Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Clinical Characteristics of MFS
Detection of Disease-associated...
The correlation of...
Epigenetic Regulation of...
Abnormalities of TGF-β...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(13):2752-2766. doi:10.7150/ijms.60685 This issue Cite
Review
The Molecular Genetics of Marfan Syndrome
Qiu Du1*, Dingding Zhang1,2 ![]() *, Yue Zhuang3*, Qiongrong Xia1, Taishen Wen2, Haiping Jia4
*, Yue Zhuang3*, Qiongrong Xia1, Taishen Wen2, Haiping Jia4
1. Marfan Research Group, College of Medical Technology, Chengdu University of Traditional Chinese Medicine, Chengdu, 610072, Sichuan, China.
2. Sichuan Provincial Key Laboratory for Genetic Disease, Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, 611731, Sichuan, China.
3. Department of Rheumatology and Immunology, Sichuan Provincial People's Hospital, University of Electronic Science and Technology of China, Chengdu, 611731, Sichuan, China.
4. Department of Immunology, North Sichuan Medical College, Nanchong, 637100, Sichuan, China.
*Contributed equally
Received 2021-3-21; Accepted 2021-5-18; Published 2021-5-27
Abstract

Marfan syndrome (MFS) is a complex connective tissue disease that is primarily characterized by cardiovascular, ocular and skeletal systems disorders. Despite its rarity, MFS severely impacts the quality of life of the patients. It has been shown that molecular genetic factors serve critical roles in the pathogenesis of MFS. FBN1 is associated with MFS and the other genes such as FBN2, transforming growth factor beta (TGF-β) receptors (TGFBR1 and TGFBR2), latent TGF-β-binding protein 2 (LTBP2) and SKI, amongst others also have their associated syndromes, however high overlap may exist between these syndromes and MFS. Abnormalities in the TGF-β signaling pathway also contribute to the development of aneurysms in patients with MFS, although the detailed molecular mechanism remains unclear. Mutant FBN1 protein may cause unstableness in elastic structures, thereby perturbing the TGF-β signaling pathway, which regulates several processes in cells. Additionally, DNA methylation of FBN1 and histone acetylation in an MFS mouse model demonstrated that epigenetic factors play a regulatory role in MFS. The purpose of the present review is to provide an up-to-date understanding of MFS-related genes and relevant assessment technologies, with the aim of laying a foundation for the early diagnosis, consultation and treatment of MFS.
Keywords: Marfan syndrome, diagnose, genetics, technology
Introduction
Marfan syndrome (MFS), a complicated genetic connective tissue disorder named after Antoine-Bernard Marfan in 1896, presents with striking pleiotropism and clinical variability. Patients with MFS exhibit a wide range of clinical symptoms, including abnormalities in the ocular, skeletal and cardiovascular systems. Aortic pathology is the predominant cause of death in patients with MFS [1]. Mild cases may only have isolated MFS characteristics, whereas severe cases have rapidly progressive lesions involving multiple systems during the neonatal period, and may succumb to the disease within 2 years after birth [2,3]. MFS affects ~1/5,000 individuals in the general population [4]. The prevalence of MFS in the Danish population in 2015 was 6.5/100,000, which was 41% higher compared with 20 years ago [5]. The average lifespan of Danish patients with MFS was 50 years [6]. The average lifespan of MFS patients was 32 years in 1970s. With the development of aortic root replacement treatment, the mean lifespan expectancy of MFS increased to 41 years old in 1995 [7], even nearly 72 years old with proper management [8]. Notably, Veiga-Fernández et al showed that 34.54% of early-onset MFS (EOMS) cases were suspected based on prenatal ultrasound anomalies, and 65.45% of EOMS cases were diagnosed after birth. The mortality rate in the first 15 months was 73.68% in cases with prenatal suspicion, and 61.1% of patients that were postnatally diagnosed died within the first 5 years [3]. Of note, MFS not only has a high mortality rate among newborns, but also seriously affects the quality of life of surviving patients.
