Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Cholesterol increases...
Cholesterol affects...
HDL and SR-B1 play a role in the...
Inspiration for treatment
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(15):3533-3543. doi:10.7150/ijms.62021 This issue Cite
Review
Possible mechanisms of cholesterol elevation aggravating COVID-19
Yan Tang, UGS1,5#, Longtai Hu, UGS1,4#, Yi Liu, UGS1,5, Bangyi Zhou, UGS1,5, Xiaohuan Qin, UGS1,5, Jujian Ye, UGS1,5, Maoze Shen, MD2, Zhijian Wu, MD3 ![]() , Peidong Zhang, MD1
, Peidong Zhang, MD1 ![]()
1. Department of Cardiology, Heart Center, Zhujiang Hospital, Southern Medical University, 235 Industrial Avenue, Guangzhou, 510282, Guangdong, People's Republic of China.
2. Department of Cardiology, Raoping County People's Hospital, 161 Caichang Street, Huanggang Town, Chaozhou, 515700, Guangdong, People's Republic of China.
3. Department of Cardiology, Affiliated Boai Hospital of Zhongshan, Southern Medical University, No. 6, Chenggui Road, East District, Zhongshan, 528403, Guangdong, People's Republic of China.
4. School of Traditional Chinese Medicine, Southern Medical University, No. 6, Chenggui Road, East District, Zhongshan, 528403, Guangdong, People's Republic of China.
5. Zhujiang Hospital, Southern Medical University/The Second School of Clinical Medicine, Southern Medical University, No. 6, Chenggui Road, East District, Zhongshan, 528403, Guangdong, People's Republic of China.
UGS: undergraduate students; MD: medical doctor.
#These authors contributed equally to this work.
Received 2021-4-25; Accepted 2021-8-4; Published 2021-8-21
Abstract

Importance: Despite the availability of a vaccine against the severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2), humans will have to live with this virus and the after-effects of the coronavirus disease 2019 (COVID-19) infection for a long time. Cholesterol plays an important role in the infection and prognosis of SARS-CoV-2, and the study of its mechanism is of great significance not only for the treatment of COVID-19 but also for research on generic antiviral drugs.
Observations: Cholesterol promotes the development of atherosclerosis by activating NLR family pyrin domain containing 3 (NLRP3), and the resulting inflammatory environment indirectly contributes to COVID-19 infection and subsequent deterioration. In in vitro studies, membrane cholesterol increased the number of viral entry sites on the host cell membrane and the number of angiotensin-converting enzyme 2 (ACE2) receptors in the membrane fusion site. Previous studies have shown that the fusion protein of the virus interacts with cholesterol, and the spike protein of SARS-CoV-2 also requires cholesterol to enter the host cells. Cholesterol in blood interacts with the spike protein to promote the entry of spike cells, wherein the scavenger receptor class B type 1 (SR-B1) plays an important role. Because of the cardiovascular protective effects of lipid-lowering therapy and the additional anti-inflammatory effects of lipid-lowering drugs, it is currently recommended to continue lipid-lowering therapy for patients with COVID-19, but the safety of extremely low LDL-C is questionable.
Conclusions and Relevance: Cholesterol can indirectly increase the susceptibility of patients to SARS-CoV-2 and increase the risk of death from COVID-19, which are mediated by NLRP3 and atherosclerotic plaques, respectively. Cholesterol present in the host cell membrane, virus, and blood may also directly participate in the virus cell entry process, but the specific mechanism still needs further study. Patients with COVID-19 are recommended to continue lipid-lowering therapy.
Keywords: cholesterol, LDL-C, COVID-19, membrane fusion, lipid-lowering drugs
Introduction
The ongoing coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) poses a huge challenge to the global medical system [1]. According to WHO, as of 16:22 (Central European Time) on February 18, 2021, the worldwide confirmed cases of COVID-19 were 109,594,835, including 2,424,060 deaths. Although COVID-19 vaccine research has rapidly advanced in the past few months, a number of scientific concerns such as optimization of vaccination regimens, enhanced doses, relevance of protection, vaccine effectiveness, and safety and enhanced surveillance still need addressing to improve vaccine efficacy [2]. It appears that SARS-CoV-2 will continue to coexist with humans for a long time. Therefore, further research into the mechanisms of blocking viruses from infecting host cells is critically important.
Studies have shown that patients with underlying cardiovascular disease (CVD) are more susceptible to develop severe COVID-19 or even die from it [3]. Cholesterol is one of the risk factors for CVD, and CVD may reflect the indirect mechanism between elevated cholesterol and increased susceptibility to COVID-19. In addition, cholesterol is directly related to the infection mechanism of coronavirus, via membrane fusion, endocytosis, migration, and other processes. In this article, we review both the indirect and direct mechanisms of cholesterol elevation that seem to aggravate COVID-19, in which we find that the link between cholesterol and cell entry [4], and the pathways of viral entry into cells being interesting subjects for targeted therapy of SARS‐CoV‐2 [5].
Cholesterol increases susceptibility to COVID-19 by causing atherosclerosis
Increased cholesterol levels are associated with high CVD risk in both old [6] and young [7] adults, and a confirmed diagnosis of CVD is a prognostic factor for both mortality and severity in COVID‑19 [8]. Therefore, elevated cholesterol may indirectly aggravate the infection severity in COVID-19, and possibly even mortality, by causing cardiovascular disease.
Inflammation may play an important role in CVD and COVID-19
Cardiovascular disease is an umbrella term for conditions that affect the heart or blood vessels. The Framingham Heart Study is one of the original epidemiological observations of the relationship between elevated blood cholesterol levels and future atherosclerotic cardiovascular disease (ASCVD) [9]. Cholesterol levels play an important role in the development and regression of atherosclerosis. A hallmark of ASCVD is the accumulation of cholesterol in arterial macrophages [10]. During the development of atherosclerotic plaques, cholesterol content positively correlates with markers of plaque vulnerability, and negatively correlates with stability markers [11]. To further clarify the process of plaque rupture and/or erosion, which is considered a major cause of cardiovascular events, a study showed that during cholesterol crystallization, the volume of the plaque increases rapidly and the sharp crystals pass through the biofilm of vascular epithelial cells in their path [12]. Saturation of free cholesterol can result in cholesterol crystallization, which is proven to be able to induce inflammation and instability of atherosclerotic plaques in arteries [13]. Cholesterol crystals (CCs) or oxidized low-density lipoprotein (ox-LDL) can be engulfed by macrophages to form CCs in the phagolysosome [14]. Recent studies have shown that free cholesterol may form metastable structures that evolve into persistent flat plate CCs and are prone to inflammation and traumatic injury [15].
An autopsy study showed a higher density of macrophages and dendritic cells in atherosclerotic plaques in patients with systemic infection; greater infiltration of macrophages and T-cell lymphocytes in the adventitia of coronary arteries; and more periadventitial fat in deceased patients than those who did not die from infection [16]. SARS-CoV-2 itself is an infectious factor, which, like other infectious factors, increases plaque instability, thereby increasing the risk of cardiovascular events and, to some extent, mortality in COVID-19 patients. Moreover, we noticed that inflammation may play an important role in CVD and COVID-19. Inflammation is an important component of atherosclerosis [17, 18] and a cytokine storm is responsible for the severity of COVID-19 and also an important cause of COVID-19-related death. Further, the impaired acquired immune response and the uncontrolled innate inflammatory response may be related to the cytokine storm mechanism in COVID-19 [19].
