453 | |
| coronary artery disease (P<1×10-10) | rs67180937 | G | T | 0.078807 | 0.0110551 | 42457 | 1.01E-12 | 0.001 | 50.816 |
| rs7528419 | G | A | -0.11453 | 0.011482 | 42457 | 1.97E-23 | 0.002 | 99.495 | |
| rs9970807 | T | C | -0.12575 | 0.016695 | 42457 | 5.00E-14 | 0.001 | 56.734 | |
| rs115654617 | A | C | 0.137846 | 0.0158314 | 42457 | 3.12E-18 | 0.002 | 75.814 | |
| rs12202017 | G | A | -0.066813 | 0.0099612 | 42457 | 1.98E-11 | 0.001 | 44.988 | |
| rs55730499 | T | C | 0.316641 | 0.0242403 | 42457 | 5.39E-39 | 0.004 | 170.631 | |
| rs186696265 | T | C | 0.550351 | 0.0481949 | 42457 | 3.35E-30 | 0.003 | 130.400 | |
| rs9349379 | G | A | 0.131836 | 0.0096527 | 42457 | 1.81E-42 | 0.004 | 186.539 | |
| rs2107595 | A | G | 0.073415 | 0.0112951 | 42457 | 8.05E-11 | 0.001 | 42.246 | |
| rs11556924 | T | C | -0.072569 | 0.0110605 | 42457 | 5.34E-11 | 0.001 | 43.048 | |
| rs2891168 | G | A | 0.193401 | 0.0091877 | 42457 | 2.29E-98 | 0.010 | 443.102 | |
| rs2487928 | A | G | 0.062633 | 0.0095049 | 42457 | 4.41E-11 | 0.001 | 43.422 | |
| rs1870634 | G | T | 0.075878 | 0.0097113 | 42457 | 5.55E-15 | 0.001 | 61.049 | |
| rs1412444 | T | C | 0.066812 | 0.0096809 | 42457 | 5.15E-12 | 0.001 | 47.630 | |
| rs2128739 | C | A | -0.065565 | 0.0100568 | 42457 | 7.05E-11 | 0.001 | 42.503 | |
| rs2681472 | G | A | 0.074114 | 0.0113331 | 42457 | 6.17E-11 | 0.001 | 42.766 | |
| rs4468572 | C | T | 0.077234 | 0.0095277 | 42457 | 4.44E-16 | 0.002 | 65.711 | |
| rs4420638 | G | A | 0.091906 | 0.0140977 | 42457 | 7.07E-11 | 0.001 | 42.500 | |
| rs56289821 | A | G | -0.13361 | 0.0170415 | 42457 | 4.44E-15 | 0.001 | 61.470 | |
| rs28451064 | A | G | 0.127571 | 0.015952 | 42457 | 1.33E-15 | 0.002 | 63.955 | |
| genus Lachnoclostridium id.11308 (P<1×10-5) | rs12566975 | T | C | -0.0468097 | 0.0105787 | 14306 | 9.57194E-06 | 0.001 | 19.580 |
| rs1528479 | A | G | 0.0497799 | 0.0111919 | 14306 | 9.63984E-06 | 0.001 | 19.783 | |
| rs615997 | T | C | 0.0511752 | 0.0106491 | 14306 | 2.0268E-06 | 0.002 | 23.094 | |
| rs62285313 | A | G | 0.0864203 | 0.0181565 | 14306 | 1.58332E-06 | 0.002 | 22.655 | |
| rs1031599 | T | G | 0.078627 | 0.0175644 | 14306 | 6.31379E-06 | 0.001 | 20.039 | |
| rs3821998 | C | A | -0.0864066 | 0.0192519 | 14306 | 6.72048E-06 | 0.001 | 20.144 | |
| rs4738679 | A | G | 0.0520267 | 0.011404 | 14306 | 4.41754E-06 | 0.001 | 20.813 | |
| rs1997204 | C | T | 0.108075 | 0.0242022 | 14306 | 5.97077E-06 | 0.001 | 19.941 | |
| rs62028349 | G | C | 0.0469989 | 0.0105971 | 14306 | 9.17044E-06 | 0.001 | 19.670 | |
| rs72829893 | G | T | 0.117472 | 0.0268103 | 14306 | 5.57763E-06 | 0.001 | 19.198 | |
| rs78068103 | A | G | 0.0886199 | 0.0194248 | 14306 | 3.66522E-06 | 0.001 | 20.814 | |
| rs2385421 | A | G | 0.0746186 | 0.0180734 | 14306 | 7.13724E-06 | 0.001 | 17.046 | |
| rs789029 | C | T | -0.0641288 | 0.0137974 | 14306 | 3.75327E-06 | 0.002 | 21.603 | |
| rs6112314 | A | C | -0.0561715 | 0.0108174 | 14306 | 2.43215E-07 | 0.002 | 26.964 | |
| genus Streptococcus id.1853 (P<1×10-5) | rs11720390 | G | A | 0.107024 | 0.0228121 | 14306 | 3.59484E-06 | 0.002 | 22.011 |
| rs6806351 | T | C | -0.0633829 | 0.0136647 | 14306 | 4.93867E-06 | 0.002 | 21.515 | |
| rs57646748 | G | A | -0.0907696 | 0.0200344 | 14306 | 5.47545E-06 | 0.001 | 20.527 | |
| rs10028567 | C | T | -0.0921167 | 0.0191881 | 14306 | 7.30348E-06 | 0.002 | 23.047 | |
| rs395407 | C | G | 0.0792781 | 0.0173697 | 14306 | 4.36506E-06 | 0.001 | 20.832 | |
| rs77558518 | A | G | -0.103999 | 0.0229714 | 14306 | 4.70858E-06 | 0.001 | 20.497 | |
| rs11764382 | A | G | -0.0695345 | 0.0143671 | 14306 | 1.28632E-06 | 0.002 | 23.424 | |
| rs17708276 | A | G | -0.0793955 | 0.0170628 | 14306 | 3.04096E-06 | 0.002 | 21.652 | |
| rs10448310 | A | G | -0.0517935 | 0.0111324 | 14306 | 3.30704E-06 | 0.002 | 21.646 | |
| rs71481756 | T | G | 0.0931048 | 0.0207949 | 14306 | 6.51478E-06 | 0.001 | 20.046 | |
| rs7916711 | A | G | 0.102891 | 0.0217362 | 14306 | 0.000002717 | 0.002 | 22.407 | |
| rs1918540 | A | G | -0.059639 | 0.0128148 | 14306 | 2.44068E-06 | 0.002 | 21.659 | |
| rs11110281 | T | C | -0.137519 | 0.0227398 | 14306 | 2.58315E-09 | 0.003 | 36.572 | |
| rs2370083 | G | T | -0.0816836 | 0.0185851 | 14306 | 9.75237E-06 | 0.001 | 19.317 | |
| rs72739637 | A | G | 0.0959942 | 0.0193213 | 14306 | 1.03307E-06 | 0.002 | 24.684 | |
| rs6563952 | C | G | -0.0827344 | 0.0180035 | 14306 | 5.8213E-06 | 0.001 | 21.118 | |
| rs4968759 | A | G | -0.0515109 | 0.0112068 | 14306 | 3.7812E-06 | 0.001 | 21.127 | |
| rs9903102 | C | A | -0.0709483 | 0.0155275 | 14306 | 4.17994E-06 | 0.001 | 20.878 | |
