Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The upstream mechanism of...
Factors secreted by activated...
Prospects and potential...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(14):3059-3065. doi:10.7150/ijms.61153 This issue Cite
Review
The role and mechanisms of Microglia in Neuromyelitis Optica Spectrum Disorders
Wenqun Li1,2, Jiaqin Liu1,2, Wei Tan3,4, Yedi Zhou3,4 ![]()
1. Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, 410011, Hunan, China.
2. Institute of Clinical Pharmacy, Central South University, Changsha, 410011, Hunan, China.
3. Department of Ophthalmology, The Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China.
4. Hunan Clinical Research Center of Ophthalmic Disease, Changsha, Hunan 410011, China.
Received 2021-4-1; Accepted 2021-6-3; Published 2021-6-16
Abstract

Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune neurological disease that can cause blindness and disability. As the major mediators in the central nervous system, microglia plays key roles in immunological regulation in neuroinflammatory diseases, including NMOSD. Microglia can be activated by interleukin (IL)-6 and type I interferons (IFN-Is) during NMOSD, leading to signal transducer and activator of transcription (STAT) activation. Moreover, complement C3a secreted from activated astrocytes may induce the secretion of complement C1q, inflammatory cytokines and progranulin (PGRN) by microglia, facilitating injury to microglia, neurons, astrocytes and oligodendrocytes in an autocrine or paracrine manner. These processes involving activated microglia ultimately promote the pathological course of NMOSD. In this review, recent research progress on the roles of microglia in NMOSD pathogenesis is summarized, and the mechanisms of microglial activation and microglial-mediated inflammation, and the potential research prospects associated with microglial activation are also discussed.
Keywords: microglia, neuromyelitis optica spectrum disorder, demyelination, microglial activation
Introduction
Neuromyelitis optica (NMO) is a demyelinating autoimmune disease in which the optic nerves and spinal cord are most commonly involved [1], and was first described by Eugène Devic's team in 1894 [2]. Some NMO patients will eventually become blind and disabled that even need to be wheelchair-bound [3], which will greatly reduce quality of life and cause a great economic burden.
Similar to multiple sclerosis (MS), NMO is also an inflammatory demyelinating disease. However, in NMO, astrocytes are the targets of immune attack, rather than oligodendrocytes, as occurs in in MS. These two diseases could be reliably distinguished by the detection of the biomarker aquaporin-4 (AQP4)-IgG, which is present in the majority of NMO patients but usually absent in cases of MS [4]. On this basis, the concept of neuromyelitis optica spectrum disorder (NMOSD) was first proposed and defined in 2007 [5]. In 2015, the latest diagnostic guidelines included NMO in NMOSD and divided NMOSD into AQP4-IgG-positive and AQP4-IgG-negative forms according to the expression of the AQP4 antibody [6]. In addition to AQP4-IgG, another biomarker, myelin oligodendrocyte glycoprotein (MOG)-IgG, has been identified and used for the diagnosis of NMOSD; AQP4-IgG has been detected in approximately 70% of NMOSD patients globally, and MOG-IgG has been detected in approximately 40% of AQP4-Ab-negative NMOSD patients [7]. According to many epidemiological studies, NMOSD has great importance in the prevalence in different regions and racial groups worldwide [8, 9]. Although the prevalence of NMOSD is limited, severe autoimmune attacks may cause spinal cord and optic nerve involvement, which leads to paralysis and blindness in the patients [10, 11].
Immunosuppressive therapies have been used to prevent relapses and decrease attack severity in patients with NMOSD [12]. For instance, because the repopulation of B cells is relevant to the relapse rate of NMOSD, immunotherapies targeting B cells and related proteins were demonstrated to be effective in treating NMOSD [13, 14], and several drugs have been licenced and applied as clinical therapies [15, 16]. In addition to B cell depletion therapy, numerous kinds of treatments have been demonstrated to be effective in treating NMOSD, such as interleukin (IL)-6 receptor antagonists, complement blockers and other monoclonal antibodies (such as aquaporumab, bevacizumab and ublituximab) [17]. The efficacy of novel therapies (including eculizumab to target the complement system, satralizumab to target the IL-6 receptor, and inebilizumab to target B cells) has been assessed by randomized controlled trials of patients with NMOSD, and the results show that these therapies were beneficial in preventing autoimmune attacks, but the studies also show various efficacy, safety, tolerability, and practical considerations associated with these drugs [18].