NLR family pyrin domain containing 3 (NLRP3) may mediate the process of cholesterol aggravation of COVID-19
Members of the interleukin (IL) family can be expressed and/or affected by all cells of the innate immune system; of these, IL-1 is a central mediator of innate immunity and inflammation [20]. The NLRP3 inflammasome is an innate immune signaling complex that has been shown to be a key mediator in the production of IL-1 cytokines in atherosclerosis. In atherosclerotic lesions, ox-LDL, CCs, and other rich endogenous danger signals can activate NLRP3 [14].
Interestingly, acute respiratory distress syndrome (ARDS) was observed as a characteristic feature of the rapid progression of severe lung injury among those that died from severe COVID-19 infection, and one of its pathological features is activation of the NLRP3 inflammatory pathway [21]. In another review, patients with reduced immune fitness may exhibit inactivity of NLRP3 inflammasomes, leading to severe COVID-19 tissue damage and cytokine storm [22]. Furthermore, some researchers hypothesize that excessive activation of the NLRP3 inflammasome may be one of the main factors leading to serious complications in COVID-19 in patients with underlying uncontrolled diabetes [23]. A similar conclusion was reported in another review on obesity: the initial deficiency in defense mechanisms that was reflected in the increased susceptibility to SARS-CoV-2 in obesity-related metabolic disorders is most likely due to elevated systemic metabolic inflammation [24], which is closely related to the NLRP3 inflammasome.
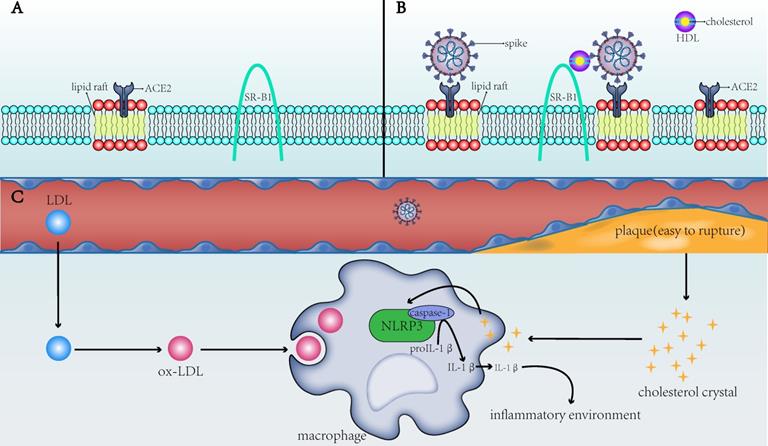
To summarize, ox-LDL and CCs in macrophages of patients with CVD activate the NLRP3 inflammasome to promote the development of atherosclerosis, which, in turn, induces a systemic inflammatory state that provides a favorable scenario for infection with SARS‐CoV‐2 (Figure 1C). This not only partially explains the susceptibility to SARS-CoV-2 in patients with underlying CVD but also explains the dysregulated lipid metabolism in patients with a history of SARS-CoV infection [25]. The activated NLRP3 in patients with SARS-CoV can contribute to vascular injury and even CVD after recovery from SARS-CoV. In COVID-19, the chronic damage to the cardiovascular system [26], which is similar to that due to SARS-CoV, may also be found; therefore, cardiovascular protection should be paid attention to in the treatment of COVID-19 [27]. NLRP3 is not the only pathway through which atherosclerosis and COVID-19 interact. Other possible associations remain [28], but NLRP3 is a good explanation for cholesterol's role in COVID-19 susceptibility.
The direct and indirect mechanisms by which cholesterol affects the infectivity of SARS-CoV-2 and the severity of COVID-19. A) ACE2 is the receptor for SARS-CoV-2 to enter the host cell and is present in the lipid rafts of the cell membrane; SR-B1 is the membrane receptor of HDL. B) When the membrane cholesterol increases, the number of lipid rafts on the cell membrane and on the ACE2 receptor in the lipid rafts also increases. SR-B1 mediates the interaction of cholesterol with the spike of SARS-CoV-2, thereby facilitating the entry of SARS-CoV-2 into the cell. C) When blood cholesterol levels increase, the formed cholesterol crystals will make the plaques unstable and prone to rupture, increasing risks to the thromboembolism in patients. After the CCs and ox-LDL are endocytosed by macrophages into the cells, they activate the NLRP3 inflammasome to produce interleukins (e.g., IL-1β) and create a favorable inflammatory environment for viruses to invade cells. SARS-CoV-2: severe acute respiratory syndrome-coronavirus-2; ACE2: angiotensin-converting enzyme 2; SR-B1: scavenger receptor B type 1; HDL: high-density lipoprotein; ox-LDL: oxidized low-density lipoprotein; NLRP3: NLR family pyrin domain containing 3; IL-1β: interleukin-1 beta.
Cholesterol affects susceptibility by influencing the process of membrane fusion
The entry of nucleocapsid into the host cell is the first step in coronavirus infection, which in turn depends on the fusion of their envelope with the host cell membrane [29]. The glycoproteins in the virus spikes (S) mediate the entry of the coronavirus [30]. The spike has two subunits - S1 and S2 [31] - which mediate receptor binding and membrane fusion, respectively [1]. Further, other studies have supported the important role of cholesterol engagement in cell entry [32]. Interestingly, membrane cholesterol has been shown to alter the oligomer status of membrane-bound peptides and the effect of altered peptide binding on depth-dependent membrane tissue and kinetics [33]. Cholesterol is ubiquitous in the plasma membrane of eukaryotes [34], so this could provide a potential target against coronavirus infection.
Cholesterol increases the density of ACE2 receptors on host cell membranes
Cholesterol is an important component of lipid rafts. Cholesterol can fill the gap between the associated sphingolipids, resulting in a tighter alignment of cholesterol and sphingolipids, which makes the lipid raft more resistant to washing than the non-lipid raft. The binding of cholesterol and saturated lipids promotes the formation of lipid rafts, which are relatively dense membrane domains and have a higher affinity for certain proteins and lipids [35]. A study showed that the ACE2 receptor which is on the host cell membrane mediates the entry of SARS-CoV-2, and then the transmembrane serine protease transmembrane protease, serine 2 (TMPRSS2) activates the S protein [36], while ACE2 co-localizes with cholesterol and sphingolipid-rich lipid raft microdomains in the plasma membrane of pneumocytes [37]. The role of lipid rafts in cell entry has been summarized in a previous review, which reported that although many studies have analyzed the influence of cholesterol consumption on viral entry, cholesterol dependence does not necessarily imply involvement of membrane rafts [38]. Hence, more research is required to verify these findings.
One study on SARS-CoV suggested that cholesterol-rich microdomains provide a platform for promoting effective interaction of S protein with cellular receptor ACE2 [39]. Wang et al. found that using cholesterol from serum to increase the cholesterol in cell membranes could enhance the invasion of pseudo-SARS-CoV-2 and the ability of the virus to infect, and both the number and apparent diameter of monosialotetrahexosylganglioside (GM1) lipid rafts (the number of viral entry points of ACE2) on cholesterol-loaded cells increased [40]. Wang et al. also found that cholesterol can simultaneously transport ACE2 to the endocytic entry point that the virus might dock, thus effectively utilizing it to enter cells [40]. In other words, cholesterol may increase the entry of pseudo-SARS-CoV-2 by increasing the number of endocytic entry points and transporting ACE2 to the endocytic entry point (Figure 1A,B). It should be noted that this observation is only from an in vitro study and cannot fully reflect the actual situation.
