Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. The Energy Metabolism of...
2. The Relationship Between...
3. Conclusion and Prospect
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2024; 21(2):369-375. doi:10.7150/ijms.89370 This issue Cite
Review
NAD+-A Hub of Energy Metabolism in Heart Failure
Yaoxin Wu1, Zuowei Pei1,2 ![]() , Peng Qu1
, Peng Qu1 ![]()
1. Faculty of Medicine, Dalian University of Technology, 116024, Dalian, China.
2. Department of Cardiology, Central Hospital of Dalian University of Technology, Dalian, 116033, China.
Received 2023-8-22; Accepted 2023-11-27; Published 2024-1-1
Abstract

Heart failure is a condition where reduced levels of adenosine triphosphate (ATP) affect energy supply in myocardial cells. Nicotinamide adenine dinucleotide (NAD+) plays a crucial role as a coenzyme for electron transfer in energy metabolism. Decreased NAD+ levels in myocardial cells lead to inadequate ATP production and increased susceptibility to heart failure. Researchers are exploring ways to increase NAD+ levels to alleviate heart failure. Targets such as sirtuin2 (sirt2), sirtuin3 (sirt3), Poly (ADP-ribose) polymerase (PARP), and diastolic regulatory proteins are being investigated. NAD+ supplementation has shown promise, even in heart failure with preserved ejection fraction (HFpEF). By focusing on NAD+ as a central component of energy metabolism, it is possible to improve myocardial activity, heart function, and address energy deficiency in heart failure.
Keywords: NAD+, Energy Metabolism, Heart Failure, Mitochondria
1. The Energy Metabolism of Myocardial Cells and Heart Failure
1.1. Energy Metabolism in the Heart
The heart is one of the important metabolic organs in the human body, which promotes the blood circulation of the whole body through its relaxation and contraction. Like other cells, the source of energy for healthy human myocardial cells is ATP. However, for myocardial cells, fatty acids are the primary substrate for ATP production in myocardial cells, contributing 50%-70% of the ATP demands [1], other substrates such as lactate, ketone bodies, glucose, and branched-chain amino acids (BCAAs) are utilized by myocardial cells for ATP production as well [2]. Glucose is also an important energy material for the heart. In myocardial cells, a small amount of ATP and pyruvic acid can be produced by cytoplasmic glycolysis, and pyruvic acid enters the mitochondria to produce more ATP. Under the condition of increased circulating lactic acid levels, lactic acid could also be used as an important energy substrate for the heart. The heart takes up lactic acid through the monocarboxylic acid anion transporter (MCT4) and converts it into pyruvic acid using lactate dehydrogenase (LDH); the pyruvic acid is then utilized in mitochondrial metabolism to produce ATP [3, 4]. Ketone bodies are easily metabolized by the heart. When the level of ketone bodies increases in the circulation, the heart switches to using ketone bodies as its primary fuel source [5], this process is achieved by regulating the phosphorylation level in the heart and the concentration of mitochondrial NADH [6]. A growing body of research supports the notion that ketone bodies play a significant role as energy substrates for the heart [7, 8]. Amino acid oxidation is considered a significant potential source of ATP production in myocardial cells, with branched-chain amino acids (BCAAs) being particularly optimal in this regard [9]. Therefore, in terms of energy metabolism, the heart has extremely high flexibility, which can flexibly select among different substrates under different conditions to ensure adequate ATP supply, thereby providing sufficient conditions for the heart to complete its functions.