As an essential inflammatory cytokine, IL-6 has been reported to be highly expressed in the serum and cerebrospinal fluid (CSF) of NMO patients, and IL-6 levels were positively associated with disease severity [19]. It is reported that Th1 and Th17 cytokines/chemokines are upregulated in NMO [20], and the dominant Th17-related response in NMO patients is positively correlated with neurological disability [21]. Moreover, the serum concentrations of CXCL6/GCP-2, Midkine and C5/C5a are significantly altered between AQP4-positive NMO and MOG-positive NMO cases, which indicates different immunological mechanisms of NMO pathogenesis that are associated with these two kinds of antibodies [22].
As native macrophages in the central nervous system (CNS), microglia plays diverse roles and has phagocytic and pro- and anti-inflammatory functions in numerous neurological diseases [23, 24]. Microglia also plays a key role in the immunoregulation of neuroinflammatory diseases [25]. However, in recent years, investigations on NMO have often focused on astrocytes, while the regulatory roles of microglia have been largely neglected [26]. Histopathological analysis demonstrates prominent infiltration and activation of macrophages/microglia in NMO lesions [27]. Microglia are the major mediators of CNS inflammation, and AQP4-IgG can induce cytokine production by activating astrocytes and lead to bystander activation towards microglia [28]. As one of the major antibody-mediated effects of NMOSD, microglial activation has both protective and disruptive effects, while the regulation of these two opposing effects remains to be further studied [29]. Chen et al. indicate that with the involvement of complement, astrocyte-microglia crosstalk plays a critical role in promoting the development of NMO [30]. Therefore, microglia are thought to be potential therapeutic targets in treating NMO [26].
In this review, we summarize the recent research progress on the roles of microglia in NMOSD pathogenesis, and the mechanisms of microglial activation and microglial-mediated inflammation are also discussed. We also suggest the potential research prospects of microglial activation in the context of basic research and clinical practice.
The upstream mechanism of microglial activation in NMOSD
IL-6
Activated microglia can recruit macrophages and neutrophilic granulocytes to the NMO lesions, induce the production of IL-1β, IL-6, IL-17, and high-mobility group box 1 protein (HMGB1), which might be involved in NMO pathogenesis [31]. IL-17/IL-8 axis activation in cerebrospinal fluid (CSF) and the increase in HMGB1 in the plasma of NMOSD patients have been determined [32, 33]. More importantly, CSF levels of IL-6 in NMOSD patients are much higher compared to MS patients, and there is strong evidence showing that IL-6 may play a key role in the pathogenesis of NMOSD [34].
As a potent regulator of cellular communication, the inflammatory factor IL-6 is responsible for both innate and adaptive inflammatory responses by interacting with IL-6 receptor-α (IL-6R) [35]. Many CNS-resident cells can produce IL-6, but IL-6R is expressed on a limited subset of cells. IL-6R is mainly found on the surface of microglia but not on the surface of astrocytes, oligodendrocytes, endothelial cells, or neurons [36]. IL-6 binds to membrane-bound IL-6R on microglia, triggering gp130 homodimerization to form a functional receptor complex. The homodimerization of the receptor complex activates Janus kinase 1 (JAK1) and JAK2, leading to the activation of signal transducers and activators of transcription (STAT) 3 [37]. Thus, activated STAT3 may translocate into the nuclei and mediate the expression of IL-6-regulated genes (Figure 1).
The upstream mechanism of microglial activation in NMOSD. (1) IL-6 activates microglia by directly binding to IL-6R/gp130 (classical signalling) or sIL-6R (trans-signalling), leading to STAT3 activation and ultimately promoting the gene expression of iNOS, IL-1α, IL-1β and TNF-α. (2) In response to IFN-I, STAT1 is activated in microglia, resulting in the gene expression of 2'5'-OAS, CD86, MHC-I and MHC-II. IL-6, interleukin-6; IFN-I: type I interferon; sIL-6R, soluble IL-6R; JAK, Janus kinase; STAT, signal transducer and activator of transcription; MHC, major histocompatibility complex.