| genus Actinomyces id.423 (P<1×10-5) | rs71315246 | A | G | -0.0969809 | 0.021925 | 14306 | 9.82969E-06 | 0.001 | 19.566 |
| rs34583783 | G | T | 0.126596 | 0.0268461 | 14306 | 4.48528E-06 | 0.002 | 22.237 | |
| rs4073240 | G | A | 0.0749687 | 0.0167368 | 14306 | 7.94273E-06 | 0.001 | 20.064 | |
| rs35011108 | A | G | 0.232634 | 0.0512044 | 14306 | 6.33826E-06 | 0.001 | 20.641 | |
| rs4146653 | G | A | 0.0985224 | 0.0214182 | 14306 | 4.49645E-06 | 0.001 | 21.159 | |
| rs10787984 | G | C | 0.094316 | 0.0213513 | 14306 | 9.62299E-06 | 0.001 | 19.513 | |
| rs7915461 | C | T | -0.18776 | 0.0401636 | 14306 | 5.91984E-06 | 0.002 | 21.855 | |
| rs2715439 | T | C | -0.0746684 | 0.0164822 | 14306 | 6.27004E-06 | 0.001 | 20.523 | |
| family Streptococcaceae id.1850 (P<1×10-5) | rs77968078 | G | A | -0.0993013 | 0.0224788 | 14306 | 7.93341E-06 | 0.001 | 19.515 |
| rs76717940 | T | A | 0.150606 | 0.0334079 | 14306 | 3.08937E-06 | 0.001 | 20.323 | |
| rs6806351 | T | C | -0.0619209 | 0.0135744 | 14306 | 6.93793E-06 | 0.001 | 20.808 | |
| rs10028567 | C | T | -0.0934027 | 0.0190343 | 14306 | 3.72495E-06 | 0.002 | 24.079 | |
| rs57646748 | G | A | -0.088021 | 0.019876 | 14306 | 7.88352E-06 | 0.001 | 19.612 | |
| rs395407 | C | G | 0.0826855 | 0.0172536 | 14306 | 1.32559E-06 | 0.002 | 22.967 | |
| rs77558518 | A | G | -0.104239 | 0.022806 | 14306 | 3.72195E-06 | 0.001 | 20.891 | |
| rs957755 | T | G | -0.0642449 | 0.0142702 | 14306 | 7.41515E-06 | 0.001 | 20.268 | |
| rs2952251 | G | A | 0.0639298 | 0.0126525 | 14306 | 3.72237E-07 | 0.002 | 25.530 | |
| rs28718126 | A | G | 0.109069 | 0.0246868 | 14306 | 9.41044E-06 | 0.001 | 19.520 | |
| rs7916711 | A | G | 0.0959639 | 0.021545 | 14306 | 6.32732E-06 | 0.001 | 19.839 | |
| rs16950051 | A | G | 0.107008 | 0.0236973 | 14306 | 5.33814E-06 | 0.001 | 20.391 | |
| rs11110281 | T | C | -0.130554 | 0.0225943 | 14306 | 1.40136E-08 | 0.002 | 33.387 | |
| rs2370083 | G | T | -0.0842751 | 0.0184509 | 14306 | 4.25667E-06 | 0.001 | 20.862 | |
| rs72739637 | A | G | 0.0927983 | 0.0192021 | 14306 | 1.82163E-06 | 0.002 | 23.355 | |
| rs6563952 | C | G | -0.0801931 | 0.0178576 | 14306 | 8.70583E-06 | 0.001 | 20.166 | |
| rs35344081 | G | A | 0.0609349 | 0.0129703 | 14306 | 2.63846E-06 | 0.002 | 22.072 | |
| rs9903102 | C | A | -0.0693015 | 0.0154096 | 14306 | 4.91802E-06 | 0.001 | 20.226 | |
| rs4968759 | A | G | -0.0544035 | 0.0111271 | 14306 | 8.91887E-07 | 0.002 | 23.905 | |
| unknown genus id.2041 (P<1×10-5) | rs1032598 | G | A | -0.0886426 | 0.0189039 | 14306 | 4.07587E-06 | 0.002 | 21.988 |
| rs16843660 | A | G | 0.234697 | 0.04907 | 14306 | 1.75344E-06 | 0.002 | 22.876 | |
| rs11941716 | A | G | 0.101243 | 0.0224414 | 14306 | 9.0663E-06 | 0.001 | 20.353 | |
| rs249459 | A | G | 0.0737341 | 0.0165018 | 14306 | 8.12307E-06 | 0.001 | 19.965 | |
| rs553072 | G | A | 0.109193 | 0.0230198 | 14306 | 3.69097E-06 | 0.002 | 22.500 | |
| rs1962916 | G | A | -0.0737876 | 0.0162232 | 14306 | 6.13847E-06 | 0.001 | 20.687 | |
| rs35703006 | G | T | 0.0926669 | 0.0190779 | 14306 | 9.00762E-07 | 0.002 | 23.593 | |
| rs921383 | G | A | 0.0723153 | 0.0159706 | 14306 | 7.72894E-06 | 0.001 | 20.503 | |
| rs2651663 | A | G | -0.0762556 | 0.0168635 | 14306 | 5.64144E-06 | 0.001 | 20.448 | |
| rs2336448 | T | C | 0.0773742 | 0.0160883 | 14306 | 1.42899E-06 | 0.002 | 23.130 | |
| rs7187855 | A | C | 0.199941 | 0.0418308 | 14306 | 2.20602E-06 | 0.002 | 22.846 | |
| rs6514318 | T | C | 0.128198 | 0.0281765 | 14306 | 5.37675E-06 | 0.001 | 20.701 | |
| genus Oxalobacter id.2978 (P<1×10-5) | rs4428215 | G | A | 0.130293 | 0.0242237 | 14306 | 7.51069E-08 | 0.002 | 28.931 |
| rs36057338 | G | T | 0.207847 | 0.0421439 | 14306 | 8.79812E-07 | 0.002 | 24.323 | |
| rs1569853 | T | C | -0.138078 | 0.0296981 | 14306 | 3.64502E-06 | 0.002 | 21.617 | |
| rs6993398 | G | A | 0.127217 | 0.0278855 | 14306 | 7.12771E-06 | 0.001 | 20.813 | |
| rs10464997 | G | A | 0.137691 | 0.0294804 | 14306 | 3.29754E-06 | 0.002 | 21.814 | |
| rs12002250 | A | C | 0.217122 | 0.0466317 | 14306 | 1.41504E-06 | 0.002 | 21.679 | |
| rs736744 | T | C | -0.117882 | 0.0211262 | 14306 | 2.57472E-08 | 0.002 | 31.135 | |
| rs3862635 | C | T | -0.172142 | 0.0394026 | 14306 | 9.18692E-06 | 0.001 | 19.086 | |
| rs11108500 | A | G | -0.199099 | 0.0427327 | 14306 | 3.74283E-06 | 0.002 | 21.708 | |
| rs111966731 | T | C | 0.213114 | 0.047162 | 14306 | 7.29861E-06 | 0.001 | 20.419 | |
| rs6071435 | T | A | -0.105512 | 0.021489 | 14306 | 1.07431E-06 | 0.002 | 24.109 | |