1.2. Energy Metabolism of Myocardial Cells During Heart Failure
Heart failure (HF) refers to a disease of heart circulation disorder caused by heart systolic and/or diastolic dysfunction, resulting in blood stasis in the venous system and insufficient blood perfusion in the arterial system and massive venous return to the heart. It is generally divided into heart failure with reduced ejection fraction (HFrPH), HFpEF and heart failure with mid-range ejection fraction (HFmrEF) according to left ventricular ejection fraction [10]. HFpEF represents approximately 50% of all cases of heart failure, and its morbidity and mortality rates are comparable to those of heart failure with reduced ejection fraction, but the underlying mechanisms and pathogenesis of HFpEF are not yet well understood [11, 12]. For heart failure, on the one hand, it loses metabolic activity, and on the other hand, it becomes energy deficient due to the decreased ability to produce ATP. Existing studies have shown that the above energy deficiency may be due to the decreased mitochondrial oxidation ability in heart failure. The common causes of mitochondrial injury in heart failure include the following: (1) Increased production of reactive oxygen species (ROS) and disruption of mitochondrial calcium (Ca2+) homeostasis; (2) Mitochondrial kinetic injury, persistent mitosis, autophagy of myocardial cells, and elevated cell death; (3) Alterations in the transcriptional regulation of mitochondrial proteins and increased post-translational protein modifications [2]. Although various abnormalities related to calcium homeostasis, contractile protein function, energy metabolism, cytoskeletal rearrangements, and loss of viable muscle cells have been observed, the fundamental molecular and cellular mechanisms underlying the pathogenesis of heart failure are not yet well understood [13]. It should also be noted that, as mentioned above, myocardial cells have great flexibility in selecting substrates for energy metabolism. Studies have shown that in the early stage of HF, the selection of myocardial substrates is relatively normal; However, in the late stage, fatty acid oxidation is down-regulated, glycolysis and glucose oxidation are increased, respiratory chain activity is reduced, and mitochondrial oxidation flux reserve is damaged [14], which are the reasons for insufficient myocardial energy supply during HF.
1.3. NAD(H) of Energy Metabolism in Heart Failure
Intracellular NAD exists in both oxidized form (NAD+) and reduced form (NADH), and it serves as the primary electron carrier coenzyme during energy metabolism processes such as glycolysis, oxidative phosphorylation, and the tricarboxylic acid cycle (TCA) [15]. Studies have demonstrated that reductions in NAD or NAD+/NADH levels are observed in heart failure [16, 17]. Additionally, relevant studies have confirmed the beneficial effects of providing NAD+ precursors in clinical patients with heart failure as well as in preclinical models [18]. Numerous studies indicate that the metabolic level of NAD+ in heart failure is associated with sirtuins, PARP (poly (ADP-ribose) polymerase), and cyclic ADP-ribose synthase (CD38) [19, 20]. These enzymes use NAD+ as metabolic substrate in heart failure. Increasing the level of NAD+ has emerged as a new research direction for alleviating and treating heart failure. Two key issues in this area include: (1) understanding the impact of low heart NAD+ levels on the pathogenesis of heart failure, and (2) elucidating the mechanisms by which elevated NAD+ levels provide benefits in heart failure treatment.
1.3.1. Research Status of NAD+
Increasing NAD+ levels is considered a promising strategy for treating heart failure, although the mechanistic details may vary. Various compounds, including NAD+ precursors or nicotinamide phosphotransferase (Nampt) activators, have shown potential in this regard [21]. However, there is currently a lack of information regarding the pharmacokinetics and drug tolerance of these compounds in patients. Diguet et al. developed and published the dynamics of NAD+ metabolomics in human subjects after administering a single dose of Nicotinamide ribonucleoside (NR) [22]. Another study investigated the pharmacokinetics of NR and its impact on blood NAD+ levels in healthy volunteers over a nine-day treatment period [23]. The above research undoubtedly made great progress in the treatment of heart failure with NAD+, but the current research has always had limitations. One point is that the current detection method is to measure the levels of NAD+ in blood, and the changes of NAD+ levels in heart and other metabolic conditions cannot be determined by NAD+ levels in blood alone. While many preclinical studies have taken a precautionary approach by inducing heart failure and expanding the NAD+ pool simultaneously, for clinical applications, it is more appropriate to treat individuals who already have heart dysfunction [21]. The focus shifts to administering NAD+-boosting therapies to subjects with existing heart dysfunction for potential clinical benefits.