Although sporadic cases of MFS account for 25-30% of those diagnosed [9], relevant clinical data provide evidence that MFS is primarily an autosomal dominant disease. Additionally, there are a few cases of MFS families with a reported autosomal recessive inheritance model [10, 11]. Several patients with MFS can be traced based on their family history. Manifestations of MFS may worsen with age, although early-stage symptoms are not apparent/visible. For mild cases, early-stage diagnosis may be delayed due to a lack of symptoms. Certain manifestations of MFS also overlap with other diseases, such as Loeys-Dietz syndrome (LDS) and Shprintzen-Goldberg syndrome (SGS) [12]. Additionally, the molecular pathogenesis of MFS has not been fully established.
Aortic development in patients with MFS presents sexual dimorphism. Aortic aneurysms in patients with MFS are more severe in males compared with females [13]. Early diagnosis of MFS is useful in preventing cardiovascular symptoms and may encourage early treatment of the disease, thereby improving the quality of life of the patients and potentially prolonging their lifespan. Diagnostic criteria involving multiple system manifestation scores and updated diagnostic criteria in 2010 emphasized the importance of fibrillin-1 (FBN1) genetic testing [14]. Currently, specific gene mutations, including FBN2, transforming growth factor beta (TGF-β) receptors (TGFBR1 and TGFBR2), latent TGFβ-binding protein 2 (LTBP2) and SKI, have been identified in their associated syndromes by several molecular genetic techniques, including linkage analysis, scanning methods and specific identification techniques. The present review focuses on the genetic aspects of MFS and the screening of MFS-associated genes.
Clinical Characteristics of MFS
MFS involves abnormalities of multiple systems, including the cardiovascular, ocular and skeletal systems, and include lung, skin and central nervous system disorders [15]. Patients with MFS may also develop sleep disorders and depressive symptoms due to long-term severe physical pain [16]. Compared to the abnormalities of the cardiovascular system in patients with MFS, loss of vision or skeletal anomalies are more likely to be discovered earlier.
Effects on the cardiovascular system
Pathological changes to the cardiovascular system are common amongst patients with MFS. The typical clinical manifestation of MFS is dilation of the aortic root, and the atrioventricular valves are the most likely affected tissues. The thickening of atrioventricular valves is often associated with atrioventricular valves prolapse [17]. Rybczynski et al reported that the incidence of mitral valve prolapse (MVP), regurgitation and endocarditis was 42.6, 56.5 and 0.92% among 204 patients with MFS at 30 years of age [18]. Furthermore, patients with MFS who appeared to be asymptomatic during childhood and adolescence, particularly female patients, gradually developed mitral valve dysfunction and aortic abnormalities at a constant rate between the ages of 5 and 20 years [19]. The dysfunction of mitral valve causes left ventricle volume overload then evolving towards systolic and diastolic dysfunction of left ventricle [20]. Moreover, in children with severe or EOMS, abnormal mitral valve function may cause regurgitation and lead to congestive heart failure, pulmonary hypertension and even death [21].
Aortic tears or ruptures due to continuous enlargement of the aorta were the primary causes of death. Dilation often occurs in the first part of the ascending aorta and progresses to an aortic aneurysm, which then forms a type A aortic dissection [22]. Hascoet et al found that children who exhibited a Valsalva sinus Z-score increase of ≥0.1 per year or a Valsalva sinus Z-score ≥3 before the age of 16 years, had a higher risk of cardiovascular events [23]. The prevalence of ascending aorta dilation increases with age, and ~96% of 965 patients with MFS developed ascending aorta dilation by the age of 60 years [24]. The prevalence of aortic root dilatation, aortic valve regurgitation and MVP was similar amongst children and adults, whereas mitral valve regurgitation, pulmonary artery dilatation, aneurysms of the aortic arch, descending thoracic aorta and abdominal aorta were found predominantly in adults [25]. Moreover, MFS have a high risk of recurrent aortic dissection, and the descending aortic size of patients with recurrent aortic dissection is greater than those with initial aortic dissection [26]. MFS patients who receive the prophylactic aortic surgery have a substantial risk of type B aortic dissection [27]. Dissection or aneurysm of the thoracic aorta was associated with degradation of support structures of the aortic wall, spine tortuosity and disturbed luminal blood flow caused by the tortuousness of the aorta [28]. Jondeau et al reported that the risk of death or aortic dissection was associated with a 4-fold increase at the aortic diameter ≥50 mm, which was considered a threshold for surgery [29]. European Society of Cardiology (ESC) recommended that surgical indications in MFS patients should be with maximal aortic diameter ≥50 mm, or MFS patients with maximal aortic diameter ≥45 mm and additional risk factors (e.g.family history of aortic dissection at a low diameter, progressive aortic regurgitation, desire for pregnancy, aortic diameter increase >3 mm/year) [30].