Cholesterol in the virus may interact with the fusion protein
A lot of current research has focused on cholesterol dependence during membrane fusion, but it should also be considered whether cholesterol binds to the proteins involved in this process. The cholesterol recognition/interaction amino acid consensus (CRAC) domain is one of proteins' structural properties that makes proteins preferentially bind to those high cholesterol domains [41]. The role of CRAC in fusion protein gp41 of human immunodeficiency virus (HIV) has been extensively studied [42-44], and the CRAC motif in a flexible domain of the outer membrane domain prior to the transmembrane segment determines the binding of gp41 to cholesterol [45]. Interestingly, in a study of the primary sequence of SARS-2-S, some CRAC sequences and its inverted sequence CARC were also found [46]. In addition, the entry of HIV-1 requires not only envelope glycoproteins to be redistributed into clusters but also cholesterol in the viral membrane. A residue in the cytoplasmic tail of gp41 directly interacts with cholesterol in the viral cell membrane [47]. There is evidence that the C-terminal of the spike's cytoplasmic tail of SARS-CoV-2 is the key to retention of membrane-mediated spike in the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) [48].
Ebola fusion glycoprotein (GP) is associated with disengagement of infected cells, ultimately leading to leaky blood vessels and hemorrhagic fever. The results show that cholesterol and transmembrane domains mediate full-length GP1, 2, or GP2-induced detachment [49]. A study conducted on Ebola virus showed that the detachment induced by full-length GP1, 2, or subunit GP 2 depends on cholesterol and the structure of the transmembrane domain[50]. Transmembrane domains (TMDs) of viral fusion proteins or amino acid residues adjacent to TMDs may be involved in the cell fusion process of viruses [51], and the trimerization process of the SARS-CoV-2 spike glycoprotein is induced by the fusion of TMDs and the receptor binding domain [52]. The similarity between SARS-CoV-2 and SARS-CoV is that their TMD is very conservative, and both are essential for membrane fusion and intercellular fusion activity [52].
Currently, there are relatively few studies on the role and molecular mechanism of the spike and cholesterol in SARS-CoV-2. However, it is of great significance to further elucidate this mechanism in order to reduce coronavirus infection and drug development. A recent study has shown that the spike requires membrane cholesterol without the need for raft engagement [4]. Based on this, future in-depth studies should be conducted.
HDL and SR-B1 play a role in the infection process
Patients with COVID-19 have significant dyslipidemia, which is closely related to cardiovascular disease. The concentration of HDL, LDL-C, and total cholesterol (TC) in COVID-19 patients was significantly reduced. HDL is significantly decreased in critically ill patients, while LDL and TC are decreased but still dominant [53], indicating an increased risk of cardiovascular disease in COVID-19 patients. Patients with COVID-19 are more prone to systemic inflammatory responses and cytokine storms [54]. Studies have shown that patients with diseases such as systemic lupus erythematosus and HIV infection, which are associated with inflammation, have significantly lower levels of HDL than other patients [55, 56]. Inflammation seems to cause a decrease in HDL levels, although the mechanism by which HDL levels are reduced is still not clear. Therefore, dyslipidemia that is caused by SARS-CoV-2 infection may be related to the systemic inflammatory response caused by COVID-19. The main function of HDL is to promote the reverse transport of cholesterol from the liver to the periphery, and it also has immunomodulatory, antithrombotic, and antioxidant effects [57]. When HDL antioxidant function is impaired, it causes lipid oxidation, which worsens the body's inflammatory response. An antioxidant enzyme in HDL - paraoxonase-1 (PON1) - is also inactivated in response to inflammation-induced oxidative stress, which further impairs HDL function [58]. PON1 activity is related to the prognosis of CVD as described above, and low PON1 activity leads to poor prognosis of CVD [59, 60].
SARS-CoV-2 binds to ACE2 through the C-terminal domain of S1 subunit (RBD) [61]. Wei et al.[62] found that HDL-scavenger receptor B type 1 (SR-B1) can help S1 subunit to bind to cholesterol, especially HDL, and help SARS-CoV-2 to enter into the host cells (Figure 1A,B). Inhibiting the function of SR-B1 or blocking the cholesterol binding site of the S1 subunit can inhibit SARS-CoV-2 infection. One of the main functions of SR-B1 is to promote the transfer of cholesteryl esters (CEs) from HDL or other lipoproteins to cells through selective uptake mechanisms, and SR-B1 has been shown to be a key receptor for HCV (hepatitis C virus) entry into the body [63]. Additionally, the expression of SR-B1 protein in the liver of mice fed a high fat and high sugar diet was 1.2-1.3 times higher than that of normal mice, suggesting that the high cholesterol level caused by chronic human diseases may promote the overexpression of SR-B1, thereby promoting SARS-CoV-2 infection.
Inspiration for treatment
Large amounts of clinical data have shown that COVID-19 patients had abnormal cholesterol levels after infection [64, 65]. Further results have shown that patients with cardiovascular disease and other underlying conditions are more likely to suffer from SARS-CoV-2, which leads to worsening of the patient's condition and poor prognosis [66, 67]. Therefore, controlling the changes in cholesterol in the body may affect the development of COVID-19 disease.
The cut-offs at a normal blood test of the lipid balance for each component
Blood lipids are the lipids in the blood. Blood tests usually include total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and triglycerides (TG). It is generally believed that the critical value of each component is shown in Table 1, which refers to the standards of the Mayo Clinic.
The critical value of the lipid balance of each component in a normal blood test
| Category | U.S. and some other countries | Canada and most of Europe |
|---|---|---|
| TC | <200 mg/dL | <5.18 mmol/L |
| LDL-C | <100 mg/dL | <2.6 mmol/L |
| HDL-C | >60 mg/dL | >1.5 mmol/L |
| TG | <150 mg/dL | <1.7 mmol/L |
TC: total cholesterol; LDL-C: LDL cholesterol; HDL-C: HDL cholesterol; TG: Triglycerides.
It is noteworthy that the critical values shown in Table 1 are only approximate values [68], which are not exactly the same in the guidelines of different regions [69, 70]. In the United States, cholesterol levels are measured in milligrams per deciliter (mg/dL), while in Canada and many European countries it is millimoles per liter (mmol/L). Normal concentration of each lipid component in plasma contribute to the low risk of cardiovascular diseases. In fact, the target value of blood lipid is not always the same among people of different ages, genders, and cardiovascular risks. It is generally believed that total cholesterol should be less than 200 mg/dL (5.18 mmol/L) and HDL-C should be higher than 60 mg/dL (1.5 mmol/L). The situation with LDL-C is a little more complicated. Lowering LDL-C is one of the few established and proven principles for the prevention and treatment of atherosclerosis [71], so the guidelines regard it as the goal of lowering lipids. According to different cardiovascular risk levels, patients are divided into low-risk, medium-risk, high-risk, and extremely high-risk [70]. Current guidelines recommend the lowering of LDL-C to 115 mg/dl (3 mmol/l) in patients with low and moderate risk; the LDL-C treatment target is <100 mg/dl (2.6 mmol/l) for patients at high risk and <70 mg/dl (1.8 mmol/l) for patients at very high risk [71]. TG is not used as a strong indicator, because studies have shown that the causal influence of TG-rich lipoproteins and their residues on ASCVD risk depends on the circulating concentration of ApoB particles, rather than the TG content itself [68, 72].
LDL-C-lowering therapies
Whether it is for COVID-19 patients or non-COVID-19 people, it is beneficial to maintain the LDL-C in the blood within the normal range. On the one hand, it can reduce the population's susceptibility to COVID-19, and on the other hand, it can also reduce the cardiovascular complications of COVID-19 patients. LDL-C is the main indicator for lowering blood lipids. Lipid-lowering treatment mainly includes lifestyle intervention and drug treatment. For patients who have difficulty in lowering blood lipids, lipid apheresis technology is also available.