2. The Relationship Between NAD+ and Heart Failure
2.1. Role of NAD+ Consumption and Synthesis Pathway in the Heart
It is well known that the intracellular NAD pool consists of two forms, the oxidized form (NAD+) and the reduced form (NADH), which will carry electrons from substrate decomposition for oxidative phosphorylation and biosynthesis. Intracellular NAD+ serves as a synergistic substrate for PARPs, sirtuins, and CD38, and during these enzymatic reactions, nicotinamide (NAM) is produced [24]. PARP (poly (ADP-ribose) polymerase) is a multifunctional DNA-binding enzyme found in various nuclei, including myocardial cells [25]. It can be activated by a single-strand DNA cleavage reaction, which may result from oxidative cell injury and the presence of free radicals. Mild activation of PARP (poly (ADP-ribose) polymerase) under physiological conditions regulates several cellular processes including DNA repair, gene expression, cell cycle progression, cell survival, chromatin remodeling, and maintenance of genomic stability [26]. However, excessive activation of PARP (poly (ADP-ribose) polymerase) leads to the depletion of cellular NAD+ content [7]. This, in turn, inhibits other NAD+-dependent cellular processes. Sirtuins are a crucial class of deacetylating proteins involved in numerous physiological processes. There are several types of sirtuins, including sirtuin 1-7 (sirt1-7), each playing distinct roles in cellular regulation [27]. Endogenous sirtuin 1 (sirt1) plays a critical role in mediating cell death an [28]. Sirtuin 3 (Sirt3) protects myocardial cells from aging, oxidative stress, and inhibits heart hypertrophy [29]. Sirt1 regulates mitochondrial function through acetylated nuclear proteins, while sirt3 regulates mitochondrial function through acetylated mitochondrial proteins [30]. Eukaryotes typically synthesize NAD+ through the ab initio pathway using tryptophan or salvage it from NAM. In the salvage pathway, NAM is converted to nicotinamide mononucleotide (NMN) by the enzyme Nampt. Additionally, phosphorylation of nicotinamide ribonucleoside kinase (Nrk) can generate NMN from NR [31].
Biosynthesis and consumption pathway of NAD+.
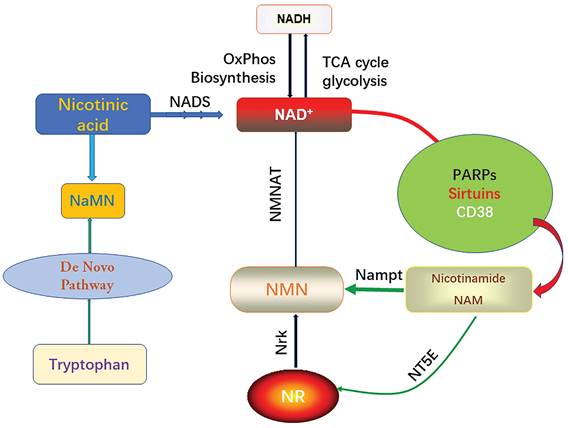
2.2 Heart Changes Caused by Reduced NAD+ Levels
In cell metabolism, the activities of acyl-coenzyme-A dehydrogenase and 3-hydroxyacyl coenzyme-A dehydrogenase are influenced by flavin adenine dinucleotide (FAD) / reduced flavin adenine dinucleotide (FADH2) and the ratio of NAD+/NADH, respectively. These factors, in turn, impact oxidative phosphorylation and ATP synthesis. Based on existing research, both human and HFpEF rat studies have shown preserved expression of Nampt in the heart [2]. This suggests that the decrease in NAD+ content may be due to increased NAD+ consumption or a low level of substrate nicotinamide available for Nampt. In SRFHKO mice with heart failure, the NMRk2 pathway is activated. This activation leads to an increase in the expression of NT5E, which is the extracellular enzyme responsible for hydrolyzing extracellular NAD+ and NMN to Nicotinamide nucleoside (NR) [27]. In addition, in the tissue biopsy of heart failure, since the increase of active oxygen and oxygen free radicals, the expression and activity of PARP1 are increased. On the one hand, PARP1 can degrade its substrate NAD+ leading to a reduction in NAD+ levels. On the other hand, PARP1 inhibits the phosphoinositide 3 (PI3)-kinase-Akt signaling pathway, which hampers Akt's ability to inhibit muscle cell death-promoting enzymes such as c-Jun N-terminal kinase (JNK) and P38. Conversely, inhibiting PARP-1 can enhance Akt activation, thereby mitigating heart injury, reducing fibrosis, and protecting heart function [32]. Studies have found that the reduced level of NAD+ in cell is actually the cause of PARP-mediated cardiomyocyte death. Reduced activity of the sirtuin isoform, sirt2 deacetylase, contributes to PARP-mediated myocyte death. However, activation of this enzyme can prevent PARP-mediated NAD+ depletion and subsequent cell injury [33].