Welby et al confirmed that the rate of carotid artery tortuosity was 88% in patients with MFS and an increased prevalence of retrojugular course was associated with increased occurrence of aortic dissection [31]. Moreover, visceral arterial tortuosity, the aortic tortuosity index (ATI) and vertebral tortuosity index (VTI) were found to be significantly increased in patients with MFS. ATI indicates the risk of severe aortic phenotypes that are classed as type B aortic dissection as well as the aortic volume expansion rate, whereas VTI is related to early-onset dissection and death. Both are potential markers of aortic involvement [32-34].
Ophthalmological effects
Ocular defects, another major pathological manifestation of MFS, may precede cardiovascular system symptoms. Approximately 50-80% of patients with MFS exhibit ectopia lentis (EL). Compared with non-patients with MFS, additional manifestations amongst patients with MFS include an elongated axial length of the eyes, reduced visual acuity, flatter corneas, greater corneal astigmatism, a thinner central cornea, lower intraocular pressure, iris transillumination defects and lower values of sagittal height in the central cornea, corneal periphery and the sclera [35, 36]. Wang et al suggested that the mean keratometry combined with central corneal thickness can be considered as a screening index for MFS [37]. Patients with MFS are also at high risk of developing cataracts [38]. Iris irradiance defects, EL and myopia are characteristic manifestations in childhood and adolescent MFS [39]. The incidence of EL during the early stages of MFS varies widely, from 63-75% for children aged <10 years, to 15-57% for children aged <17 years [38]. Therefore, ophthalmic testing should be performed in patients with MFS even during childhood, before any cardiovascular manifestations develop.
Skeletal effects
It is generally hypothesized that the most common skeletal feature of patients with MFS is elongated bones, which may be characterized by a ratio of arm span to stature of >1.05. The overgrowth of ribs leads to pectus carinatum or pectus excavatum, and long finger and loose joints cause the wrist sign [17]. Arachnodactyly and the thumb sign are typical symptoms. Pes planus is caused by the increased ligamentous laxity and varies from mild to severe deformity [17, 40]. The scoliosis of a curve of 30°or greater is typical in MFS patients and more than 50°is severe, and the thoracic curve is usually convex to the right [40]. The other skeletal symptoms include dural ectasia, acetabular protrusion, decreased bone mineral density and etc. [40]. Sponseller et al found that ~27% of patients with MFS had skeletal lesions, 19% had <1 skeletal manifestation, and the common physical features amongst the patients were craniofacial characteristics, thumb and wrist signs, pectus excavatum and severe hindfoot valgus [41]. There is no difference in the imaging characteristics of adolescents between MFS and idiopathic scoliosis, however, scoliosis-like symptoms in patients with MFS progresses more rapidly. The incidence of cerebrospinal fluid leakage and dual expansion in patients with MFS are higher compared with non-patients [42, 43]. Thumb and wrist signs are common in children with MFS. Symptoms that involve the upper and lower limb extremities in patients with MFS typically appear first. Chest and spinal deformities may be less frequent in adolescents. The occurrence of pectus deformities, wrist signs and scoliosis as a result of MFS increases with age, whereas the prevalence of hypermobility and pes planus decreases with age [44, 45]. Moreover, total body bone mineral content and muscle mass worsen with age in patients with MFS [46].
Diagnostic criteria
Diagnostic criteria of MFS primarily depend on pathological manifestations of multiple systems and family history. Cardiovascular symptoms of MFS are emphasized in the revised Ghent criteria. In cases without a family history of MFS, patients with aortic root dissection or dilatation (Z-score ≥2) and one of the three MFS-related manifestations (EL, FBN1 mutation, the system score ≥7) can be diagnosed with MFS, and MFS diagnosis is also confirmed by the presence of EL and FBN1 mutation associated with aortic disease. Moreover, in cases with a family history of MFS, the presence of EL, or the systemic score ≥7 points or aortic root dilatation (Z-score ≥2 in individuals above 20 years old or Z-score ≥3 in individuals below 20 years old) are sufficient for an MFS diagnosis [14]. In addition, when clinical manifestations do not meet the diagnostic criterion, gene mutation detection is a suitable means of early diagnosis [47].