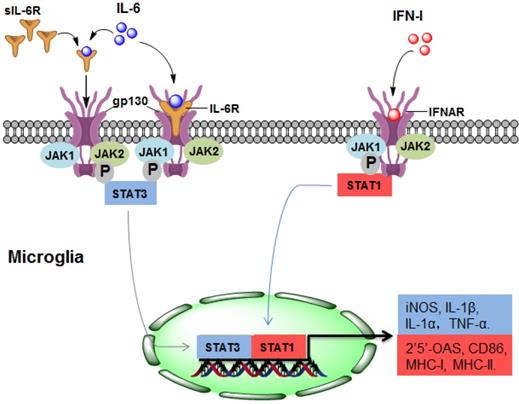
During the pathogenesis of NMOSD, the promoting effect of IL-6 may be mainly due to IL-6-mediated activation of microglia through binding with IL-6R. In CNS cells lacking IL-6R, soluble IL-6R (sIL-6R) mediates the response to IL-6, which is called trans-signalling [38]. Compared with IL-6R, sIL-6R lacks cytoplasmic and transmembrane regions, binds to IL-6 and activates downstream signalling via ubiquitously expressed gp130 on the cell surface [39], which may also contribute to the pathological process of NMOSD (Figure 1).
Type I interferon (IFN-I)
Type I interferons (IFN-Is), which are group of polypeptides associated with intracellular antimicrobial programmes, are crucial in mediating innate and adaptive immune responses [40]. It has been determined that the chronic production of IFN-Is in the CNS is the causal factor driving the development of NMOSD (Figure 1). The IFN-I family consists of many subtypes, the human IFN-I family includes IFN-α, IFN- β, IFN- ε, IFN-κ, and IFN-ω, and the mouse origin includes IFN-α, IFN-β, IFN-ε, IFN-κ, and IFN-ζ [25, 41]. The serum level of IFN-α in patients with NMOSD is increased, which is positively correlated with disease severity [42]. IFN-β is widely used for treating MS, but it has no treatment effect in NMOSD, and even aggravates the severity of the disease [43].
Almost every CNS-resident cell can express and secrete IFN-I, including microglia, neurons, astrocytes and endothelial cells. IFNAR1 and IFNAR2, the IFN-I receptor genes, are also widely expressed in the CNS [25]. Therefore, IFN-I binds to IFNAR and activates downstream signalling which may regulate 2'5'-OAS, CD86, MHC-Ⅰ and MHC-Ⅱ, and this effect may be widespread in different cell types of the CNS [25] (Figure 1). Importantly, the response of microglia to IFN-I is more pronounced than that of astrocytes or neurons [44], indicating that microglia may account for the major cell population that responds to IFN-I in NMOSD. A recent study determined that IFN-β treatment exacerbates the severity of NMOSD and induces microglial activation, as indicated by the expansion of a CD11c+ subset of microglia, suggesting that IFNI-activated microglia play a pathologic role in NMOSD [43].
Factors secreted by activated microglia are involved in the pathogenesis of NMOSD
Complement
AQP4 is mainly distributed on astrocyte foot processes located in the blood-brain barrier. AQP4-specific antibody (NMO-IgG) supports the cooperation between the cellular and humoral arms of adaptive immunity in NMOSD pathogenesis [45] which is capable of activating complement. Complement proteins are involved in astrocyte destruction and secondary demyelination during the pathological process of NMOSD [27]. Therefore, NMOSD is also considered a complement-mediated astrocytopathy.
It is accepted that NMO-IgG can induce massive production of C3 by astrocytes [46], and C3a receptor (C3aR) is mostly expressed on microglia [47]. Whether in healthy or pathological conditions, astrocytes and microglia coordinate their functions [48]. We may speculate that astrocytes could promote microglial activation through the C3 cleavage product C3a. A recent study showed that NMO-IgG binds to AQP4 on astrocyte endfoot processes, resulting in AQP4 internalization, decreased cell surface AQP4 expression, and astrocyte activation [30]. Elevated C3 produced by activated astrocytes is cleaved to form C3a and C3b. Secreted C3a binds with C3aR on resting microglia, which promotes microglial activation, the production of C1q, and the convergence towards astrocytes [30]. C1q derived from activated microglia facilitates localized injury to neurons and oligodendrocytes [30] (Figure 2). In addition, C1q may activate the classic complement cascade, leading to sustained CNS tissue damage in NMOSD [26].
Factors secreted by activated microglia are involved in the pathogenesis of NMOSD. NMO-IgG binds to AQP4 on astrocytes, resulting in astrocyte activation. Activated astrocytes secrete complement C3a, which binds with C3aR on resting microglia, leading to microglial activation. The secretion of complement C1q, inflammatory cytokines and PGRN by activated microglia is increased. These secretions further promote microglial activation in an autocrine manner and facilitate localized injury to neurons, astrocytes and oligodendrocytes in a paracrine manner. AQP4, aquaporin 4; NMO-IgG, neuromyelitis optica immunoglobulin; PGRN, progranulin.