2.3 Improvement of Heart Failure and its Mechanisms by Increasing NAD+ Levels
As mentioned above, ATP is in short supply in heart failure, and the key reason for ATP deficiency is the low level of NAD+. Therefore, supplementing and increasing NAD+ level has become a feasible scheme to treat heart failure and alleviate heart injury. As a direct precursor in the synthesis of NAD+, NMN plays a direct role in improving the level of NAD+. A study using KFL4 deficient mice (with decreased NAD+ level) shows that NMN can protect the ultrastructure of mitochondria, reduce reactive oxygen species, prevent the death of heart cells, and increase the oxidation of long-chain fatty acids, thereby increasing the NAD+ level [34]. Another experiment using p32cKO mice as a heart failure model shows that NMN can also alleviate heart failure by improving the function of lysosomes and reducing autophagy [35]. NMN has the same effect on HFpEF. Studies have shown that oral supplementation of its precursor nicotinamide to increase NAD+ can improve diastolic dysfunction induced by aging (in 2-year-old C57BL/6J mice), hypertension (in Dahl salt-sensitive rats) or cardiac metabolic syndrome (in ZSF1 obese rats) [36]. Another study on Nampt shows that as a rate-limiting enzyme for synthesizing NAD+ from NMN, its activity is very important for the remediation of NAD+ through NMN [37]. The latest clinical study on NMN can directly show that NMN can increase the level of NAD+ and help restore energy metabolism [38]. Another clinical study also shows that oral NMN is a safe and practical strategy to improve the level of NAD+ in human body [39]. Due to the reaction that NR is phosphorylated by Nrk to produce NMN, NR supplementation has been mainly used in the existing studies [40]. NR increases the levels of heart nicotinic acid adenine dinucleotide, a sensitive biomarker of increased NAD+ metabolism, and methyl NAM (MeNAM), which is oxidized by aldehyde oxidase (AOX1) to N1- methyl -4- pyridone -5- carboxamide (Me4PY) and releases hydrogen peroxide [41]. NR has the potential to improve both congenital and acquired heart failure [42]. Furthermore, NR may also have systemic effects, particularly in liver metabolism, insulin antagonism, and skeletal muscle function, which can be altered in conjunction with heart defects [43]. When NR treatment is performed, the induction level of Myh7 in SRFHKO mice is limited, and Myh7 induction is a signal of heart stress and metabolic remodeling in heart failure [44]. NR also increases mitochondrial citrate synthase (CS) and TP- citrate lyase (ACL) activity, leading to increased nucleoprotein acetylation (Acetyl coenzyme A precursor citrate
citrate Acetyl coenzyme A) [45]. Due to the reduced concentration of NAD+ in muscle cells, PARP1 can attenuate the activity of sirts deacetylase in muscle cells. On the contrary, overexpression of sirt1 by increasing the level of NAD+ prevents PARP-1-induced depletion of NAD+, while the deacetylation of sirts inhibits the effects of P53 and Ku70 [46]. In studies conducted on the heart, analysis of acetylated proteins revealed that supplementation with nicotinamide resulted in the deacetylation of two diastolic regulatory proteins, titin and SERCA2a [47]. SERCA2a is a direct substrate for sirt1 and P300 (histone acetyltransferase P300). sirt1 is activated by the metabolic activator β-lapachone (β-lap) of sirt1, which significantly reduces acetylation and recovers the function of SERCEAA, thereby alleviating heart failure. In addition, as mentioned above, ROS are increased in heart failure, while supplementation of NAD+ by NR has been shown to reduce mtROS of different tissue types [48-52].
Acetyl coenzyme A) [45]. Due to the reduced concentration of NAD+ in muscle cells, PARP1 can attenuate the activity of sirts deacetylase in muscle cells. On the contrary, overexpression of sirt1 by increasing the level of NAD+ prevents PARP-1-induced depletion of NAD+, while the deacetylation of sirts inhibits the effects of P53 and Ku70 [46]. In studies conducted on the heart, analysis of acetylated proteins revealed that supplementation with nicotinamide resulted in the deacetylation of two diastolic regulatory proteins, titin and SERCA2a [47]. SERCA2a is a direct substrate for sirt1 and P300 (histone acetyltransferase P300). sirt1 is activated by the metabolic activator β-lapachone (β-lap) of sirt1, which significantly reduces acetylation and recovers the function of SERCEAA, thereby alleviating heart failure. In addition, as mentioned above, ROS are increased in heart failure, while supplementation of NAD+ by NR has been shown to reduce mtROS of different tissue types [48-52].