The prevalence and average age of MFS diagnosis are increasing [5]. This can be attributed to several factors: First, patients with MFS symptoms gradually worsen and do not reach the diagnostic criteria until an older age; thus, more people are now being diagnosed, although the actual true incidence may not have increased substantially; the mortality of MFS decreases with timely treatment, and the increasing number of patients with MFS is partially caused by a decrease in the relative risk of death; an increase in awareness of MFS has enabled the diagnosis of several cases in the adult population; finally, patients who live longer may increase the possibility of the transmission of mutants [5].
Detection of Disease-associated Genes in MFS
Multiple gene loci related to MFS have been discovered through linkage analysis and other techniques, making the study of molecular genetic mechanisms underlying the development of MFS possible. In addition, detection of specific sequences reveals potential pathogenic mutations, and can advance diagnosis and targeted therapy for patients with MFS. Detection of FBN1 mutation has been included in the revised Ghent criteria, before which only 56% of MFS children could be diagnosed without an FBN1 mutation test, whereas 85% of children currently meet the criteria following molecular testing [48]. These technologies are further discussed below.
Linkage analysis for loci exploration
Linkage analysis focuses on pedigrees and is primarily used for single-gene diseases [49]. Detection rates with linkage analysis depend on the heterogeneity of the disease locus, and may be impacted by incomplete penetrance instead of allele heterogeneity within one gene [49]. Linkage analysis has been used to explore the potential pathological loci in MFS pedigrees. In 1992, Tsipouras et al mapped the single fibrillin locus of MFS on chromosome 15 in 28 families by genetic linkage analysis [50]. In 1994, Collod et al identified a second locus of MFS located at 3p24.2-p25 by linkage analysis in a large French family without EL [51]. The usage of linkage analysis in humans is limited within a finite family size and long generational times [52].
Scanning methods for related genetic change detection
Single-strand conformation polymorphism (SSCP) analysis
The SSCP process includes PCR amplification of the target fragment. In the subsequent electrophoretic process, mutant and wild-type single strands fold into 3D shapes, showing various electrophoretic mobilities and bands in a non-denaturing gel [53]. Since the mutation contains 60% G+C of 100-300 bp, it is easily detected, but the sensitivity of SSCP is affected by multiple factors, such as the gel matrix used, size of the DNA fragments and temperature setting during electrophoresis, amongst other factors [53, 54]. Thus, there is the limitation of the size of the fragments in SSCP. Wang et al detected a 13 bp deletion (gccTc Tgcaccca) located on exon 25 of FBN1 in 9 MFS families by SSCP using direct sequencing technology [55]. Rommel et al found 7 missense mutations, 3 splice site alterations and 1 indel mutation of FBN1 in 76 patients with MFS or related symptoms by SSCP and sequencing [56].
Conformation sensitive gel electrophoresis (CSGE)
CSGE is used to scan PCR amplification products. Conformational changes of products caused by single-base mismatches are amplified by a denaturing solvent and hence increase the migration difference of single-base mismatched double strands [57]. CSGE is widely used in mutation screening of genetic diseases and detection of single-nucleotide polymorphisms, but can only detect single base changes, small insertions and deletions, with an optimal length of 200-500 bp [58]. The detection rates of FBN1 mutations by CSGE is >90% [59]. Loeys et al performed an initial mutation analysis on the cDNA and gDNA of 93 patients with MFS by CSGE and SSCP analysis. In this analysis, 73 mutations were identified, amongst which, 52 were present in the gDNA and 21 were present in the cDNA [60]. Uyeda et al reported 3 mutations of FBN1 (c.719 C>T, c.4229 T>C and c.8121 G>C), and 10 FBN3 single nucleotide polymorphisms in 12 Japanese patients with MFS by CSGE and direct sequencing [61].