Lifestyle intervention for lowering lipids
Lifestyle factors that can be intervened to improve overall lipoprotein distribution include weight control [73], physical exercise [74], dietary fat control, reasonable intake of dietary carbohydrates and fiber, smoking cessation and alcohol restriction. Studies have shown that when weight loss is 5 to 8 kg, HDL-C increases by 2-3 mg/dL, and LDL-C decreases by 2-5 mg/dL [75]. In addition, adding three weekly aerobic and resistance-training sessions to the dieting program may deliver better outcomes [76]. Studies have also shown that physical exercise reduced the atherogenic burden as experienced by the reduction in apoB or apoB/apoA-I levels, but not by LDL-C in healthy middle-aged men [77]. In terms of dietary fat control, it is recommended to avoid any intake of trans fatty acids, and the intake of saturated fat is supposed to be less than 10% of the total caloric intake, and should be further reduced in the presence of hypercholesterolemia (<7% of energy) [69]. Compared with high MUFA (oleic acid) diets, the high saturated fat in butter or cream has a worse overall effect on blood lipids; the overall effect of marine omega-3 EPA and DHA on blood lipids seems to be protective, while omega-6 polyunsaturated fatty acids may increase the risk of coronary heart disease [78]. Dietary fiber (especially soluble fiber) has the effect of lowering cholesterol, and the adverse effects of high carbohydrate diet on other lipoproteins should be minimized [79]. Lifestyle intervention is very important, because in addition to lowering lipids, it also has an additional benefit on enhancing immunity. A case report has shown that a patient with Hodgkin's lymphoma who relapses with COVID-19 during chemotherapy may be the result of a weakened immune system [80].
Lipid-lowering drug therapy
If lifestyle intervention fails to improve hyperlipidemia, then lipid-lowering medications are needed. Dyslipidemia is very closely related to inflammation in many pathological conditions, for example, dyslipidemia and inflammation are closely related in their effects on atherosclerosis [81]. The results of a meta-analysis suggest that in clinical practice, the anti-inflammatory effects of most LDL lowering therapies are related to the magnitude of change in LDL [82]. There are many studies and discussions about whether COVID-19 patients should lower lipids. At present, more opinions are that statin therapy should be continued in COVID-19 patients because patients can benefit from it [83-85]. For patients who have not reached the LDL-C treatment goals with high-intensity statins, and patients who are intolerant to statins, ezetimibe and PCSK9 (Proprotein convertase subtilisin/kexin type 9) inhibitors are recommended for treatment [86].
When using medications, not only should pay attention to the improvement of blood lipids, but also the possible toxic and side effects of lipid-lowering treatment [87]. Once signs of liver and kidney damage and other adverse reactions appear, the use of lipid-lowering drugs should be suspended in time. Remdesivir and some statins (atorvastatin, simvastatin, and lovastatin) are metabolized in the same way, so simultaneous use should be avoided; tocilizumab interferes with the metabolic pathway of statins, so it is recommended to temporarily stop statin treatment when using tocilizumab [88]. The expert team from HEART UK has summarized in detail the possible interactions between drugs used to treat COVID-19 infection and lipid-lowering drugs, as well as drug recommendations [88], which has great reference value in the management of COVID-19 patients' lipid lowering.
Both statins and PCSK9 inhibitors have been shown to have additional anti-inflammatory effects, which can reduce inflammation, oxidative stress and improve vascular inflammation, which is also one of the reasons why lipid-lowering therapy should be retained in COVID-19 patients [83]. Statins refer to 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors as the most commonly used lipid-lowering agents and the most effective hypolipidemic compounds for reducing mortality in patients with coronary heart disease [89]. HMG-CoA reductase produces mevalonate and is the rate limiting enzyme for hepatic cholesterol biosynthesis, which is competitively and reversibly inhibited by statins through both the lactone ring and the side chain, aiding in its binding to the active site of the enzyme [90]. The anti-inflammatory effects of statins, whether through the clearance of proinflammatory modified LDL from the arterial wall, or through the inherent non LDL anti-inflammatory properties of isoprenoids, are well known [91].
Statins can reduce CRP in several ways: reducing LDL-C while reducing the production of ox- LDL, thereby reducing inflammation [92]; reducing Rac-1 geranylgeranylation to reduce IL-6-induced serine phosphorylation of the transcription factor STAT3; inhibiting the expression of vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and E-selectin; increasing the rate of CRP catabolism [93]. In addition, statins may also serve anti-inflammatory purposes through their effects on endothelial NO (nitric oxide). Statins can negatively regulate endothelial nitric oxide synthase (eNOS) expression by altering NO release through multiple mechanisms, such as inhibiting the production of mevalonate and important isoprenoid intermediates, thereby preventing the prenylation of the small GTPase Rho [94]. NO inhibits NF-κB by inducing and stabilizing I-κBα, thereby exerting anti-inflammatory effects [95]. One study investigated whether statin treatment in the population was associated with differences in oxidative generated nucleotide damage and chronic inflammation, and the relationship between nucleotide damage and chronic inflammation, drawing from the results that statin users had a lower burden of oxidative generated DNA damage and inflammation compared with nonusers [96].
The anti-inflammatory versus antioxidant effects exerted by statins in the clinic suggested to us that it might also play the same role in COVID-19 to mitigate part of the adverse effects resulting from inflammation. Statins have previously been shown to be promising in a variety of viral infections, such as avian, H1N1 pandemic [97-99]. Statins have anti-inflammatory and antithrombotic effects, including enhancing ACE2 expression, and in addition statins may also improve endothelial function in patients with COVID-19 infection, and these effects suggest that statins have potential as adjunctive therapy to attenuate endothelial dysfunction and dysregulated inflammation in covid-19-infected patients [100]. There is literature discussing in greater detail the possible underlying molecular mechanisms involved in the action of statins in COVID-19: SARS-CoV-2 enters cells by binding to ACE2. Once inside the cell, SARS-CoV-2 causes ACE2 downregulation, which reduces its conversion to angiotensin mediated protection by angiotensin II. Statins upregulate ACE2 [98, 99, 101, 102]. It is also known that the coronavirus induces a pro-inflammatory host response through the activation of the TLR-MYD88-NF-κB pathway, which can be inhibited by statins. Finally, statins can also reduce viral entry by depleting the cholesterol content of the cell membrane, but this effect remains controversial [103].
In terms of lipid lowering, new therapies with PCSK9 inhibitors that lower LDL to a greater extent have led to new breakthroughs in the treatment of atherosclerotic cardiovascular disease [104]. PCSK9 degrades low-density lipoprotein cholesterol (LDL) receptors, which in turn elevate serum LDL cholesterol. Clinical trials have shown that inhibition of PCSK9 is effective in lowering LDL cholesterol levels and reducing cardiovascular events [105]. Recent studies have shown that PCSK9 is involved in the inflammatory pathway of atherosclerosis. Although trials with PCSK9 inhibitors have not shown any alterations in plasma C-reactive protein levels, there is accumulating evidence of a reduced inflammatory response in the arterial wall, which may attenuate atherosclerotic plaque development beyond the established LDL lowering effect of PCSK9 inhibitors [106]. In conclusion, the available human studies show beneficial effects of statins and PCSK9 inhibitors on pneumonia and sepsis. These agents may act as immunomodulators in COVID-19 and prevent major complications such as acute respiratory distress syndrome and cytokine release syndrome [107].