Although there are few studies on HFpEF at present, it is still feasible to treat HFpEF by increasing NAD+. On the one hand, NR has the ability to directly enhance the passive stiffness of myocardial cells and improve active calcium-dependent relaxation. This is achieved by increasing the deacetylation of titin, which impacts passive stiffness, and the sarcoplasmic reticulum calcium-ATPase 2a (SERCA2a) enzyme, which influences active relaxation [53]. On the other hand, sirt3 is related to ATP synthase under normal mitochondrial membrane potential, but it is released to activate the TCA when the mitochondrial membrane potential is reduced. However, the positive feedback loop needs sirt3 to activate NMNAT3 [54]. Therefore, increasing the NAD+ level can increase sirt3 activity and alleviate the hyperacetylation of the related mitochondrial enzymes. Studies have found that the increased acetylation of very-long chain acyl-CoA dehydrogenase deficiency (VLCAD) and hydratase subunit A (HADHA) can decrease the fatty acid oxidation (FAO) of HFpEF-derived myocardial cells, and the supplementation of NAD+ can effectively reduce the FAO defect [10]. In another study on HFpEF, it was found that NR supplementation can normalize NAD+/NADH ratio, down-regulate acetylation level, improve mitochondrial function and improve HFpEF phenotype [55]. In addition to NR, NAM can also increase the level of NAD+. It has been found that NAM can improve the systemic HFpEF risks of ZSF1 obese rats, including obesity and hypertension. Similarly, NAM may also improve diastolic function through the deacetylation of titin and SERCA2a. Titin improve the passive stiffness of myocardial cells while SERCA 2a improve the dependent active relaxation [36].
In addition to promoting the synthesis of NAD+ by increasing the precursor of NAD+, the existing research shows that this goal can be achieved by directly using NAD+. In a study on the regulation of NAD+ on neuroinflammation, the level of NAD+ can also be increased by direct intraperitoneal injection of NAD+ instead of its precursor [56]. In another study on the mouse model with high-fat diet, the researchers also adopted the experimental method of intravenous injection. The experimental results showed that intravenous injection of NAD+ could save the cells in hypothalamic neurons from NAD+ depletion and its induced inhibition of PER1 transcription activity [57]. Another study on the energy supply system of left and right ventricular myocardium after using intravenous injection of NAD+ to restore short-term circulatory disorder shows that intravenous injection of NAD+ is helpful to oxidative phosphorylation of myocardial cells [58]. However, it should be noted that the structure of NAD+ itself is unstable and it cannot be taken orally, so the direct injection of NAD+ is still under study, especially about HF.
Relevant mechanisms of increasing NAD+ levels alleviating heart failure. In mitochondria, NR increased the activities of CS and ACL, increased CoA, and acetylated nucleoprotein. NR limited induction levels of Myh7; NR increases the NAD+ level to inhibit the activity of PARP1, and increases the activity of sirt1 and sirt3, which in turn inhibits P53 and Ku70, to deacetylate titin and SERCA2α, two relaxation regulatory proteins. The increase in NAD+ level also decreases mitochondrial ROS. NR, nicotinamide ribonucleoside; CS, mitochondrial citrate synthase; ACL, TP- citrate lyase; CoA, acetyl-coA, mtROS, mitochondrial reactive oxygen species; PARP, poly (ADP- ribose) polymerase.