| rs6000536 | C | T | -0.130992 | 0.0253804 | 14306 | 2.06054E-07 | 0.002 | 26.637 | |
| genus Turicibacter id.2162 (P<1×10-5) | rs149744580 | A | G | 0.169883 | 0.0315478 | 14306 | 7.00971E-08 | 0.002 | 28.998 |
| rs4869133 | G | A | 0.131186 | 0.027197 | 14306 | 2.5537E-06 | 0.002 | 23.267 | |
| rs2221441 | G | C | 0.0710364 | 0.015343 | 14306 | 3.45669E-06 | 0.001 | 21.436 | |
| rs3734633 | G | A | -0.120957 | 0.02683 | 14306 | 5.31912E-06 | 0.001 | 20.325 | |
| rs55756211 | T | C | -0.115115 | 0.0240708 | 14306 | 2.8053E-06 | 0.002 | 22.871 | |
| rs2952020 | A | G | 0.0759019 | 0.0165764 | 14306 | 5.63313E-06 | 0.001 | 20.966 | |
| rs61265175 | G | C | -0.0858591 | 0.0185778 | 14306 | 4.13676E-06 | 0.001 | 21.359 | |
| rs11054680 | T | C | -0.104751 | 0.0226997 | 14306 | 2.30978E-06 | 0.001 | 21.295 | |
| rs4247078 | G | C | -0.0710377 | 0.0155221 | 14306 | 5.46072E-06 | 0.001 | 20.945 | |
| rs11649454 | G | C | 0.0950891 | 0.0203433 | 14306 | 3.26625E-06 | 0.002 | 21.848 | |
| rs7199484 | G | A | -0.0731428 | 0.0160172 | 14306 | 5.7666E-06 | 0.001 | 20.853 | |
| rs12603364 | T | C | 0.110861 | 0.0225598 | 14306 | 8.66603E-07 | 0.002 | 24.148 | |
| rs11666533 | C | T | -0.111689 | 0.0248436 | 14306 | 7.37106E-06 | 0.001 | 20.211 | |
| rs2834977 | T | C | -0.0959995 | 0.0208261 | 14306 | 3.95585E-06 | 0.001 | 21.248 | |
| genus Butyricicoccus id.2055 (P<1×10-5) | rs12034718 | G | A | -0.0701199 | 0.0158213 | 14306 | 9.57679E-06 | 0.001 | 19.643 |
| rs10084203 | G | A | -0.0549699 | 0.0123563 | 14306 | 8.58638E-06 | 0.001 | 19.791 | |
| rs56221232 | T | C | 0.0828027 | 0.0167401 | 14306 | 7.61939E-07 | 0.002 | 24.467 | |
| rs2017189 | T | G | 0.0506956 | 0.011024 | 14306 | 3.87258E-06 | 0.001 | 21.148 | |
| rs62478070 | T | G | 0.224039 | 0.0494959 | 14306 | 5.93772E-06 | 0.001 | 20.488 | |
| rs4962426 | T | G | -0.0614216 | 0.0135979 | 14306 | 7.38482E-06 | 0.001 | 20.403 | |
| rs7322368 | C | T | -0.0815733 | 0.0183167 | 14306 | 5.51785E-06 | 0.001 | 19.834 | |
| rs12585793 | T | C | -0.262206 | 0.0564729 | 14306 | 5.79189E-06 | 0.002 | 21.558 | |
| rs75238760 | T | A | 0.0619423 | 0.0139942 | 14306 | 6.79704E-06 | 0.001 | 19.592 | |
| unknown genus id.2071 (P<1×10-5) | rs4644504 | T | C | -0.0969321 | 0.0216146 | 14306 | 5.81969E-06 | 0.001 | 20.111 |
| rs11809762 | G | A | -0.0934634 | 0.0190198 | 14306 | 1.68287E-06 | 0.002 | 24.147 | |
| rs11904514 | A | G | 0.109498 | 0.0249839 | 14306 | 7.89951E-06 | 0.001 | 19.208 | |
| rs1809136 | C | G | -0.0994594 | 0.0228638 | 14306 | 8.36989E-06 | 0.001 | 18.923 | |
| rs16823675 | C | T | -0.0767515 | 0.0149973 | 14306 | 2.33346E-07 | 0.002 | 26.191 | |
| rs11684166 | A | G | -0.0769635 | 0.0168349 | 14306 | 3.49116E-06 | 0.001 | 20.900 | |
| rs10200320 | T | C | -0.0641139 | 0.0142769 | 14306 | 5.6607E-06 | 0.001 | 20.167 | |
| rs2898979 | G | C | 0.0901515 | 0.0202199 | 14306 | 7.67291E-06 | 0.001 | 19.879 | |
| rs35740166 | C | T | -0.112246 | 0.0226824 | 14306 | 8.39982E-07 | 0.002 | 24.489 | |
| rs17086536 | C | A | -0.100851 | 0.022432 | 14306 | 3.3638E-06 | 0.001 | 20.213 | |
| rs34985298 | G | A | -0.0623526 | 0.013772 | 14306 | 8.33758E-06 | 0.001 | 20.498 | |
| rs1455639 | A | G | -0.0760307 | 0.0169831 | 14306 | 7.83899E-06 | 0.001 | 20.042 | |
| rs11195523 | C | A | -0.0689278 | 0.0145351 | 14306 | 2.40121E-06 | 0.002 | 22.488 | |
| rs2939766 | A | G | -0.0591611 | 0.013042 | 14306 | 7.01148E-06 | 0.001 | 20.577 | |
| rs76532867 | T | C | 0.112353 | 0.0242364 | 14306 | 2.55859E-06 | 0.001 | 21.490 | |
| rs56975773 | T | A | 0.113282 | 0.0248859 | 14306 | 7.65491E-06 | 0.001 | 20.721 | |
| rs12147596 | C | T | -0.0719818 | 0.0141609 | 14306 | 2.86207E-07 | 0.002 | 25.838 | |
| rs72700702 | T | C | -0.091726 | 0.0189005 | 14306 | 1.59272E-06 | 0.002 | 23.553 | |
| rs72707147 | C | T | 0.110109 | 0.0244644 | 14306 | 6.84022E-06 | 0.001 | 20.257 | |
| rs6007642 | C | T | -0.0791412 | 0.0177958 | 14306 | 9.95543E-06 | 0.001 | 19.777 | |
| family Clostridiales vadin BB60 group id.11286 | rs7538034 | T | G | -0.078598 | 0.0165982 | 14306 | 2.36706E-06 | 0.002 | 22.423 |
| (P<1×10-5) | rs6588624 | A | G | 0.0662317 | 0.0138147 | 14306 | 1.79287E-06 | 0.002 | 22.985 |
| rs13409132 | A | G | -0.165419 | 0.0352154 | 14306 | 4.3723E-06 | 0.002 | 22.065 | |
| rs2191834 | T | G | -0.0746375 | 0.0159136 | 14306 | 2.50196E-06 | 0.002 | 21.998 | |
| rs6755871 | C | G | -0.0613825 | 0.0138976 | 14306 | 9.33061E-06 | 0.001 | 19.508 | |
| rs989682 | A | G | 0.070194 | 0.0155364 | 14306 | 6.84715E-06 | 0.001 | 20.413 | |
| rs10517600 | G | T | -0.0626993 | 0.0139364 | 14306 | 6.82763E-06 | 0.001 | 20.241 | |