Lipid apheresis
Simply put, lipoprotein apheresis is a therapy that filters out some lipid components in the blood to achieve the purpose of lowering blood lipids [108]. Since it was first introduced, it has been the “last resort” to treat uncontrollable dyslipidemia [109]. When neither diet nor medication can improve the hyperlipidemia, lipoprotein apheresis technology is a better choice. It works well but is expensive and time-consuming. In particular, for patients with familial hypercholesterolemia, lipoprotein apheresis technology is more widely used. Moreover, patients with familial hypercholesterolemia (FH) have a higher risk of CVD than normal people regardless of their age, so FH patients will have an increased risk of experiencing a severe course of COVID-19 [86]. Lipoprotein apheresis is currently the only truly safe and effective treatment for homozygous FH, and it is recommended that children with homozygous FH should consider lipoprotein apheresis from the age of 6 to achieve early control of cholesterol and reduce cardiovascular risk [110, 111]. Lipoprotein apheresis is usually carried out once 1-2 weeks. If there is no way to achieve it, it can be postponed for two weeks with intensive lipid-lowering treatment and close monitoring of symptoms [86].
The goal of lowering lipid
For COVID-19 patients, there is currently no study to determine the blood LDL-C cutoff value that is beneficial to the prognosis of SARS-Cov-2 infection. Therefore, the current lipid-lowering goals of COVID-19 patients are still the same as those of the non-COVID-19 population, that is, starting from reducing the risk of cardiovascular events and other complications, the treatment target value of LDL-C is drawn up according to different risk groups. As mentioned above, the current guidelines recommend reducing the LDL-C of low-risk and intermediate-risk patients, high-risk patients, and very high-risk patients to 115 mg/dl (3 mmol/l), 100 mg/dl (2.6 mmol/l) and 70 mg/dl (1.8 mmol/l), respectively [71]. Even for people with low CVD risk, the LDL-C level should be maintained at a low level for a long time (LDL-C ≤ 115 mg/dL [3 mmol/L]) [112]. Studies have shown that patients with a higher baseline LDL-C level benefit more from lipid-lowering therapy[113]. When the baseline LDL-C level is less than 100 mg/dL, there is no correlation between the reduction in LDL-C and the reduction in the risk of total mortality and cardiovascular mortality [113]. It is worth noting that most of the lipid-lowering goals set the upper limit of LDL-C without mentioning the lower limit, mainly because the lipid-lowering ability of lipid-lowering drugs before cannot reduce the absolute value of LDL-C to a very low level, and the safety of very low cholesterol has not been evaluated [114]. It is currently believed that LDL-C reaching 40-50 mg/dL seems to be safe, while the data below 25 mg/dL is limited, and more in-depth research and post-mortem analysis are needed in the future [115].
But for COVID-19 patients, cholesterol is not as low as possible. Studies have shown that in COVID-19 patients, low LDL-c serum levels (LDL-c ≤69 mg/dl) are independently associated with higher 30-day mortality [116]. For patients with severe symptoms of COVID-19 who cannot take oral medications in a serious condition, lipid-lowering drugs may be temporarily discontinued [88]. And some studies have shown that COVID-19 patients will have hypolipemia as early as when the symptoms are mild, and the degree of hypolipemia is positively correlated with the severity of the disease [64, 117]. The current speculation is that the decrease in lipid levels is probably caused by SARS-COV-2 infection. The possible mechanisms include liver damage, acute inflammation, increased free radicals, and changes in vascular permeability [64]. Compared with patients with high LDL-C levels, patients with low LDL-C levels are more likely to have immune and inflammatory dysfunction, renal dysfunction, liver dysfunction, and cardiac insufficiency on admission [118]. It is still uncertain whether the secondary hypocholesterolemia of COVID-19 patients will participate in other pathophysiological mechanisms that are not conducive to the prognosis [119], so the lipid-lowering goal for COVID-19 patients may not be as low as possible. In particular, when COVID-19 patients are combined with other diseases related to hypoproteinemia, such as tuberculosis [120, 121], careful consideration should be given to whether to carry out lipid-lowering treatment. A recent study with a large sample size showed that in the Chinese population, low LDL-C levels are associated with an increased risk of all-cause, CVD, ischemic stroke, hemorrhagic stroke, and cancer mortality [122]. The safety of low LDL-C needs further research.
Conclusion
Cholesterol in the macrophages of CVD patients (as ox-LDL and CCs) activates NLRP3 and promotes the development of atherosclerosis, which also induces a systemic inflammatory state that provides favorable conditions for SARS-CoV-2 infection and indirectly leads to a poor prognosis in COVID-19 patients. Cholesterol can also directly affect the entry of viruses into host cells. First, in vitro studies showed that membrane cholesterol can increase the number of endocytic entry points on the host cell membrane and transport ACE2 to the endocytic entry points, thereby increasing the entry of pseudoSARS-CoV-2. Second, previous studies showed that viral fusion proteins or spikes have receptors cholesterol, and recent studies on SARS-CoV-2 have shown that spikes require cholesterol to enter cells. In addition, SR-B1 can help the spike's S1 subunit to bind to HDL cholesterol in the blood and promote the entry of SARS-CoV-2 into the host cells. However, more detailed molecular mechanisms remain to be studied. It is currently recommended to maintain the original lipid-lowering treatment for COVID-19, but very low LDL-C levels are not recommended. Specific lipid-lowering programs and precautions can refer to the current guidelines.
Acknowledgements
Funding
Guangdong Medical Science and Technology Research Foundation (A2020162, B2021182), Guangdong Basic and Applied Basic Research Foundation (2020A1515010288), Scientific Research Project of Chaozhou Health Bureau (2019110), Higher Education Teaching Reform Project of Southern Medical University (JG2019126, 2018JG43, JG2019130, JG201984).
Author Contributions
P.D. Zhang and Z.J. Wu conceptualized and designed the study. Yan Tang and Longtai Hu performed the literature search and drafted the manuscript. All authors critically revised the manuscript and approved the final version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tang T, Bidon M, Jaimes JA, Whittaker GR, Daniel S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020;178:104792
2. Kim JH, Marks F, Clemens JD. Looking beyond COVID-19 vaccine phase 3 trials. Nature Medicine. 2021;27:205-11
3. Bae S, Kim SR, Kim M-N, Shim WJ, Park S-M. Impact of cardiovascular disease and risk factors on fatal outcomes in patients with COVID-19 according to age: a systematic review and meta-analysis. Heart. 2021;107:373-80
4. Sanders DW, Jumper CC, Ackerman PJ, Bracha D, Donlic A, Kim H. et al. SARS-CoV-2 Requires Cholesterol for Viral Entry and Pathological Syncytia Formation. bioRxiv. 2020. 2020 12.14.422737
5. Seyedpour S, Khodaei B, Loghman AH, Seyedpour N, Kisomi MF, Balibegloo M. et al. Targeted therapy strategies against SARS-CoV-2 cell entry mechanisms: A systematic review of in vitro and in vivo studies. Journal of Cellular Physiology. 2021;236:2364-92
6. Corti M-C, Guralnik JM, Salive ME, Harris T, Ferrucci L, Glynn RJ. et al. Clarifying the direct relation between total cholesterol levels and death from coronary heart disease in older persons. Annals of Internal Medicine. 1997;126:753-60
7. Jeong SM, Choi S, Kim K, Kim SM, Lee G, Park SY. et al. Effect of Change in Total Cholesterol Levels on Cardiovascular Disease Among Young Adults. Journal of the American Heart Association. 2018;7:e008819
8. Hatmi ZN. A Systematic Review of Systematic Reviews on the COVID-19 Pandemic. SN Comprehensive Clinical Medicine. 2021