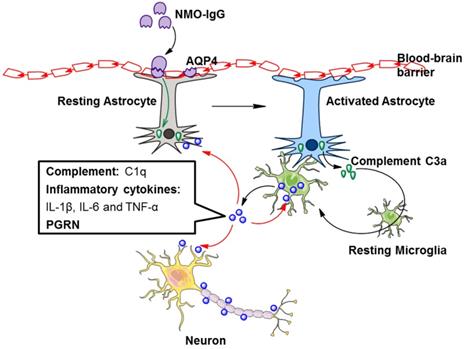
In addition to complement, TGF-β derived from astrocytes induces neuronal release of complement, which ultimately acts upon microglia [48]. Astrocyte-derived IL-1 could change the permeability of the blood-brain barrier, accordingly allowing microglial activation [48]. Astrocytes and microglia directly affect each other via molecules such as IL-1a, TNF, and IL-33 [48]. Hence, astrocyte-microglial communication may be a target of NMOSD therapy (Figure 2).
Inflammatory cytokines
Microglial activation is a two-edged sword in NMOSD. Microglial activation may lead to demyelination [49], while it has also been reported to contribute to remyelination [50]. Microglia is capable to secrete both pro- and anti-inflammatory cytokines in response to external stimuli [51]. Correspondingly, activated microglia can be mainly polarized into two functional phenotypes: M1 and M2 phenotype [52-55].
In the initial stage of CNS diseases, activated microglia are dominated by the M2 phenotype, which is related to the release of several anti-inflammatory cytokines, including IL-10, IL-4, IL-13 and TGF-β, and proinflammatory cytokine production is suppressed [56]. Meanwhile M1 phenotype activation is connected with the release of proinflammatory cytokines, such as IL-1β, IL-6 and TNF-α, and an excess accumulation of these factors caused by chronic activation of microglia can result in damage to the surrounding neuronal cells [51]. These proinflammatory cytokines are involved in the pathological process of NMOSD [57]. For example, IL-1β induces neuromyelitis optica-like lesions in a in vivo study [58]. Collectively, M1/M2 polarization might regulate microglial activation as a dual role in NMOSD. However, the regulatory role of inflammatory cytokines derived from microglial activation in NMOSD has not been deeply studied and might be a potential research field.
Progranulin (PGRN)
Progranulin (PGRN), a secreted glycosylated protein, was originally considered to be a growth factor that regulates immune responses and cancer growth [59]. PGRN has been reported to be highly enriched in microglia and neurons [60]. Kimura et al. reported that CSF levels of PRRN in NMOSD patients are notably higher than those in MS patients and controls without inflammation [61]. They also demonstrated that CSF levels of PGRN have a significant correlation with cell counts and protein levels in CSF. It also affects total spinal cord lesion length, which may be related to the severity of spinal cord inflammation in NMOSD [61]. Furthermore, there is a positive correlation between IL-6 and PGRN levels in patients with NMOSD and MS [61]. As IL-6 triggers PGRN expression in cancer studies [62, 63], IL-6 may serve as a crucial regulator of PGRN in response to microglial activation. It has also been indicated that PGRN enhances IL-6 expression in adipocytes [64]. Therefore, CSF PGRN in NMOSD may be mainly derived from microglia and may have interactive regulation with IL-6.
A growing body of evidence suggests that PGRN functions as an autocrine neuroprotective factor by modulating neuroinflammation [59]. PGRN-deficient mice are hypersusceptible to neuroinflammation and neuronal loss in response to the injury, and PGRN exhibits a neuroprotective effect by decreasing proinflammatory cytokines and increasing anti-inflammatory cytokines [60, 65-67]. In view of this evidence and recent research advances on the role of PGRN in NMOSD, PGRN derived from activated microglia may provide negative feedback in NMOSD (Figure 2).
Prospects and potential applications
Recently, microglia has been identified as potential modulators and targets for NMOSD diagnosis and therapy because of their important roles in neurological disorders.