Denaturing high-performance liquid chromatography (DHPLC)
In the presence of mutations, the mutant single strand and the wild-type single strand form a heteroduplex, and complementary mutant single strands or two wild-type single strands form a homoduplex, each with a different residence time on a reverse phase chromatography column. DHPLC has a high detection sensitivity of 0.5%, the minimum detection amount of the total analyzed DNA [62]. The combination of DHPLC and direct sequencing is fast and cost-effective, but cannot detect the overall deletion and duplication of the entire exon [63]. Huang et al identified a small insertion mutation (4307insTCGT) and a missense mutation (5309G>A) of FBN1 in 9 patients with MFS by DHPLC and sequencing [64]. Kosaki et al designed an automated and economical 'COPPER plate' to simultaneously detect all exons of one gene by PCR and DHPLC and, subsequently, the COPPER plate was developed for the detection of FBN1 [65].
Specific techniques for identification of MFS-associated genes
Whole exome sequencing (WES)
WES is a type of high-throughput sequencing method that functions by capturing DNA from the exonic region of the whole genome. The rare genetic events and novel mutations that are disease-associated (particularly for monogenic diseases) and potential disease-causing mutations due to incomplete penetrance can be detected by WES. Thus, it is widely used to diagnostically evaluate a patient's genetic condition [66, 67]. Moreover, WES can improve the detection rate of prenatal genetic abnormalities and more medical prediction information can be provided based on whether the mutation occurs in an important functional area of the gene [68]. The success of WES depends on mutations being compassed in the captured portion of genome and the ability to identify the pathogenic variant among many thousands of new variants [69]. Yang et al performed WES to provide a molecular diagnosis for 25% of 3,386 patients with suspected genetic conditions [66]. LaDuca et al reported that 99.7% of 153,300 pathogenic variants were detectable by WES, and 98.6% of 93,062,298 pathogenic variants presented an adequate depth for potential detection [70]. Aubart et al found five relevant variants (c.1286 G>C in FBN1, c.304 G>A in SMAD3, c.1588 C>T, c.329 T>C and c.3164 C>T in COL4A1) in 51 patients with severe MFS by WES [71]. However, 5-10% of genes (low-quality sequences) are rarely covered in WES, and sequences with high CG content are not easily captured [72, 73]. Unfortunately, the detection of differences in copy number variation (CNV), translocations, repeat expansions, and tandem repeat size are unavailable by WES [68].
Whole genomic sequencing (WGS)
WGS is used to detect variants across the whole genome with the most continuous coverage, hence detecting coding and non-coding region variants of nuclear and mitochondrial genomes and increasing the detection ability of CNV and the mapping of genome-wide dense homozygosity [74, 75]. However, the sequencing read length of short-read WGS may result in incomplete coverage of some sequences with mappability <1, and raw-reads of long-read WGS have a high error rate, not to mention that the cost of WGS can be prohibitive [74]. Moreover, due to the limitations of databases (uncomprehensive and conflicting data) and knowledge about genic effects, it produces great difficulties for interpreting all of these sequencing data [76]. Benke et al found a 31,956 bp deletion of FBN1 by WGS in a female MFS patient, and the deletion was also confirmed by multiplex ligation-dependent probe amplification (MLPA) and Sanger sequencing [77]. Yuan et al reported a new preimplantation genetic testing (PGT) method combined with WGS for de novo mutations, and a paternal MFS family with a de novo mutation (c.4952_4955delAATG) in FBN1 successfully had a healthy newborn infant by PGT and chromosomal balanced translocations [78].
Targeted genome sequencing (TGS)
TGS is used to screen a panel of markers known with clinical relevance by selective enrichment of the genomic areas where comprise these markers [79, 80]. Compared with WES and WGS, TGS not only has greater sequencing depth but also the reduced overall costs and data burden [79]. Li et al used panel-based targeted next-generation sequencing to analyze the FBN1, TGFBR1 and TGFBR2 genes in 123 Chinese with MFS or related disease and found that 97 cases had at least one pathogenic mutation [81]. Wei et al combined the targeted DNA-HiSeq and next-generation sequencing to design an array-based gene chip for the detection of the exons of 193 genes associated with 103 genetic diseases [82]. However, TGS has low ability to detect structural rearrangements or CNV [76].