9. Risk Factors in Coronary Heart Disease. Annals of Internal Medicine. 1964; 61: 888-99.
10. Oram JF, Vaughan AM. ATP-Binding Cassette Cholesterol Transporters and Cardiovascular Disease. Circulation Research. 2006;99:1031-43
11. Chen Z, Ichetovkin M, Kurtz M, Zycband E, Kawka D, Woods J. et al. Cholesterol in human atherosclerotic plaque is a marker for underlying disease state and plaque vulnerability. Lipids in Health and Disease. 2010;9:61
12. Abela GS, Aziz K. Cholesterol crystals rupture biological membranes and human plaques during acute cardiovascular events-a novel insight into plaque rupture by scanning electron microscopy. Scanning. 2006;28:1-10
13. Janoudi A, Shamoun FE, Kalavakunta JK, Abela GS. Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. European Heart Journal. 2015;37:1959-67
14. Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circulation Research. 2018;122:1722-40
15. Nidorf M, Fiolet A, Abela GS. Viewing atherosclerosis through a crystal lens: how the evolving structure of cholesterol crystals in atherosclerotic plaque alters its stability. Journal of Clinical Lipidology. 2020
16. Madjid M, Vela D, Khalili-Tabrizi H, Casscells SW, Litovsky S. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries: clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34:11-8
17. Libby P. Inflammation in Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:2045-51
18. Grebe A, Latz E. Cholesterol Crystals and Inflammation. Current Rheumatology Reports. 2013;15:313
19. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. Journal of Medical Virology. 2021;93:250-6
20. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003-18
21. Freeman TL, Swartz TH. Targeting the NLRP3 Inflammasome in Severe COVID-19. Frontiers in Immunology. 2020 11
22. van den Berg DF, te Velde AA. Severe COVID-19: NLRP3 Inflammasome Dysregulated. Frontiers in Immunology. 2020 11
23. Lambadiari V, Kousathana F, Raptis A, Katogiannis K, Kokkinos A, Ikonomidis I. Pre-Existing Cytokine and NLRP3 Inflammasome Activation and Increased Vascular Permeability in Diabetes: A Possible Fatal Link With Worst COVID-19 Infection Outcomes? Frontiers in immunology. 2020;11:557235 -
24. Bertocchi I, Foglietta F, Collotta D, Eva C, Brancaleone V, Thiemermann C. et al. The hidden role of NLRP3 inflammasome in obesity-related COVID-19 exacerbations: Lessons for drug repurposing. British Journal of Pharmacology. 2020;177:4921-30
25. Wu Q, Zhou L, Sun X, Yan Z, Hu C, Wu J. et al. Altered Lipid Metabolism in Recovered SARS Patients Twelve Years after Infection. Scientific Reports. 2017;7:9110
26. Arcari L, Luciani M, Cacciotti L, Musumeci MB, Spuntarelli V, Pistella E. et al. Incidence and determinants of high-sensitivity troponin and natriuretic peptides elevation at admission in hospitalized COVID-19 pneumonia patients. Internal and emergency medicine. 2020;15:1467-76
27. Zheng Y-Y, Ma Y-T, Zhang J-Y, Xie X. COVID-19 and the cardiovascular system. Nature Reviews Cardiology. 2020;17:259-60
28. Grzegorowska O, Lorkowski J. Possible Correlations between Atherosclerosis, Acute Coronary Syndromes and COVID-19. Journal of Clinical Medicine. 2020;9:3746
29. Belouzard S, Millet JK, Licitra BN, Whittaker GR. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4:1011-33
30. Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. 2003;77:8801-11
31. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nature Microbiology. 2020;5:562-9
32. Hu B, Höfer CT, Thiele C, Veit M. Cholesterol Binding to the Transmembrane Region of a Group 2 Hemagglutinin (HA) of Influenza Virus Is Essential for Virus Replication, Affecting both Virus Assembly and HA Fusion Activity. Journal of Virology. 2019;93:e00555-19
33. Meher G, Bhattacharjya S, Chakraborty H. Membrane Cholesterol Modulates Oligomeric Status and Peptide-Membrane Interaction of Severe Acute Respiratory Syndrome Coronavirus Fusion Peptide. The Journal of Physical Chemistry B. 2019;123:10654-62
34. Mouritsen OG, Zuckermann MJ. What's so special about cholesterol? Lipids. 2004;39:1101-13
35. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nature Reviews Molecular Cell Biology. 2017;18:361-74
36. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271-80.e8
37. Qian K, Hu S. Ig-like ACE2 protein therapeutics: A revival in development during the COVID-19 pandemic. mAbs. 2020;12:1782600
38. Torrinhas RS, Calder PC, Lemos GO, Waitzberg DL. Parenteral fish oil: An adjuvant pharmacotherapy for coronavirus disease 2019? Nutrition. 2021;81:110900 -
39. Glende J, Schwegmann-Wessels C, Al-Falah M, Pfefferle S, Qu X, Deng H. et al. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology. 2008;381:215-21
40. Wang H, Yuan Z, Pavel MA, Hansen SB. The role of high cholesterol in age-related COVID19 lethality. bioRxiv. 2020
41. Epand RM. Cholesterol and the interaction of proteins with membrane domains. Progress in Lipid Research. 2006;45:279-94
42. Greenwood AI, Pan J, Mills TT, Nagle JF, Epand RM, Tristram-Nagle S. CRAC motif peptide of the HIV-1 gp41 protein thins SOPC membranes and interacts with cholesterol. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2008;1778:1120-30
43. Vishwanathan SA, Thomas A, Brasseur R, Epand RF, Hunter E, Epand RM. Large changes in the CRAC segment of gp41 of HIV do not destroy fusion activity if the segment interacts with cholesterol. Biochemistry. 2008;47:11869-76
44. Kwon B, Mandal T, Elkins MR, Oh Y, Cui Q, Hong M. Cholesterol interaction with the trimeric HIV fusion protein gp41 in lipid bilayers investigated by solid-state NMR spectroscopy and molecular dynamics simulations. Journal of molecular biology. 2020;432:4705-21
45. Schroeder C. Cholesterol-binding viral proteins in virus entry and morphogenesis. Subcell Biochem. 2010;51:77-108
46. Wei C, Wan L, Zhang Y, Fan C, Yan Q, Yang X. et al. Cholesterol Metabolism—Impacts on SARS-CoV-2 Infection Prognosis, Entry, and Antiviral Therapies. medRxiv. 2020. 2020 04.16.20068528
47. Nieto-Garai JA, Arboleya A, Otaegi S, Chojnacki J, Casas J, Fabriàs G. et al. Cholesterol in the Viral Membrane is a Molecular Switch Governing HIV-1 Env Clustering. Advanced Science. 2021;8:2003468
48. Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset F-L. et al. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. Journal of Biological Chemistry. 2021;296:100111
49. Hacke M, Björkholm P, Hellwig A, Himmels P, de Almodóvar CR, Brügger B. et al. Inhibition of Ebola virus glycoprotein-mediated cytotoxicity by targeting its transmembrane domain and cholesterol. Nature Communications. 2015;6:7688
50. Lee J, Kreutzberger AJB, Odongo L, Nelson EA, Nyenhuis DA, Kiessling V. et al. Ebola virus glycoprotein interacts with cholesterol to enhance membrane fusion and cell entry. Nature Structural & Molecular Biology. 2021;28:181-9
51. Corver J, Broer R, van Kasteren P, Spaan W. Mutagenesis of the transmembrane domain of the SARS coronavirus spike glycoprotein: refinement of the requirements for SARS coronavirus cell entry. Virology Journal. 2009;6:230