Regulation by noncoding RNAs
Although they are never translated into proteins, noncoding RNAs (ncRNAs) have been recognized as playing essential regulatory roles in the human CNS, as well as in neurological diseases [68]. It has been indicated that microRNAs (miRNAs) are significantly altered in the whole blood of NMOSD patients when compared to MS patients[69, 70], while long noncoding RNAs (lncRNAs), the another kind of ncRNA, are differentially expressed in peripheral blood mononuclear cells (PBMCs) of NMOSD patients compared to those of controls [71]. Circulating miRNAs from whole blood, as well as serum exosomal miRNAs, might serve as potential biomarkers for the diagnosis and prognosis of NMOSD [72, 73], and several miRNAs are also capable of discriminating NMOSD from MS or neuropsychiatric systemic lupus erythematosus (NPSLE) [74, 75]. It has been revealed that different kinds of ncRNAs (miRNAs [76], lncRNAs [77], circular RNAs (circRNAs) [78], etc.) are involved in the regulation of microglia in the pathogenesis of neurological diseases such as MS and epilepsy. The mechanisms include regulating gene expressions and microglia polarization, and acting as a molecular sponge. However, the relevance and mechanisms of microglia and ncRNAs in NMOSD remain unclear and deserve to be further investigated.
Utilization of other novel technologies
Recently, many new technologies have been used to study NMOSD and other autoimmune neurological disorders. For example, single-cell sequencing technologies have been applied to investigate B cells and antibodies in the blood and CSF of NMOSD patients [79]. Haematopoietic stem cell transplantation has been considered a possible option for treating patients with severe NMOSD [80]. To reduce brain lesions and therapeutically treat patients with CSF1R-related leukoencephalopathy, haematopoietic stem cell transplantation could renew dysfunctional microglia [81]. The effect and application of haematopoietic stem cell transplantation on microglia should be further investigated in patients with NMOSD. Although there are some limitations, such as immunogenic reactions and a lack of specificity of viral vectors, gene therapy has been applied with positive outcomes in neurological diseases, including MS [82]. Insulin-like growth factor 1 (IGF1) gene therapy could target microglia, modify the inflammatory response and improve cognitive or motor deficits [83]. Therefore, gene therapeutic approaches might be a promising choice to partially modulate the progression of NMOSD by targeting microglial activation. To identify new therapeutic and diagnostic approaches with safety and efficacy, these novel technologies should be utilized in future studies of microglial activation in NMOSD.
Conclusion
Although the functions and mechanisms of microglia remain under investigation, the activation of these immunomodulatory cells attracts the attention of researchers. Cytokines such as IL-6 and the IFN-I family, the complement system and PGRN are involved in the pathogenesis of NMOSD. M1 or M2 polarization might regulate microglial activation in NMOSD. Combined studies with ncRNAs, as well as novel technologies such as single-cell sequencing, stem cell therapy and gene therapy, are promising options for investigating the microglial activation in NMOSD.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81800855), Hunan Provincial Natural Scientific Foundation (No. 2019JJ50849) and Scientific Research Project of Hunan Provincial Health Commission (No. 202113050843).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. 1999;53:1107-14
2. Jarius S, Wildemann B. The history of neuromyelitis optica. J Neuroinflammation. 2013;10:8
3. Mao-Draayer Y, Thiel S, Mills EA, Chitnis T, Fabian M, Katz Sand I. et al. Neuromyelitis optica spectrum disorders and pregnancy: therapeutic considerations. Nat Rev Neurol. 2020;16:154-70
4. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473-7
5. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805-15
6. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T. et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177-89
7. Thoman ME, McKarns SC. Metabolomic Profiling in Neuromyelitis Optica Spectrum Disorder Biomarker Discovery. Metabolites. 2020 10