Multiplex ligation-dependent probe amplification (MLPA)
MLPA is used to detect the deletions or duplications of specific genes and the presence of abnormal DNA methylation in diseases [83]. Contamination of genomic DNA samples by PCR may lead to a false-positive result of MLPA [84]. Yang et al found five novel large deletions in FBN1 in 5 patients with MFS by MLPA. One of the deletions encompassed exons 44-66 in FBN1, and others encompassed exons 43, 56, 54 and 50 [85]. Furtado et al reported two novel large deletions in 4 patients with MFS by MLPA, including an FBN1 deletion which encompassed exons 1-5, and a 542 Kb deletion in chromosome 15 that spanned the entire FBN1 gene and another 5 genes [86]. Moreover, Li et al demonstrated that in-frame deletions between exons 24-53 were related to severe clinical phenotypes. Patients with mild MFS showed an exon 6 deletion, and the classic MFS was linked to deletions of exons 1-36 by MLPA analysis [87]. These studies not only supplement the FBN1 mutation data, but also highlight the importance of detecting large FBN1 deletions in patients with MFS.
Single cell sequencing (SCS)
SCS technologies explore the gene expression heterogeneity between cells by analyzing the genetic information in a single cell. One of the most commonly used SCS methods is single-cell RNA sequencing (scRNA-seq), which is used to reveal the regulation between genes and track the trajectories of cell lineages during development [88]. Pedroza et al found a cluster of modulated smooth muscle cells (SMCs) in aortic aneurysm tissue of an adult FBN1C1041G/+ mouse model by scRNA seq, and upregulated activity of TGF-β signaling, and expression of Kruppel-like factor 4 (Klf4) may be a potential upstream driver that promotes SMCs modulation [89].
Genes contribute to MFS and related diseases
MFS and related disorders including LDS, SGS, MASS phenotype etc., display a significant overlap. These diseases and nonsyndromic aneurysmal syndromes are associated with the abnormal of TGF-β signaling [12]. Multiple genes contribute to MFS and related diseases, such as the FBN-encoding genes FBN1 and FBN2, and genes encoding signaling molecules of the TGF-β pathway, such as TGFBR1/2, LTBPs and SKI. Mutations of FBN1 were observed in >90% of cases of MFS [90] (Table 1).
Genes associated with MFS and related diseases
| Gene | Chromosomal region | Functions | Involvement in disease | Roles in TGF-β pathway | Reference |
|---|---|---|---|---|---|
| FBN1 | 15q21.1 | A component of calcium-binding microfibrils, provide force-bearing structural support in connective tissue | MFS, MASS, EL, WMS, SGS, NPS | Maintain matrix structure and function, participate in the matrix sequestering of TGF-β | [91-93] |
| FBN2 | 5q23.3 | A component of connective tissue microfibrils, assembled into elastic fiber | CCA | Maintain matrix structure and function, participate in the matrix sequestering of TGF-β | [98-100] |
| TGFBR1 | 9q22.33 | Transduce the TGF-β signal | LDS | Transduce of TGF-β pathway signaling from the cell surface to the cytoplasm | [101-103, 109] |
| TGFBR2 | 3p24.1 | Phosphorylates proteins and regulates the transcription of genes | MFS, LDS, tumors | Transduce of TGF-β pathway signaling from the cell surface to the cytoplasm | [101-103, 109] |
| LTBP-1 | 2p22.3 | Targets the TGF-β to extracellular matrix | N/A | Regulate the concentration of TGF-β | [112-115] |
| LTBP-2 | 14q24.3 | A component of TGF-β latent complex, a structural component of microfibrils | PCG, MSPKA, WMS3 | Regulate the concentration of TGF-β | [118,119] |
| LTBP-3 | 11q13.1 | Combines with TGF-β, a structural component of extracellular matrix | DASS, GPHYSD3 | Regulate the concentration of TGF-β | [112,113] |
| SKI | 1p36.33-p36.32 | A repressor of TGF-β signaling, regulates the neural tube development and muscle differentiation | SGS | Inhibit the phosphorylation of Smad2 and Smad3 | [126,127] |
Abbreviations: CCA, Congenital Contractural Arachnodactyly; DASS, Dental anomalies and short stature; EL, ectopia lentis syndrome; GPHYSD3, Geleophysic dysplasia 3; LDS, Loeys-Dietz syndrome; MASS, Mitral valve, Aorta, Skeleton, Skin involvement; MFS, Marfan syndrome; MSPKA, Microspherophakia; NPS, Neonatal Progeroid syndrome; PCG, Primary Congenital Glaucoma; SGS: Shprintzen-Goldberg syndrome; WMS, Weill-Marchesani syndrome; N/A, not applicable.