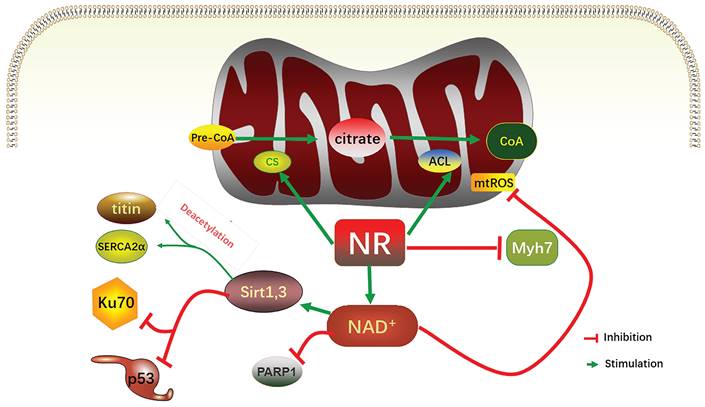
3. Conclusion and Prospect
Although the mechanism of changes about NAD(H) levels in heart failure is not fully understood, the action targets for increasing NAD+ levels to treat heart failure are not clear. On the basis of existing studies, such as involving relevant mechanisms as sirts, PARPs, troponin, and contraction/relaxation protein, as well as supplementing information on pharmacokinetics and drug toxicology, NAD+ as a therapeutic direction for heart failure will further prove to be completely feasible. In conclusion, the NAD+ therapy is an effective idea for treating heart failure and alleviating heart injury. It is believed that with further research and further supplement of relevant information, the mechanism and efficacy under relevant scenarios will be better understood.
Acknowledgements
This work was supported by Liaoning Province Applied Basic Research Program (grant number: 2023JH2/101300098).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ. et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation. 2016;133:706-16
2. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac Energy Metabolism in Heart Failure. Circ Res. 2021;128:1487-513
3. Lee S, Choi Y, Jeong E, Park J, Kim J, Tanaka M. et al. Physiological significance of elevated levels of lactate by exercise training in the brain and body. J Biosci Bioeng. 2023;135:167-75
4. Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC. et al. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science. 2020;370:364-8
5. Ho KL, Karwi QG, Wagg C, Zhang L, Vo K, Altamimi T. et al. Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc Res. 2021;117:1178-87
6. Kim DK, Heineman FW, Balaban RS. Effects of beta-hydroxybutyrate on oxidative metabolism and phosphorylation potential in canine heart in vivo. Am J Physiol. 1991;260:H1767-73
7. Puchalska P, Crawford PA. Metabolic and Signaling Roles of Ketone Bodies in Health and Disease. Annu Rev Nutr. 2021;41:49-77
8. Karwi QG, Biswas D, Pulinilkunnil T, Lopaschuk GD. Myocardial Ketones Metabolism in Heart Failure. J Card Fail. 2020;26:998-1005
9. Fillmore N, Wagg CS, Zhang L, Fukushima A, Lopaschuk GD. Cardiac branched-chain amino acid oxidation is reduced during insulin resistance in the heart. Am J Physiol Endocrinol Metab. 2018;315:E1046-e52
10. Tong D, Schiattarella GG, Jiang N, Altamirano F, Szweda PA, Elnwasany A. et al. NAD(+) Repletion Reverses Heart Failure With Preserved Ejection Fraction. Circ Res. 2021;128:1629-41
11. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2017;14:591-602
12. Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA. et al. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation. 2016;134:73-90
13. Strømland Ø, Diab J, Ferrario E, Sverkeli LJ, Ziegler M. The balance between NAD(+) biosynthesis and consumption in ageing. Mech Ageing Dev. 2021;199:111569
14. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093-129
15. Cantó C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31-53
16. Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA. et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation. 2016;134:883-94
17. Yoshino J, Baur JA, Imai SI. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018;27:513-28
18. Zhou B, Wang DD, Qiu Y, Airhart S, Liu Y, Stempien-Otero A. et al. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J Clin Invest. 2020;130:6054-63
19. Verdin E. NAD⁺ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208-13
20. Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB. et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016 2
21. Walker MA, Tian R. Raising NAD in Heart Failure: Time to Translate? Circulation. 2018;137:2274-7
22. Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW. et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948
23. Airhart SE, Shireman LM, Risler LJ, Anderson GD, Nagana Gowda GA, Raftery D. et al. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One. 2017;12:e0186459
24. Zapata-Pérez R, Wanders RJA, van Karnebeek CDM, Houtkooper RH. NAD(+) homeostasis in human health and disease. EMBO Mol Med. 2021;13:e13943
25. Mashimo M, Kita M, Nobeyama A, Nomura A, Fujii T. PARP1 is activated by membrane damage and is involved in membrane repair through poly(ADP-ribosyl)ation. Genes Cells. 2022;27:305-12
26. Tiwari P, Khan H, Singh TG, Grewal AK. Poly (ADP-ribose) polymerase: An Overview of Mechanistic Approaches and Therapeutic Opportunities in the Management of Stroke. Neurochem Res. 2022;47:1830-52
27. Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N. et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation. 2018;137:2256-73
28. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
29. Shi X, Pi L, Zhou S, Li X, Min F, Wang S. et al. Activation of Sirtuin 1 Attenuates High Glucose-Induced Neuronal Apoptosis by Deacetylating p53. Front Endocrinol (Lausanne). 2018;9:274
30. Tomczyk MM, Cheung KG, Xiang B, Tamanna N, Fonseca Teixeira AL, Agarwal P. et al. Mitochondrial Sirtuin-3 (SIRT3) Prevents Doxorubicin-Induced Dilated Cardiomyopathy by Modulating Protein Acetylation and Oxidative Stress. Circ Heart Fail. 2022;15:e008547
31. Chen J, Chen S, Zhang B, Liu J. SIRT3 as a potential therapeutic target for heart failure. Pharmacol Res. 2021;165:105432
32. Zhou S, Dai Z, Wang L, Gao X, Yang L, Wang Z. et al. MET inhibition enhances PARP inhibitor efficacy in castration-resistant prostate cancer by suppressing the ATM/ATR and PI3K/AKT pathways. J Cell Mol Med. 2021;25:11157-69
33. Maugeri A, Russo C, Musumeci L, Lombardo GE, De Sarro G, Barreca D. et al. The Anticancer Effect of a Flavonoid-Rich Extract of Bergamot Juice in THP-1 Cells Engages the SIRT2/AKT/p53 Pathway. Pharmaceutics. 2022 14
34. Zhang R, Shen Y, Zhou L, Sangwung P, Fujioka H, Zhang L. et al. Short-term administration of Nicotinamide Mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J Mol Cell Cardiol. 2017;112:64-73
35. Yagi M, Do Y, Hirai H, Miki K, Toshima T, Fukahori Y. et al. Improving lysosomal ferroptosis with NMN administration protects against heart failure. Life Sci Alliance. 2023 6
36. Abdellatif M, Trummer-Herbst V, Koser F, Durand S, Adão R, Vasques-Nóvoa F. et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci Transl Med. 2021 13
37. Yano M, Akazawa H, Oka T, Yabumoto C, Kudo-Sakamoto Y, Kamo T. et al. Monocyte-derived extracellular Nampt-dependent biosynthesis of NAD(+) protects the heart against pressure overload. Sci Rep. 2015;5:15857
38. Yi L, Maier AB, Tao R, Lin Z, Vaidya A, Pendse S. et al. The efficacy and safety of β-nicotinamide mononucleotide (NMN) supplementation in healthy middle-aged adults: a randomized, multicenter, double-blind, placebo-controlled, parallel-group, dose-dependent clinical trial. Geroscience. 2023;45:29-43
39. Okabe K, Yaku K, Uchida Y, Fukamizu Y, Sato T, Sakurai T. et al. Oral Administration of Nicotinamide Mononucleotide Is Safe and Efficiently Increases Blood Nicotinamide Adenine Dinucleotide Levels in Healthy Subjects. Front Nutr. 2022;9:868640
40. Wang YJ, Paneni F, Stein S, Matter CM. Modulating Sirtuin Biology and Nicotinamide Adenine Diphosphate Metabolism in Cardiovascular Disease-From Bench to Bedside. Front Physiol. 2021;12:755060
41. Sung JW, Yun HY, Park S, Kim YJ, Yee J, Lee KE. et al. Population Pharmacokinetics of Sulindac and Genetic Polymorphisms of FMO3 and AOX1 in Women with Preterm Labor. Pharm Res. 2020;37:44