8. Hor JY, Asgari N, Nakashima I, Broadley SA, Leite MI, Kissani N. et al. Epidemiology of Neuromyelitis Optica Spectrum Disorder and Its Prevalence and Incidence Worldwide. Front Neurol. 2020;11:501
9. Papp V, Magyari M, Aktas O, Berger T, Broadley SA, Cabre P. et al. Worldwide Incidence and Prevalence of Neuromyelitis Optica: A Systematic Review. Neurology. 2021;96:59-77
10. Ciccarelli O, Cohen JA, Reingold SC, Weinshenker BG, International Conference on Spinal Cord I, Imaging in Multiple S. et al. Spinal cord involvement in multiple sclerosis and neuromyelitis optica spectrum disorders. Lancet Neurol. 2019;18:185-97
11. Gospe SM 3rd, Chen JJ, Bhatti MT. Neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disorder-optic neuritis: a comprehensive review of diagnosis and treatment. Eye (Lond). 2021;35:753-68
12. Paul F, Murphy O, Pardo S, Levy M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin Investig Drugs. 2018;27:265-71
13. Nosadini M, Alper G, Riney CJ, Benson LA, Mohammad SS, Ramanathan S. et al. Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2016;3:e188
14. Kim SH, Hyun JW, Kim HJ. Individualized B cell-targeting therapy for neuromyelitis optica spectrum disorder. Neurochem Int. 2019;130:104347
15. Graf J, Mares J, Barnett M, Aktas O, Albrecht P, Zamvil SS. et al. Targeting B cells to modify MS, NMOSD, and MOGAD: Part 2. Neurol Neuroimmunol Neuroinflamm. 2021 8
16. Graf J, Mares J, Barnett M, Aktas O, Albrecht P, Zamvil SS. et al. Targeting B Cells to Modify MS, NMOSD, and MOGAD: Part 1. Neurol Neuroimmunol Neuroinflamm. 2021 8
17. Xie Q, Sun M, Sun J, Zheng T, Wang M. New progress in the treatment of neuromyelitis optica spectrum disorder with monoclonal antibodies (Review). Exp Ther Med. 2021;21:148
18. Levy M, Fujihara K, Palace J. New therapies for neuromyelitis optica spectrum disorder. Lancet Neurol. 2021;20:60-7
19. Icoz S, Tuzun E, Kurtuncu M, Durmus H, Mutlu M, Eraksoy M. et al. Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int J Neurosci. 2010;120:71-5
20. Matsushita T, Tateishi T, Isobe N, Yonekawa T, Yamasaki R, Matsuse D. et al. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One. 2013;8:e61835
21. Linhares UC, Schiavoni PB, Barros PO, Kasahara TM, Teixeira B, Ferreira TB. et al. The ex vivo production of IL-6 and IL-21 by CD4+ T cells is directly associated with neurological disability in neuromyelitis optica patients. J Clin Immunol. 2013;33:179-89
22. Ai N, Liu H, Zhou H, Lin D, Wang J, Yang M. et al. Cytokines and chemokines expression in serum of patients with neuromyelitis optica. Neuropsychiatr Dis Treat. 2019;15:303-10
23. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23:1018-27
24. Qin C, Zhou LQ, Ma XT, Hu ZW, Yang S, Chen M. et al. Dual Functions of Microglia in Ischemic Stroke. Neurosci Bull. 2019;35:921-33
25. West PK, Viengkhou B, Campbell IL, Hofer MJ. Microglia responses to interleukin-6 and type I interferons in neuroinflammatory disease. Glia. 2019;67:1821-41
26. Moinfar Z, Zamvil SS. Microglia complement astrocytes in neuromyelitis optica. J Clin Invest. 2020;130:3961-4
27. Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol. 2014;24:83-97
28. Chen T, Bosco DB, Ying Y, Tian DS, Wu LJ. The Emerging Role of Microglia in Neuromyelitis Optica. Front Immunol. 2021;12:616301
29. Ramakrishnan P. Could Galectin-3 be a key player in the etiology of neuromyelitis optica spectrum disorder? Med Hypotheses. 2021;146:110450
30. Chen T, Lennon VA, Liu YU, Bosco DB, Li Y, Yi MH. et al. Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J Clin Invest. 2020;130:4025-38
31. Bradl M, Kanamori Y, Nakashima I, Misu T, Fujihara K, Lassmann H. et al. Pain in neuromyelitis optica-prevalence, pathogenesis and therapy. Nat Rev Neurol. 2014;10:529-36
32. Ishizu T, Osoegawa M, Mei FJ, Kikuchi H, Tanaka M, Takakura Y. et al. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain. 2005;128:988-1002
33. Wang KC, Tsai CP, Lee CL, Chen SY, Chin LT, Chen SJ. Elevated plasma high-mobility group box 1 protein is a potential marker for neuromyelitis optica. Neuroscience. 2012;226:510-6
34. Uzawa A, Mori M, Ito M, Uchida T, Hayakawa S, Masuda S. et al. Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J Neurol. 2009;256:2082-4
35. Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175:3463-8
36. Hsu MP, Frausto R, Rose-John S, Campbell IL. Analysis of IL-6/gp130 family receptor expression reveals that in contrast to astroglia, microglia lack the oncostatin M receptor and functional responses to oncostatin M. Glia. 2015;63:132-41
37. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16:448-57
38. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. 2012;8:1237-47
39. Rose-John S. The soluble interleukin-6 receptor and related proteins. Best Pract Res Clin Endocrinol Metab. 2015;29:787-97
40. Blank T, Prinz M. Type I interferon pathway in CNS homeostasis and neurological disorders. Glia. 2017;65:1397-406
41. Owens T, Khorooshi R, Wlodarczyk A, Asgari N. Interferons in the central nervous system: a few instruments play many tunes. Glia. 2014;62:339-55
42. Asgari N, Voss A, Steenstrup T, Kyvik KO, Stenager E, Lillevang ST. Interferon alpha association with neuromyelitis optica. Clin Dev Immunol. 2013;2013:713519
43. Wlodarczyk A, Khorooshi R, Marczynska J, Holtman IR, Burton M, Jensen KN. et al. Type I interferon-activated microglia are critical for neuromyelitis optica pathology. Glia. 2021;69:943-53
44. Li W, Viengkhou B, Denyer G, West PK, Campbell IL, Hofer MJ. Microglia have a more extensive and divergent response to interferon-alpha compared with astrocytes. Glia. 2018;66:2058-78
45. Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, Cree BA. et al. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol. 2012;72:53-64
46. Howe CL, Kaptzan T, Magana SM, Ayers-Ringler JR, LaFrance-Corey RG, Lucchinetti CF. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia. 2014;62:692-708
47. Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26:201-11
48. Vainchtein ID, Molofsky AV. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020;43:144-54
49. Liu Y, Given KS, Harlow DE, Matschulat AM, Macklin WB, Bennett JL. et al. Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol Commun. 2017;5:25
50. Liu Y, Given KS, Owens GP, Macklin WB, Bennett JL. Distinct patterns of glia repair and remyelination in antibody-mediated demyelination models of multiple sclerosis and neuromyelitis optica. Glia. 2018;66:2575-88
51. Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. 2019;94:112-20
52. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173:649-65
53. Pan J, Jin JL, Ge HM, Yin KL, Chen X, Han LJ. et al. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. J Neuroinflammation. 2015;12:51
54. Liu LR, Liu JC, Bao JS, Bai QQ, Wang GQ. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front Immunol. 2020;11:1024
55. Jin X, Liu MY, Zhang DF, Zhong X, Du K, Qian P. et al. Natural products as a potential modulator of microglial polarization in neurodegenerative diseases. Pharmacol Res. 2019;145:104253
56. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19:987-91
57. Tradtrantip L, Asavapanumas N, Verkman AS. Emerging therapeutic targets for neuromyelitis optica spectrum disorder. Expert Opin Ther Targets. 2020;24:219-29
58. Kitic M, Hochmeister S, Wimmer I, Bauer J, Misu T, Mader S. et al. Intrastriatal injection of interleukin-1 beta triggers the formation of neuromyelitis optica-like lesions in NMO-IgG seropositive rats. Acta Neuropathol Commun. 2013;1:5
59. Kao AW, McKay A, Singh PP, Brunet A, Huang EJ. Progranulin, lysosomal regulation and neurodegenerative disease. Nat Rev Neurosci. 2017;18:325-33
60. Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T. et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117-28
61. Kimura A, Takemura M, Saito K, Serrero G, Yoshikura N, Hayashi Y. et al. Increased cerebrospinal fluid progranulin correlates with interleukin-6 in the acute phase of neuromyelitis optica spectrum disorder. J Neuroimmunol. 2017;305:175-81
62. Liu F, Zhang W, Yang F, Feng T, Zhou M, Yu Y. et al. Interleukin-6-stimulated progranulin expression contributes to the malignancy of hepatocellular carcinoma cells by activating mTOR signaling. Sci Rep. 2016;6:21260
63. Frampton G, Invernizzi P, Bernuzzi F, Pae HY, Quinn M, Horvat D. et al. Interleukin-6-driven progranulin expression increases cholangiocarcinoma growth by an Akt-dependent mechanism. Gut. 2012;61:268-77