52. Azad T, Singaravelu R, Crupi MJF, Jamieson T, Dave J, Brown EEF. et al. Implications for SARS-CoV-2 Vaccine Design: Fusion of Spike Glycoprotein Transmembrane Domain to Receptor-Binding Domain Induces Trimerization. Membranes. 2020;10:215
53. Kimhofer T, Lodge S, Whiley L, Gray N, Loo RL, Lawler NG. et al. Integrative Modeling of Quantitative Plasma Lipoprotein, Metabolic, and Amino Acid Data Reveals a Multiorgan Pathological Signature of SARS-CoV-2 Infection. Journal of Proteome Research. 2020;19:4442-54
54. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nature Reviews Immunology. 2020;20:363-74
55. Feingold KR, Grunfeld C. Effect of inflammation on HDL structure and function. Current opinion in lipidology. 2016;27:521-30
56. Feingold KR, Grunfeld C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, et al. editors. Endotext. South Dartmouth (MA): MDText.com, Inc.Copyright © 2000-2021, MDText.com, Inc. 2000
57. Karathanasis SK, Freeman LA, Gordon SM, Remaley AT. The Changing Face of HDL and the Best Way to Measure It. Clinical Chemistry. 2017;63:196-210
58. Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S. et al. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. Journal of immunology (Baltimore, Md: 1950). 2007;178:496-502
59. Farid AS, Horii Y. Modulation of paraoxonases during infectious diseases and its potential impact on atherosclerosis. Lipids Health Dis. 2012;11:92
60. Bayrak A, Bayrak T, Bodur E, Kılınç K, Demirpençe E. The effect of HDL-bound and free PON1 on copper-induced LDL oxidation. Chemico-biological interactions. 2016;257:141-6
61. Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181:894-904 e9
62. Wei C, Wan L, Yan Q, Wang X, Zhang J, Yang X. et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nature Metabolism. 2020;2:1391-400
63. Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK. et al. Role of Scavenger Receptor Class B Type I in Hepatitis C Virus Entry: Kinetics and Molecular Determinants. Journal of Virology. 2010;84:34-43
64. Wei X, Zeng W, Su J, Wan H, Yu X, Cao X. et al. Hypolipidemia is associated with the severity of COVID-19. Journal of clinical lipidology. 2020;14:297-304
65. Fan J, Wang H, Ye G, Cao X, Xu X, Tan W. et al. Letter to the Editor: Low-density lipoprotein is a potential predictor of poor prognosis in patients with coronavirus disease 2019. Metabolism: clinical and experimental. 2020;107:154243
66. Li B, Yang J, Zhao F, Zhi L, Wang X, Liu L. et al. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clinical Research in Cardiology. 2020;109:531-8
67. Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW. et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. Jama. 2020;323:2052-9
68. Aberra T, Peterson ED, Pagidipati NJ, Mulder H, Wojdyla DM, Philip S. et al. The association between triglycerides and incident cardiovascular disease: What is “optimal”? Journal of Clinical Lipidology. 2020;14:438-47.e3
69. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L. et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). European Heart Journal. 2019;41:111-88
70. Atar D, Jukema JW, Molemans B, Taub PR, Goto S, Mach F. et al. New cardiovascular prevention guidelines: How to optimally manage dyslipidaemia and cardiovascular risk in 2021 in patients needing secondary prevention? Atherosclerosis. 2021;319:51-61
71. März W, Scharnagl H, Gouni-Berthold I, Silbernagel G, Dressel A, Grammer TB. et al. LDL-Cholesterol: Standards of Treatment 2016: A German Perspective. American journal of cardiovascular drugs: drugs, devices, and other interventions. 2016;16:323-36
72. Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ. et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019;321:364-73
73. Hasan B, Nayfeh T, Alzuabi M, Wang Z, Kuchkuntla AR, Prokop LJ. et al. Weight Loss and Serum Lipids in Overweight and Obese Adults: A Systematic Review and Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism. 2020;105:3695-703
74. Igarashi Y, Nogami Y. Response of lipids and lipoproteins to regular aquatic endurance exercise: a meta-analysis of randomized controlled trials. Journal of atherosclerosis and thrombosis. 2018: 42937.
75. Jensen MD, Ryan DH, Apovian CM, Ard JD, Comuzzie AG, Donato KA. et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129:S102-38
76. Said MA, Abdelmoneem M, Alibrahim MC, Elsebee MA, Kotb AAH. Effects of diet versus diet plus aerobic and resistance exercise on metabolic syndrome in obese young men. Journal of Exercise Science & Fitness. 2020;18:101-8
77. Holme I, Høstmark A, Anderssen S. ApoB but not LDL-cholesterol is reduced by exercise training in overweight healthy men. Results from the 1-year randomized Oslo Diet and Exercise Study. Journal of internal medicine. 2007;262:235-43
78. DiNicolantonio JJ, O'Keefe JH. Effects of dietary fats on blood lipids: a review of direct comparison trials. Open Heart. 2018;5:e000871
79. Riccardi G, Vaccaro O, Costabile G, Rivellese AA. How Well Can We Control Dyslipidemias Through Lifestyle Modifications? Curr Cardiol Rep. 2016;18:66
80. Luciani M, Bentivegna E, Spuntarelli V, Lamberti PA, Cacioli G, Del Porto F. et al. Recurrent COVID-19 pneumonia in the course of chemotherapy: Consequence of a weakened immune system? J Med Virol. 2021;93:1882-4
81. Tuñón J, Bäck M, Badimón L, Bochaton-Piallat ML, Cariou B, Daemen MJ. et al. Interplay between hypercholesterolaemia and inflammation in atherosclerosis: Translating experimental targets into clinical practice. European journal of preventive cardiology. 2018;25:948-55
82. Kinlay S. Low-density lipoprotein-dependent and-independent effects of cholesterol-lowering therapies on C-reactive protein: a meta-analysis. Journal of the American College of Cardiology. 2007;49:2003-9
83. Katsiki N, Banach M, Mikhailidis DP. Lipid-lowering therapy and renin-angiotensin-aldosterone system inhibitors in the era of the COVID-19 pandemic. Archives of medical science: AMS. 2020;16:485-9
84. Morawietz H, Julius U, Bornstein SR. Cardiovascular diseases, lipid-lowering therapies and European registries in the COVID-19 pandemic. Cardiovascular Research. 2020;116:e122-e5
85. Lee H-Y, Ahn J, Park J, Kang CK, Won S-H, Kim DW. et al. Beneficial Effect of Statins in COVID-19–Related Outcomes—Brief Report. Arteriosclerosis, Thrombosis, and Vascular Biology. 2021;41:e175-e82
86. Banach M, Penson PE, Fras Z, Vrablik M, Pella D, Reiner Ž. et al. Brief recommendations on the management of adult patients with familial hypercholesterolemia during the COVID-19 pandemic. Pharmacological Research. 2020;158:104891
87. Wilkinson MJ, Laffin LJ, Davidson MH. Overcoming toxicity and side-effects of lipid-lowering therapies. Best Practice & Research Clinical Endocrinology & Metabolism. 2014;28:439-52
88. Iqbal Z, Ho JH, Adam S, France M, Syed A, Neely D. et al. Managing hyperlipidaemia in patients with COVID-19 and during its pandemic: An expert panel position statement from HEART UK. Atherosclerosis. 2020;313:126-36
89. Stancu C, Sima A. Statins: mechanism of action and effects. Journal of cellular and molecular medicine. 2001;5:378-87
90. Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circulation research. 2017;120:229-43
91. Rocha VZ, Santos RD. Cholesterol and inflammation: the lesser the better in atherothrombosis. Oxford University Press. 2018
92. Hileman CO, Turner R, Funderburg NT, Semba RD, McComsey GA. Changes in oxidized lipids drive the improvement in monocyte activation and vascular disease after statin therapy in HIV. AIDS (London, England). 2016;30:65
93. Arévalo-Lorido JC. Clinical relevance for lowering C-reactive protein with statins. Annals of medicine. 2016;48:516-24
94. Laufs U. Beyond lipid-lowering: effects of statins on endothelial nitric oxide. European Journal of Clinical Pharmacology. 2003;58:719-31
95. Peng H-B, Libby P, Liao JK. Induction and Stabilization of IκBα by Nitric Oxide Mediates Inhibition of NF-κB ∗. Journal of Biological Chemistry. 1995;270:14214-9
96. Sørensen AL, Hasselbalch HC, Nielsen CH, Poulsen HE, Ellervik C. Statin treatment, oxidative stress and inflammation in a Danish population. Redox Biology. 2019;21:101088
97. Vandermeer ML, Thomas AR, Kamimoto L, Reingold A, Gershman K, Meek J. et al. Association Between Use of Statins and Mortality Among Patients Hospitalized With Laboratory-Confirmed Influenza Virus Infections: A Multistate Study. The Journal of Infectious Diseases. 2011;205:13-9
98. Fedson DS. Treating influenza with statins and other immunomodulatory agents. Antiviral Research. 2013;99:417-35
99. Fedson DS, Opal SM, Rordam OM. Hiding in Plain Sight: an Approach to Treating Patients with Severe COVID-19 Infection. mBio. 2020;11:e00398-20
100. Lee KCH, Sewa DW, Phua GC. Potential role of statins in COVID-19. International Journal of Infectious Diseases. 2020;96:615-7
101. Fajgenbaum DC, Khor JS, Gorzewski A, Tamakloe MA, Powers V, Kakkis JJ. et al. Treatments Administered to the First 9152 Reported Cases of COVID-19: A Systematic Review. Infectious diseases and therapy. 2020;9:435-49
102. Zeiser R. Immune modulatory effects of statins. Immunology. 2018;154:69-75
103. Minz MM, Bansal M, Kasliwal RR. Statins and SARS-CoV-2 disease: Current concepts and possible benefits. Diabetes & Metabolic Syndrome: Clinical Research & Reviews. 2020;14:2063-7
104. Díaz Rodríguez Á, Mantilla Morató T. LDL as a therapeutic objective. Clinica e investigacion en arteriosclerosis: publicacion oficial de la Sociedad Espanola de Arteriosclerosis. 2019;31(Suppl 2):1-15
105. Ding Z, Pothineni NVK, Goel A, Lüscher TF, Mehta JL. PCSK9 and inflammation: role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc Res. 2020;116:908-15
106. Momtazi-Borojeni AA, Sabouri-Rad S, Gotto AM, Pirro M, Banach M, Awan Z. et al. PCSK9 and inflammation: a review of experimental and clinical evidence. European heart journal Cardiovascular pharmacotherapy. 2019;5:237-45
107. Barkas F, Milionis H, Anastasiou G, Liberopoulos E. Statins and PCSK9 inhibitors: What is their role in coronavirus disease 2019? Medical hypotheses. 2021;146:110452
108. Klingel R, Mausfeld P, Fassbender C, Goehlen B. Lipidfiltration ––safe and effective methodology to perform lipid-apheresis. Transfusion and Apheresis Science. 2004;30:245-54
109. Thompson G, Parhofer KG. Current Role of Lipoprotein Apheresis. Current Atherosclerosis Reports. 2019;21:26
110. Page MM, Bell DA, Hooper AJ, Watts GF, Burnett JR. Lipoprotein apheresis and new therapies for severe familial hypercholesterolemia in adults and children. Best Practice & Research Clinical Endocrinology & Metabolism. 2014;28:387-403
111. Thompson GR. The evidence-base for the efficacy of lipoprotein apheresis in combating cardiovascular disease. Atheroscler Suppl. 2013;14:67-70
112. Penson PE, Pirro M, Banach M. LDL-C: lower is better for longer—even at low risk. BMC Medicine. 2020;18:320
113. Navarese EP, Robinson JG, Kowalewski M, Kołodziejczak M, Andreotti F, Bliden K. et al. Association Between Baseline LDL-C Level and Total and Cardiovascular Mortality After LDL-C Lowering: A Systematic Review and Meta-analysis. JAMA. 2018;319:1566-79
114. Stein EA, Raal FJ. Targeting LDL: is lower better and is it safe? Best practice & research Clinical endocrinology & metabolism. 2014;28:309-24
115. Faselis C, Imprialos K, Grassos H, Pittaras A, Kallistratos M, Manolis A. Is very low LDL-C harmful? Current pharmaceutical design. 2018;24:3658-64
116. Aparisi Á, Iglesias-Echeverría C, Ybarra-Falcón C, Cusácovich I, Uribarri A, García-Gómez M. et al. Low-density lipoprotein cholesterol levels are associated with poor clinical outcomes in COVID-19. Nutrition, Metabolism and Cardiovascular Diseases. 2021
117. Qin C, Minghan H, Ziwen Z, Yukun L. Alteration of lipid profile and value of lipids in the prediction of the length of hospital stay in COVID-19 pneumonia patients. Food Science & Nutrition. 2020;8:6144-52
118. Zhao M, Luo Z, He H, Shen B, Liang J, Zhang J. et al. Decreased Low-Density Lipoprotein Cholesterol Level Indicates Poor Prognosis of Severe and Critical COVID-19 Patients: A Retrospective, Single-Center Study. Frontiers in medicine. 2021;8:585851
119. Olsson AG, Angelin B, Assmann G, Binder CJ, Björkhem I, Cedazo-Minguez A. et al. Can LDL cholesterol be too low? Possible risks of extremely low levels. Journal of Internal Medicine. 2017;281:534-53
120. Gebremicael G, Amare Y, Challa F, Gebreegziabxier A, Medhin G, Wolde M. et al. Lipid Profile in Tuberculosis Patients with and without Human Immunodeficiency Virus Infection. International Journal of Chronic Diseases. 2017;2017:3843291
121. Luciani M, Bentivegna E, Spuntarelli V, Amoriello Lamberti P, Guerritore L, Chiappino D. et al. Coinfection of Tuberculosis Pneumonia and COVID-19 in a Patient Vaccinated with Bacille Calmette-Guérin (BCG): Case Report. SN Compr Clin Med. 2020:1-4
122. Lu J-M, Wu M-Y, Yang Z-M, Zhu Y, Li D, Yu Z-B. et al. Low LDL-C levels are associated with risk of mortality in a Chinese cohort study. Endocrine. 2021
Author contact
![]() Corresponding authors: Peidong Zhang & Zhijian Wu, Department of Cardiology, Heart Center, Zhujiang Hospital, Southern Medical University (Peidong Zhang); Department of Cardiology, Boai Hospital of Zhongshan, Southern Medical University (Zhijian Wu). 253 Industrial Avenue, Guangzhou 510282, Guangdong, People's Republic of China. E-mail: zhangpdedu.cn (Peidong Zhang); 2388743877com (Zhijian Wu); Tel: +86 020-62783686 (Peidong Zhang); +86 0760-88776622 (Zhijian Wu). ORCID ID: https://orcid.org/0000-0003-3243-691X (Peidong Zhang); https://orcid.org/0000-0001-8212-2631 (Zhijian Wu).
Corresponding authors: Peidong Zhang & Zhijian Wu, Department of Cardiology, Heart Center, Zhujiang Hospital, Southern Medical University (Peidong Zhang); Department of Cardiology, Boai Hospital of Zhongshan, Southern Medical University (Zhijian Wu). 253 Industrial Avenue, Guangzhou 510282, Guangdong, People's Republic of China. E-mail: zhangpdedu.cn (Peidong Zhang); 2388743877com (Zhijian Wu); Tel: +86 020-62783686 (Peidong Zhang); +86 0760-88776622 (Zhijian Wu). ORCID ID: https://orcid.org/0000-0003-3243-691X (Peidong Zhang); https://orcid.org/0000-0001-8212-2631 (Zhijian Wu).