| rs34088226 | A | G | -0.117807 | 0.026924 | 14306 | 7.66214E-06 | 0.001 | 19.145 | |
| rs7725895 | A | G | -0.116224 | 0.0240367 | 14306 | 3.94357E-06 | 0.002 | 23.380 | |
| rs66714985 | A | C | 0.116908 | 0.0252447 | 14306 | 4.85333E-06 | 0.001 | 21.446 | |
| rs118104867 | C | T | 0.214464 | 0.0455098 | 14306 | 3.43598E-06 | 0.002 | 22.207 | |
| rs10904722 | C | T | -0.0672314 | 0.0147123 | 14306 | 5.04836E-06 | 0.001 | 20.883 | |
| rs17121075 | G | A | 0.0769254 | 0.0172234 | 14306 | 7.91425E-06 | 0.001 | 19.948 | |
| rs55682560 | C | T | -0.131519 | 0.0261319 | 14306 | 4.97038E-07 | 0.002 | 25.330 | |
| rs28691777 | C | T | 0.137134 | 0.0266996 | 14306 | 6.95697E-07 | 0.002 | 26.380 | |
| rs7226487 | A | G | -0.0643682 | 0.0138701 | 14306 | 3.58286E-06 | 0.002 | 21.537 | |
| rs9979874 | G | C | -0.0738925 | 0.0150911 | 14306 | 1.05271E-06 | 0.002 | 23.975 | |
| unknown genus id.1000000073 (P<1×10-5) | rs6588624 | A | G | 0.0662317 | 0.0138147 | 14306 | 1.79287E-06 | 0.002 | 22.985 |
| rs7538034 | T | G | -0.078598 | 0.0165982 | 14306 | 2.36706E-06 | 0.002 | 22.423 | |
| rs2191834 | T | G | -0.0746375 | 0.0159136 | 14306 | 2.50196E-06 | 0.002 | 21.998 | |
| rs13409132 | A | G | -0.165419 | 0.0352154 | 14306 | 4.3723E-06 | 0.002 | 22.065 | |
| rs6755871 | C | G | -0.0613825 | 0.0138976 | 14306 | 9.33061E-06 | 0.001 | 19.508 | |
| rs989682 | A | G | 0.070194 | 0.0155364 | 14306 | 6.84715E-06 | 0.001 | 20.413 | |
| rs10517600 | G | T | -0.0626993 | 0.0139364 | 14306 | 6.82763E-06 | 0.001 | 20.241 | |
| rs7725895 | A | G | -0.116224 | 0.0240367 | 14306 | 3.94357E-06 | 0.002 | 23.380 | |
| rs34088226 | A | G | -0.117807 | 0.026924 | 14306 | 7.66214E-06 | 0.001 | 19.145 | |
| rs66714985 | A | C | 0.116908 | 0.0252447 | 14306 | 4.85333E-06 | 0.001 | 21.446 | |
| rs118104867 | C | T | 0.214464 | 0.0455098 | 14306 | 3.43598E-06 | 0.002 | 22.207 | |
| rs10904722 | C | T | -0.0672314 | 0.0147123 | 14306 | 5.04836E-06 | 0.001 | 20.883 | |
| rs17121075 | G | A | 0.0769254 | 0.0172234 | 14306 | 7.91425E-06 | 0.001 | 19.948 | |
| rs55682560 | C | T | -0.131519 | 0.0261319 | 14306 | 4.97038E-07 | 0.002 | 25.330 | |
| rs28691777 | C | T | 0.137134 | 0.0266996 | 14306 | 6.95697E-07 | 0.002 | 26.380 | |
| rs7226487 | A | G | -0.0643682 | 0.0138701 | 14306 | 3.58286E-06 | 0.002 | 21.537 | |
| rs9979874 | G | C | -0.0738925 | 0.0150911 | 14306 | 1.05271E-06 | 0.002 | 23.975 | |
| genus Clostridium innocuum group id.14397 | rs6577484 | G | A | 0.160425 | 0.0360857 | 14306 | 8.40601E-06 | 0.001 | 19.764 |
| (P<1×10-5) | rs1948423 | T | A | -0.108859 | 0.023425 | 14306 | 3.49406E-06 | 0.002 | 21.596 |
| rs40656 | C | T | 0.142664 | 0.0311021 | 14306 | 8.61529E-06 | 0.001 | 21.040 | |
| rs6890185 | C | T | -0.113424 | 0.0233137 | 14306 | 1.12243E-06 | 0.002 | 23.669 | |
| rs4869133 | G | A | -0.180591 | 0.0409505 | 14306 | 7.24453E-06 | 0.001 | 19.448 | |
| rs10074000 | T | C | -0.102648 | 0.0227508 | 14306 | 6.99939E-06 | 0.001 | 20.357 | |
| rs71564433 | T | A | -0.126746 | 0.0274657 | 14306 | 7.8001E-06 | 0.001 | 21.295 | |
| rs10506058 | A | G | 0.0997048 | 0.0221926 | 14306 | 8.92442E-06 | 0.001 | 20.184 | |
| rs77845139 | A | G | -0.114993 | 0.0257186 | 14306 | 8.40621E-06 | 0.001 | 19.992 | |
| rs61267978 | T | C | 0.14708 | 0.0320875 | 14306 | 5.58509E-06 | 0.001 | 21.010 | |
| rs1942371 | G | A | -0.157938 | 0.034187 | 14306 | 4.0634E-06 | 0.001 | 21.343 | |
| class Lentisphaeria id.2250 (P<1×10-5) | rs72640280 | A | G | 0.220207 | 0.0486196 | 14306 | 5.18036E-06 | 0.001 | 20.513 |
| rs73113483 | T | A | -0.131217 | 0.0288713 | 14306 | 8.66343E-06 | 0.001 | 20.656 | |
| rs2731834 | G | C | -0.109438 | 0.023693 | 14306 | 4.24356E-06 | 0.001 | 21.335 | |
| rs11770843 | C | T | 0.109431 | 0.0234879 | 14306 | 1.9073E-06 | 0.002 | 21.707 | |
| rs62570196 | C | T | -0.21635 | 0.0439866 | 14306 | 1.07924E-06 | 0.002 | 24.192 | |
| rs2031282 | A | G | 0.122368 | 0.0270329 | 14306 | 4.38258E-06 | 0.001 | 20.490 | |
| rs17114848 | G | A | 0.152377 | 0.0324332 | 14306 | 4.05864E-06 | 0.002 | 22.073 | |
| rs1002941 | A | G | -0.105025 | 0.0233484 | 14306 | 8.14836E-06 | 0.001 | 20.234 | |
| rs77599476 | A | G | 0.230292 | 0.0480168 | 14306 | 1.86132E-06 | 0.002 | 23.002 | |
| rs2825714 | A | G | -0.13741 | 0.0289246 | 14306 | 1.72211E-06 | 0.002 | 22.568 | |
| order Victivallales id.2254 (P<1×10-5) | rs72640280 | A | G | 0.220207 | 0.0486196 | 14306 | 5.18036E-06 | 0.001 | 20.513 |
| rs73113483 | T | A | -0.131217 | 0.0288713 | 14306 | 8.66343E-06 | 0.001 | 20.656 | |