64. Matsubara T, Mita A, Minami K, Hosooka T, Kitazawa S, Takahashi K. et al. PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 2012;15:38-50
65. Wang C, Zhang L, Ndong JC, Hettinghouse A, Sun G, Chen C. et al. Progranulin deficiency exacerbates spinal cord injury by promoting neuroinflammation and cell apoptosis in mice. J Neuroinflammation. 2019;16:238
66. Filiano AJ, Martens LH, Young AH, Warmus BA, Zhou P, Diaz-Ramirez G. et al. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci. 2013;33:5352-61
67. Menzel L, Kleber L, Friedrich C, Hummel R, Dangel L, Winter J. et al. Progranulin protects against exaggerated axonal injury and astrogliosis following traumatic brain injury. Glia. 2017;65:278-92
68. Salta E, De Strooper B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol. 2012;11:189-200
69. Chen J, Zhu J, Wang Z, Yao X, Wu X, Liu F. et al. MicroRNAs Correlate with Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder in a Chinese Population. Med Sci Monit. 2017;23:2565-83
70. Keller A, Leidinger P, Meese E, Haas J, Backes C, Rasche L. et al. Next-generation sequencing identifies altered whole blood microRNAs in neuromyelitis optica spectrum disorder which may permit discrimination from multiple sclerosis. J Neuroinflammation. 2015;12:196
71. Xu J, Zhang F, Gao C, Ma X, Peng X, Kong D. et al. Microarray Analysis of lncRNA and mRNA Expression Profiles in Patients with Neuromyelitis Optica. Mol Neurobiol. 2017;54:2201-8
72. Vaknin-Dembinsky A, Charbit H, Brill L, Abramsky O, Gur-Wahnon D, Ben-Dov IZ. et al. Circulating microRNAs as biomarkers for rituximab therapy, in neuromyelitis optica (NMO). J Neuroinflammation. 2016;13:179
73. Chen C, Wu Y, Li M, Cui C, Zhao Y, Sun X. et al. Different Exosomal microRNA Profile in Aquaporin-4 Antibody Positive Neuromyelitis Optica Spectrum Disorders. Front Immunol. 2020;11:1064
74. Sharaf-Eldin W, Kishk N, Sakr B, El-Hariri H, Refeat M, ElBagoury N. et al. Potential Value of miR-23a for Discriminating Neuromyelitis Optica Spectrum Disorder from Multiple Sclerosis. Arch Iran Med. 2020;23:678-87
75. Sharaf-Eldin WE, Kishk NA, Gad YZ, Hassan H, Ali MAM, Zaki MS. et al. Extracellular miR-145, miR-223 and miR-326 expression signature allow for differential diagnosis of immune-mediated neuroinflammatory diseases. J Neurol Sci. 2017;383:188-98
76. Walsh AD, Nguyen LT, Binder MD. miRNAs in Microglia: Important Players in Multiple Sclerosis Pathology. ASN Neuro. 2021;13:1759091420981182
77. Sun D, Yu Z, Fang X, Liu M, Pu Y, Shao Q. et al. LncRNA GAS5 inhibits microglial M2 polarization and exacerbates demyelination. EMBO Rep. 2017;18:1801-16
78. Xiaoying G, Guo M, Jie L, Yanmei Z, Ying C, Shengjie S. et al. CircHivep2 contributes to microglia activation and inflammation via miR-181a-5p/SOCS2 signalling in mice with kainic acid-induced epileptic seizures. J Cell Mol Med. 2020;24:12980-93
79. Zou A, Ramanathan S, Dale RC, Brilot F. Single-cell approaches to investigate B cells and antibodies in autoimmune neurological disorders. Cell Mol Immunol. 2021;18:294-306
80. Ceglie G, Papetti L, Valeriani M, Merli P. Hematopoietic Stem Cell Transplantation in Neuromyelitis Optica-Spectrum Disorders (NMO-SD): State-of-the-Art and Future Perspectives. Int J Mol Sci. 2020 21
81. Han J, Sarlus H, Wszolek ZK, Karrenbauer VD, Harris RA. Microglial replacement therapy: a potential therapeutic strategy for incurable CSF1R-related leukoencephalopathy. Acta Neuropathol Commun. 2020;8:217
82. Iqubal A, Iqubal MK, Khan A, Ali J, Baboota S, Haque SE. Gene Therapy, A Novel Therapeutic Tool for Neurological Disorders: Current Progress, Challenges and Future Prospective. Curr Gene Ther. 2020;20:184-94
83. Falomir-Lockhart E, Dolcetti FJC, Garcia-Segura LM, Herenu CB, Bellini MJ. IGF1 Gene Therapy Modifies Microglia in the Striatum of Senile Rats. Front Aging Neurosci. 2019;11:48
Author contact
![]() Corresponding author: Yedi Zhou, MD, PhD, Department of Ophthalmology, The Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China. Telephone: +86-731-85292175; E-mail: zhouyediedu.cn.
Corresponding author: Yedi Zhou, MD, PhD, Department of Ophthalmology, The Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China. Telephone: +86-731-85292175; E-mail: zhouyediedu.cn.