42. Wang DD, Airhart SE, Zhou B, Shireman LM, Jiang S, Melendez Rodriguez C. et al. Safety and Tolerability of Nicotinamide Riboside in Heart Failure With Reduced Ejection Fraction. JACC Basic Transl Sci. 2022;7:1183-96
43. Sharma C, Donu D, Cen Y. Emerging Role of Nicotinamide Riboside in Health and Diseases. Nutrients. 2022 14
44. Blottner D, Capitanio D, Trautmann G, Furlan S, Gambara G, Moriggi M. et al. Nitrosative Redox Homeostasis and Antioxidant Response Defense in Disused Vastus lateralis Muscle in Long-Term Bedrest (Toulouse Cocktail Study). Antioxidants (Basel). 2021 10
45. Park JS, Holloszy JO, Kim K, Koh JH. Exercise Training-Induced PPARβ Increases PGC-1α Protein Stability and Improves Insulin-Induced Glucose Uptake in Rodent Muscles. Nutrients. 2020 12
46. Henning RJ, Bourgeois M, Harbison RD. Poly(ADP-ribose) Polymerase (PARP) and PARP Inhibitors: Mechanisms of Action and Role in Cardiovascular Disorders. Cardiovasc Toxicol. 2018;18:493-506
47. Chen HX, Wang XC, Hou HT, Wang J, Yang Q, Chen YL. et al. Lysine crotonylation of SERCA2a correlates to cardiac dysfunction and arrhythmia in Sirt1 cardiac-specific knockout mice. Int J Biol Macromol. 2023;242:125151
48. Traba J, Kwarteng-Siaw M, Okoli TC, Li J, Huffstutler RD, Bray A. et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest. 2015;125:4592-600
49. Hong G, Zheng D, Zhang L, Ni R, Wang G, Fan GC. et al. Administration of nicotinamide riboside prevents oxidative stress and organ injury in sepsis. Free Radic Biol Med. 2018;123:125-37
50. Massudi H, Grant R, Guillemin GJ, Braidy N. NAD+ metabolism and oxidative stress: the golden nucleotide on a crown of thorns. Redox Rep. 2012;17:28-46
51. Schöndorf DC, Ivanyuk D, Baden P, Sanchez-Martinez A, De Cicco S, Yu C. et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson's Disease. Cell Rep. 2018;23:2976-88
52. Zhang Q, Liu X, Li N, Zhang J, Yang J, Bu P. Sirtuin 3 deficiency aggravates contrast-induced acute kidney injury. J Transl Med. 2018;16:313
53. Akar FG, Young LH. NAD Repletion Therapy: A Silver Bullet for HFpEF? Circ Res. 2021;128:1642-5
54. Matsushima S, Sadoshima J. The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol. 2015;309:H1375-89
55. Liu X, Zhang Y, Deng Y, Yang L, Ou W, Xie M. et al. Mitochondrial protein hyperacetylation underpins heart failure with preserved ejection fraction in mice. J Mol Cell Cardiol. 2022;165:76-85
56. Zhao Y, Zhang J, Zheng Y, Zhang Y, Zhang XJ, Wang H. et al. NAD(+) improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J Neuroinflammation. 2021;18:207
57. Roh E, Myoung Kang G, Young Gil S, Hee Lee C, Kim S, Hong D. et al. Effects of Chronic NAD Supplementation on Energy Metabolism and Diurnal Rhythm in Obese Mice. Obesity (Silver Spring). 2018;26:1448-56
58. Sukoyan GV, Andriadze NA, Guchua EI, Karsanov NV. Effect of NAD on recovery of adenine nucleotide pool, phosphorylation potential, and stimulation of apoptosis during late period of reperfusion damage to myocardium. Bull Exp Biol Med. 2005;139:46-9
Author contact
![]() Corresponding authors: Peng Qu, Faculty of Medicine, Dalian University of Technology, 116024, Dalian, China. Telephone number: 0411-84706670. E-mail: qupengedu.cn. Zuowei Pei, Department of Cardiology, Central Hospital of Dalian University of Technology, No.826 Xinan Road, 116033, Dalian, China. Telephone number: +86-0411-84412001, Fax number: +86-0411-84412001. E-mail: pzw_dlcom.
Corresponding authors: Peng Qu, Faculty of Medicine, Dalian University of Technology, 116024, Dalian, China. Telephone number: 0411-84706670. E-mail: qupengedu.cn. Zuowei Pei, Department of Cardiology, Central Hospital of Dalian University of Technology, No.826 Xinan Road, 116033, Dalian, China. Telephone number: +86-0411-84412001, Fax number: +86-0411-84412001. E-mail: pzw_dlcom.