| rs2731834 | G | C | -0.109438 | 0.023693 | 14306 | 4.24356E-06 | 0.001 | 21.335 | |
| rs11770843 | C | T | 0.109431 | 0.0234879 | 14306 | 1.9073E-06 | 0.002 | 21.707 | |
| rs62570196 | C | T | -0.21635 | 0.0439866 | 14306 | 1.07924E-06 | 0.002 | 24.192 | |
| rs2031282 | A | G | 0.122368 | 0.0270329 | 14306 | 4.38258E-06 | 0.001 | 20.490 | |
| rs1002941 | A | G | -0.105025 | 0.0233484 | 14306 | 8.14836E-06 | 0.001 | 20.234 | |
| rs17114848 | G | A | 0.152377 | 0.0324332 | 14306 | 4.05864E-06 | 0.002 | 22.073 | |
| rs77599476 | A | G | 0.230292 | 0.0480168 | 14306 | 1.86132E-06 | 0.002 | 23.002 | |
| rs2825714 | A | G | -0.13741 | 0.0289246 | 14306 | 1.72211E-06 | 0.002 | 22.568 | |
| genus Bifidobacterium id.436 (P<1×10-5) | rs12022129 | A | G | -0.0619356 | 0.0138937 | 14306 | 7.9965E-06 | 0.001 | 19.872 |
| rs1961273 | C | T | 0.0674036 | 0.0132319 | 14306 | 3.50865E-07 | 0.002 | 25.949 | |
| rs13020688 | G | A | 0.0562696 | 0.0122617 | 14306 | 4.07258E-06 | 0.001 | 21.059 | |
| rs182549 | T | C | -0.119703 | 0.0127294 | 14306 | 1.2782E-20 | 0.006 | 88.429 | |
| rs62181700 | G | A | -0.0624643 | 0.0131205 | 14306 | 2.17245E-06 | 0.002 | 22.665 | |
| rs4567981 | T | A | 0.0562084 | 0.0117923 | 14306 | 1.92832E-06 | 0.002 | 22.720 | |
| rs55888705 | A | G | 0.0546319 | 0.0121139 | 14306 | 6.67022E-06 | 0.001 | 20.339 | |
| rs4957061 | T | C | 0.0534239 | 0.0117431 | 14306 | 5.77936E-06 | 0.001 | 20.697 | |
| rs73797465 | T | G | -0.0953566 | 0.0209236 | 14306 | 4.38157E-06 | 0.001 | 20.770 | |
| rs76671854 | C | G | -0.0846055 | 0.0184003 | 14306 | 3.95667E-06 | 0.001 | 21.142 | |
| rs857444 | C | T | 0.0558234 | 0.0121219 | 14306 | 0.000003571 | 0.001 | 21.208 | |
| rs2686790 | C | T | -0.070741 | 0.0157926 | 14306 | 7.49894E-06 | 0.001 | 20.065 | |
| rs2491158 | A | G | -0.0712624 | 0.015983 | 14306 | 8.04711E-06 | 0.001 | 19.879 | |
| rs10841473 | G | C | -0.0624207 | 0.0129438 | 14306 | 1.6452E-06 | 0.002 | 23.256 | |
| rs7322849 | T | C | 0.112428 | 0.0201813 | 14306 | 1.08368E-08 | 0.002 | 31.035 | |
| rs540489 | T | G | -0.0637641 | 0.0138746 | 14306 | 5.19457E-06 | 0.001 | 21.121 | |
| rs75344046 | C | T | 0.232354 | 0.0505979 | 14306 | 4.86351E-06 | 0.001 | 21.088 | |
| rs5746486 | T | C | -0.0536216 | 0.0120801 | 14306 | 8.99953E-06 | 0.001 | 19.703 |
SNPs, single nucleotide polymorphisms; EA, effect allele; OA, other allele; Beta, effect estimate; SE, standard error
The anaerobic bacterium genus Oxalobacter, specialized in symbiosis and reliant solely on oxalic acid, was initially identified in the human gut and formally designated as Oxalobacter formigenes in 1985 [37, 38]. This bacterium has garnered significant attention in nephrolithiasis research due to correlations between heightened urinary oxalic acid excretion and the formation of oxalic acid kidney stones [39]. Distinct variations in the gut microbiome have been noted in several studies comparing individuals with type 2 diabetes (T2D) and healthy controls. Key differences include a reduction in butyrate-producing gut microbiota, diminished levels of Akkermansia muciniphila, and an increased presence of pro-inflammatory bacterial species [40]. Nonetheless, alterations in the abundance of Lachnoclostridium, Streptococcus, Actinomyces, and Streptococcaceae have been less frequently reported. Certain medications, like metformin, are known to modulate gut microbiota, thereby influencing insulin sensitivity and aiding in diabetes management. T2D may enhance the proliferation of Oxalobacter formigenes by inducing chronic intestinal inflammation and altering metabolic pathways related to oxalic acid processing [41, 42]. This condition is characterized by heightened parasympathetic activity and local ATP release into the intestinal tract [43-45]. The relatively unaffected colonization of Oxalobacter formigenes by other bacteria suggests a stable colonization characteristic of this genus [46]. Research has examined various prevalent methods and conditions pertinent to probiotic strain production, particularly highlighting the resilience of the Group I Oxalobacter strain OxCC13 in lyophilized form and when mixed in yogurt [47]. Human consumption of Oxalobacter in these forms may offer preventive benefits against CAD, although the understanding of Oxalobacter's role in CAD remains incomplete [48]. A gut microbiota-based diagnostic model suggests that increased gut colonization by Oxalobacter formigenes might elevate CAD risk [49]. This aligns with our study findings, though the underlying mechanisms require further elucidation [50].
Recent studies focusing on the interplay between T2D and gut microbiota have observed a reduction in gut microbiota species that produce butyric acid in individuals with prediabetes, aligning with previous findings [40, 41]. In the context of intestinal dysbiosis associated with T2D, metformin has been shown to enhance the production of butyric and propionic acids and improve patients' ability to break down amino acids. Additionally, metformin's role in modifying gut microbiota composition, potentially aiding in T2D prognosis through an increase in butyric acid-producing bacteria, has been highlighted [51, 52]. Past research, encompassing both animal models and epidemiological studies, has underscored the bidirectional relationship between gut microbiota and host health in the context of atherosclerotic cardiovascular disease. Notably, bacterial presence in atherosclerotic plaques has been documented [53-55]. The gut microbiota's influence on the metabolism of short-chain fatty acids (SCFAs), including Prevotellaceae, Clostridium, and Anaerostipes, has been linked to CAD, echoing findings from this study [56]. A significant observation is the decreased abundance of methanogens in individuals susceptible to CAD. Certain methanogens are known to convert Trimethylamine (TMA) into a less harmful derivative, trimethylamine N-oxide (TMAO), thus reducing TMAO production [57]. In our study, though the P-value in the IVW method for TMAO was less than 0.05, it did not pass sensitivity analyses, suggesting a potential connection between altered gut microbiota in coronary atherosclerosis patients and increased TMAO levels due to impaired metabolism.
For individuals with DCAD, long-term medication complicates the reliability of isolated gut microbiota observations. This study suggests that intestinal bacteria play a regulatory role in the development of both T2D and CAD, with implications for both elevated and reduced risk. The discovery of certain gut flora causally linked to diabetes and coronary heart disease, previously unreported, opens up new avenues for therapeutic strategies and potential targets.
The composition and activity of the gut microbiome, influenced by dietary and environmental factors, play a crucial role in the abundance and utilization of various metabolites [58]. Metabolomics research has linked bile acids, branched-chain amino acids (BCAAs), and by-products of intracellular fatty acid oxidation to diabetes, glycemic control, and insulin resistance. Despite some studies indicating a correlation between TMAO levels and an increased risk of major cardiovascular events, including CAD, other studies have not found a significant relationship between circulating TMAO concentrations and cardiovascular outcomes [59-62]. In our research, TMAO did not exhibit a notable association with CAD risk. However, we observed a positive correlation between certain lipid metabolism markers, such as phosphatidylcholine and cholesterol, including lysophosphatidylcholine (LPC18_2) and cholesterol ester (CE18_2), and CAD risk, underlining the strong connection between lipid metabolism and CAD [63-65]. Animal studies have shown that rats on a carnitine-rich diet experienced a reduction in aortic lesion size, irrespective of increased blood TMAO levels, hinting at a possible protective role of carnitine against atherosclerosis [66, 67]. This finding aligns with the results from our MR analysis, reinforcing the potential significance of carnitine in atherosclerosis prevention [68].
When evaluating the findings of this research, certain limitations must be acknowledged. Firstly, despite utilizing the most comprehensive genome-wide association study (GWAS) database currently available for gut microbiota and metabolites, the limited number of single nucleotide polymorphisms (SNPs) reaching genome-wide significance might have led to the use of weaker instrumental variables. To mitigate this, we expanded the inclusion criteria for SNPs to a statistical threshold of P < 10-5, allowing for a broader SNP inclusion. Additionally, to ascertain that these SNPs were not weak instrumental variables, they were evaluated using F statistics, ensuring values greater than 10. Secondly, given the extensive number of base pairs in the genome-wide analysis, it's challenging to completely rule out the presence of polymorphisms. Moreover, the biological implications of the selected SNPs have not been comprehensively explored. However, in our study, no horizontal pleiotropy was identified, as confirmed by the application of methods like MRPRESSO and MR Egger. Thirdly, our MR analysis was predicated on the assumption of a linear relationship between the variables of interest, hence the possibility of non-linear interactions between the exposure and outcome cannot be entirely dismissed. Finally, the metabolite database employed in our study was subject to preliminary screening. This limitation meant that a comprehensive two-way MR analysis was not feasible. Future research, ideally with more complete GWAS data, will be necessary to corroborate and expand upon our findings.
Conclusion
The MR study conducted in our research provides insights into both the positive and negative causal effects of gut microbiota composition and metabolite levels on the occurrence of T2D and coronary artery disease (CAD). Our data supports the notion that the bacterial species Oxalobacter formigenes could be a contributory factor in CAD, particularly among individuals with diabetes. This study highlights a noteworthy link between the Methanobacteria class and CAD risk, paving the way for further exploration into the roles of TMAO and the protective potential of carnitine in the development of CAD. The findings present a new viewpoint on the influence of gut microbiota in the pathogenesis of CAD, providing valuable insights that could guide therapeutic approaches and the management of CAD in patients with T2D.
Supplementary Material
Supplementary figures and tables.
Supplementary data 1.
Supplementary data 2.
Acknowledgements
Our heartfelt thanks go to Xue and colleagues for their groundbreaking T2D GWAS meta-analysis, Nikpay and team for their exhaustive CAD GWAS meta-analysis, the MiBioGen consortium for their meta-analysis of gut microbiota GWAS, and the FHS consortium for their analysis in metabolite GWAS.
Financial Support
This investigation was financially supported by several grants: from the National Natural Science Foundation of China (Grant Numbers 82271806 and 82200483), the Natural Science Foundation of Guangdong Province (Grant Numbers 2022A1515110560, 2022A1515111053 and 2023A1515011687), the Guangzhou Basic Research Program's Basic and Applied Basic Research Project (Grant Number 202201011024), Guangzhou Science and Technology Plan Project (Grant Number 2023A03J0697), and the Sun Yat-sen Memorial Hospital, Sun Yat-sen University's Scientific Research Sailing Project (Grant Number YXQH2022017).
Ethics Statement
The genome-wide association studies (GWAS) used in this study received ethical approval from their respective review committees, as indicated in their original publications. Our study, employing summary-level data, did not necessitate additional ethical approval.
Data Accessibility
The data underpinning the conclusions of this article are detailed in the main text and supplementary materials, with direct references in Supplementary Table S1.
Author contributions
The study's conceptualization and design were led by Z. Zeng, J. Qiu, J. Tao, L. Lin, and J. Zheng. D. Liang, F. Wei, Y. Fu, J. Zhang, X. Zhang and X. Wei played key roles in data analysis and interpretation. Y. Chen provided statistical expertise and editorial assistance. Z. Zeng and J. Qiu drafted the initial manuscript. J. Tao, L. Lin, and J. Zheng oversaw the project. All authors participated in a thorough review and refinement of the manuscript and approved its final version for publication.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Belkaid Y, Hand T. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121-41
2. Mazloom K, Siddiqi I, Covasa M. Probiotics: How Effective Are They in the Fight against Obesity? Nutrients. 2019 11
3. Zhao YW, Yan KX, Sun MZ, Wang YH, Chen YD, Hu SY. Inflammation-based different association between anatomical severity of coronary artery disease and lung cancer. J Geriatr Cardiol. 2022;19:575-82
4. Chen W, Zhang M, Guo Y, Wang Z, Liu Q, Yan R. et al. The Profile and Function of Gut Microbiota in Diabetic Nephropathy. Diabetes Metab Syndr Obes. 2021;14:4283-96
5. Yissachar N, Zhou Y, Ung L, Lai N, Mohan J, Ehrlicher A. et al. An Intestinal Organ Culture System Uncovers a Role for the Nervous System in Microbe-Immune Crosstalk. Cell. 2017;168:1135-48.e12
6. Montero S, Abrams D, Ammirati E, Huang F, Donker DW, Hekimian G. et al. Fulminant myocarditis in adults: a narrative review. J Geriatr Cardiol. 2022;19:137-51
7. Lin L, Lin J, Qiu J, Liufu N, Lin S, Wei F. et al. Genetic liability to multi-site chronic pain increases the risk of cardiovascular disease. Br J Anaesth. 2023
8. Bulugahapitiya U, Siyambalapitiya S, Sithole J, Idris I. Is diabetes a coronary risk equivalent? Systematic review and meta-analysis. Diabetic medicine: a journal of the British Diabetic Association. 2009;26:142-8
9. Howard B, Best L, Galloway J, Howard W, Jones K, Lee E. et al. Coronary heart disease risk equivalence in diabetes depends on concomitant risk factors. Diabetes care. 2006;29:391-7
10. Bekyarova G, Ivanova D, Madjova V. Molecular mechanisms associating oxidative stress with endothelial dysfunction in the development of various vascular complications in diabetes mellitus. Folia medica. 2007;49:13-9
11. Baig M, Panchal S. Streptozotocin-Induced Diabetes Mellitus in Neonatal Rats: An Insight into its Applications to Induce Diabetic Complications. Current diabetes reviews. 2019;16:26-39
12. Wu H, Tremaroli V, Schmidt C, Lundqvist A, Olsson L, Krämer M. et al. The Gut Microbiota in Prediabetes and Diabetes: A Population-Based Cross-Sectional Study. Cell metabolism. 2020;32:379-90.e3
13. Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA. et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535:376-81
14. Koh A, Molinaro A, Stahlman M, Khan MT, Schmidt C, Manneras-Holm L. et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell. 2018;175:947-61 e17
15. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55-60
16. Karlsson F, Tremaroli V, Nookaew I, Bergström G, Behre C, Fagerberg B. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99-103
17. Nakajima A, Mitomo S, Yuki H, Araki M, Seegers L, McNulty I. et al. Gut Microbiota and Coronary Plaque Characteristics. Journal of the American Heart Association. 2022;11:e026036
18. Mene-Afejuku TO, Jeyashanmugaraja GP, Hoq M, Ola O, Shah AJ. Determinants of mortality among seniors acutely readmitted for heart failure: racial disparities and clinical correlations. J Geriatr Cardiol. 2022;19:719-24
19. Korem T, Zeevi D, Suez J, Weinberger A, Avnit-Sagi T, Pompan-Lotan M. et al. Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science (New York, NY). 2015;349:1101-6
20. Luo FY, Bai YP, Bu HS. Protein quality control systems in hypertrophic cardiomyopathy: pathogenesis and treatment potential. J Geriatr Cardiol. 2022;19:780-4
21. Emdin C, Khera A, Kathiresan S. Mendelian Randomization. JAMA. 2017;318:1925-6
22. Zhou H, Ren J, Toan S, Mui D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res Rev. 2021;66:101250
23. Zhang Q, Zhang L, Chen C, Li P, Lu B. The gut microbiota-artery axis: A bridge between dietary lipids and atherosclerosis? Progress in lipid research. 2023;89:101209
24. Burgess S, Davey Smith G, Davies N, Dudbridge F, Gill D, Glymour M. et al. Guidelines for performing Mendelian randomization investigations. Wellcome open research. 2019;4:186
25. Skrivankova V, Richmond R, Woolf B, Yarmolinsky J, Davies N, Swanson S. et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA. 2021;326:1614-21
26. Xue A, Wu Y, Zhu Z, Zhang F, Kemper K, Zheng Z. et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nature communications. 2018;9:2941
27. Nikpay M, Goel A, Won H, Hall L, Willenborg C, Kanoni S. et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nature genetics. 2015;47:1121-30
28. Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A. et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nature genetics. 2021;53:156-65
29. Zhu H, Tan Y, Du W, Li Y, Toan S, Mui D. et al. Phosphoglycerate mutase 5 exacerbates cardiac ischemia-reperfusion injury through disrupting mitochondrial quality control. Redox Biol. 2021;38:101777
30. Rhee E, Ho J, Chen M, Shen D, Cheng S, Larson M. et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell metabolism. 2013;18:130-43
31. Chang X, Li Y, Cai C, Wu F, He J, Zhang Y. et al. Mitochondrial quality control mechanisms as molecular targets in diabetic heart. Metabolism. 2022;137:155313
32. Chang X, Toan S, Li R, Zhou H. Therapeutic strategies in ischemic cardiomyopathy: Focus on mitochondrial quality surveillance. EBioMedicine. 2022;84:104260
33. Tan Y, Zhang Y, He J, Wu F, Wu D, Shi N. et al. Dual specificity phosphatase 1 attenuates inflammation-induced cardiomyopathy by improving mitophagy and mitochondrial metabolism. Mol Metab. 2022;64:101567
34. Ma L, Zou R, Shi W, Zhou N, Chen S, Zhou H. et al. SGLT2 inhibitor dapagliflozin reduces endothelial dysfunction and microvascular damage during cardiac ischemia/reperfusion injury through normalizing the XO-SERCA2-CaMKII-coffilin pathways. Theranostics. 2022;12:5034-50
35. Zou R, Shi W, Qiu J, Zhou N, Du N, Zhou H. et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion injury through improving mitochondrial homeostasis. Cardiovasc Diabetol. 2022;21:106
36. Wang S, Zhu H, Li R, Mui D, Toan S, Chang X. et al. DNA-PKcs interacts with and phosphorylates Fis1 to induce mitochondrial fragmentation in tubular cells during acute kidney injury. Sci Signal. 2022;15:eabh1121
37. Allison M, Dawson K, Mayberry W, Foss J. Oxalobacter formigenes gen. nov, sp. nov.: oxalate-degrading anaerobes that inhabit the gastrointestinal tract. Archives of microbiology. 1985;141:1-7
38. Zou R, Tao J, Qiu J, Lu H, Wu J, Zhu H. et al. DNA-PKcs promotes sepsis-induced multiple organ failure by triggering mitochondrial dysfunction. J Adv Res. 2022;41:39-48
39. Robertson W, Peacock M, Heyburn P, Hanes F. Epidemiological risk factors in calcium stone disease. Scandinavian journal of urology and nephrology Supplementum. 1980;53:15-30
40. Zhong H, Ren H, Lu Y, Fang C, Hou G, Yang Z. et al. Distinct gut metagenomics and metaproteomics signatures in prediabetics and treatment-naïve type 2 diabetics. EBioMedicine. 2019;47:373-83
41. Lee C, Chae S, Jo S, Jerng U, Bae S. The Relationship between the Gut Microbiome and Metformin as a Key for Treating Type 2 Diabetes Mellitus. International journal of molecular sciences. 2021 22
42. Zou RJ, Tao J, He J, Wang CJ, Tan ST, Xia Y. et al. PGAM5-Mediated PHB2 Dephosphorylation Contributes to Diabetic Cardiomyopathy by Disrupting Mitochondrial Quality Surveillance. RESEARCH. 2022. 2022
43. Amin R, Sharma S, Ratakonda S, Hassan H. Extracellular nucleotides inhibit oxalate transport by human intestinal Caco-2-BBe cells through PKC-δ activation. American journal of physiology Cell physiology. 2013;305:C78-89
44. Hassan H, Cheng M, Aronson P. Cholinergic signaling inhibits oxalate transport by human intestinal T84 cells. American journal of physiology Cell physiology. 2012;302:C46-58
45. Shen Y, Peng X, Ji H, Gong W, Zhu H, Wang J. Dapagliflozin protects heart function against type-4 cardiorenal syndrome through activation of PKM2/PP1/FUNDC1-dependent mitophagy. Int J Biol Macromol. 2023;250:126116
46. Li X, Ellis M, Dowell A, Kumar R, Morrow C, Schoeb T. et al. Response of germ-free mice to colonization with O. formigenes and altered Schaedler flora. Applied and environmental microbiology. 2016;82:6952-60
47. Ellis M, Dowell A, Li X, Knight J. Probiotic properties of Oxalobacter formigenes: an in vitro examination. Archives of microbiology. 2016;198:1019-26
48. Emoto T, Yamashita T, Sasaki N, Hirota Y, Hayashi T, So A. et al. Analysis of Gut Microbiota in Coronary Artery Disease Patients: a Possible Link between Gut Microbiota and Coronary Artery Disease. Journal of atherosclerosis and thrombosis. 2016;23:908-21
49. Zheng Y, Wu T, Liu Z, Li A, Guo Q, Ma Y. et al. Gut Microbiome-Based Diagnostic Model to Predict Coronary Artery Disease. Journal of agricultural and food chemistry. 2020;68:3548-57
50. Allin K, Tremaroli V, Caesar R, Jensen B, Damgaard M, Bahl M. et al. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia. 2018;61:810-20
51. Tang W, Hazen S. The contributory role of gut microbiota in cardiovascular disease. The Journal of clinical investigation. 2014;124:4204-11
52. Zhu H, Wang J, Xin T, Chen S, Hu R, Li Y. et al. DUSP1 interacts with and dephosphorylates VCP to improve mitochondrial quality control against endotoxemia-induced myocardial dysfunction. Cell Mol Life Sci. 2023;80:213
53. Gregory J, Buffa J, Org E, Wang Z, Levison B, Zhu W. et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. The Journal of biological chemistry. 2015;290:5647-60
54. Ott S, El Mokhtari N, Musfeldt M, Hellmig S, Freitag S, Rehman A. et al. Detection of diverse bacterial signatures in atherosclerotic lesions of patients with coronary heart disease. Circulation. 2006;113:929-37
55. Zhou H, Dai Z, Li J, Wang J, Zhu H, Chang X. et al. TMBIM6 prevents VDAC1 multimerization and improves mitochondrial quality control to reduce sepsis-related myocardial injury. Metabolism. 2023;140:155383
56. Duncan S, Barcenilla A, Stewart C, Pryde S, Flint H. Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Applied and environmental microbiology. 2002;68:5186-90
57. Brugère J, Borrel G, Gaci N, Tottey W, O'Toole P, Malpuech-Brugère C. Archaebiotics: proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut microbes. 2014;5:5-10
58. Cani P, Van Hul M. Do diet and microbes really 'PREDICT' cardiometabolic risks? Nature reviews Endocrinology. 2021;17:259-60
59. Cani P, Moens de Hase E, Van Hul M. Gut Microbiota and Host Metabolism: From Proof of Concept to Therapeutic Intervention. Microorganisms. 2021 9
60. Felig P, Marliss E, Cahill G. Plasma amino acid levels and insulin secretion in obesity. The New England journal of medicine. 1969;281:811-6
61. Ferrannini E, Natali A, Camastra S, Nannipieri M, Mari A, Adam K. et al. Early metabolic markers of the development of dysglycemia and type 2 diabetes and their physiological significance. Diabetes. 2013;62:1730-7
62. Peddinti G, Cobb J, Yengo L, Froguel P, Kravić J, Balkau B. et al. Early metabolic markers identify potential targets for the prevention of type 2 diabetes. Diabetologia. 2017;60:1740-50
63. Fall T, Salihovic S, Brandmaier S, Nowak C, Ganna A, Gustafsson S. et al. Non-targeted metabolomics combined with genetic analyses identifies bile acid synthesis and phospholipid metabolism as being associated with incident type 2 diabetes. Diabetologia. 2016;59:2114-24
64. Craciun S, Balskus E. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21307-12
65. Qi J, You T, Li J, Pan T, Xiang L, Han Y. et al. Circulating trimethylamine N-oxide and the risk of cardiovascular diseases: a systematic review and meta-analysis of 11 prospective cohort studies. Journal of cellular and molecular medicine. 2018;22:185-94
66. Schiattarella G, Sannino A, Toscano E, Giugliano G, Gargiulo G, Franzone A. et al. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. European heart journal. 2017;38:2948-56
67. Meyer K, Benton T, Bennett B, Jacobs D, Lloyd-Jones D, Gross M. et al. Microbiota-Dependent Metabolite Trimethylamine N-Oxide and Coronary Artery Calcium in the Coronary Artery Risk Development in Young Adults Study (CARDIA). Journal of the American Heart Association. 2016 5
68. Collins H, Drazul-Schrader D, Sulpizio A, Koster P, Williamson Y, Adelman S. et al. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE(-/-) transgenic mice expressing CETP. Atherosclerosis. 2016;244:29-37
Author contact
![]() Corresponding authors: Jun Tao, Department of Cardiovascular Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China. Email: taoj8sysu.edu.cn. Liling Lin, Department of Anesthesiology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China. Email: Linll3sysu.edu.cn. Junmeng Zheng, Department of Cardiovascular Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China. Email: zhengjm27sysu.edu.cn.
Corresponding authors: Jun Tao, Department of Cardiovascular Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China. Email: taoj8sysu.edu.cn. Liling Lin, Department of Anesthesiology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China. Email: Linll3sysu.edu.cn. Junmeng Zheng, Department of Cardiovascular Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China. Email: zhengjm27sysu.edu.cn.