FBN1 gene in MFS
FBN1 is located on chromosome 15q21.1 and is comprised of 66 exons. It is transcribed into a 10-Kb mRNA, which is then translated into FBN1, a 2,871 amino acid long protein with a large number of cysteine repeats. FBN1 is an extracellular matrix (ECM) glycoprotein and a structural component of microfibers with a diameter of 10-12 nanometers, and contains 47 epidermal growth factor (EGF)-like modules, 7 TGF-β-binding protein-like domains and 43 EGF-like modules with calcium-binding (cb)EGF consensus sequences. FBN1 is an important component of elastic fibers in the elastic or inelastic connective tissues that provides support for the load-bearing structures and a scaffold for protein precipitation [91-93]. However, in a homozygous and heterozygous mgΔ mutant mice model, it has been shown that the primary role of FBN1 is to maintain tissue homeostasis, not to assemble the elastic matrix [94].
A total of 3,077 mutations of FBN1 in patients with MFS have been reported to date, including 2,499 (73.09%) point mutations and 51 (1.66%) large rearrangements. Missense mutations of FBN1 account for 53-56.1% of cases, 33-36.8% of truncated variants, 7.1-13% of intronic variants and 1.8-2.9% of gross genomic rearrangements [85, 90]. Mutations of FBN1 occur across almost the entire gene, and there is no obvious aggregation area and periodicity. The repetitiveness of this mutation is ~12%. Exon mutations can be found in most of the exonic regions in patients with MFS, whereas exons 45 and 57 are underrepresented and exons 13, 26, 27, 28 and 43 are overrepresented [95]. However, Groth et al reported that the total number of variants in patients with MFS (diagnosed by calculating the MFS phenotype score) accounted for only 35.8% of all registered variants [96]. They suggested that certain databases contained incorrectly interpreted conclusions of variants, and thus should only be considered as a reference for seeking information regarding the specific mutation [97].
Genes associated with the related disease
FBN2
FBN2 is located on chromosome 5q23.3, and the encoded protein, FBN2, is a component of connective tissue microfibrils. It has been shown that the developmental expression of FBN2 is earlier than that of FBN1 [98]. FBN2 is related to the formation of elastic fiber structures, whereas FBN1 primarily maintains the function of elastic structures. Therefore, the expression of FBN2 is common in elastic tissues, and FBN1 is dominant in stress and weight-bearing structures [98, 99]. Gupta et al found a mutation site of FBN2 in a female proband and her brother with congenital contractual arachnodactyly, and the proband also met the diagnostic criteria of MFS, with progressive dilatation of the aorta at the sinuses of Valsalva, however, none of them presented with MVP or regurgitation [100].
TGFBR2
TGFBR2 is located on chromosome 3p24.1. TGFBR2 is associated with MFS at the second locus for MFS [101]. TGFBR2 encodes the TGFBR2 protein, which forms a complex with the TGFBR1 and binds to TGF-β mediating protein phosphorylation, and modulating cell proliferation, cell cycle progression and ECM formation [101]. Mutations of TGFBR2 are associated with MFS without major ocular symptoms [102]. In nematode models, mutations of TGFBR2 that cause MFS or MFS-like syndromes may disrupt the structure of TGFBR2 with an exposed surface domain, alter subcellular localization patterns, and indirectly alter the trafficking of the TGFBR1 [103]. Zhang et al found a p.V453E mutation of TGFBR2 (located in the F-helix in the kinase domain) in a Chinese patient with MFS and two relatives with certain MFS-like manifestations. They demonstrated that mutations of TGFBR2 located on the F-helix in the kinase domain may be related to severe cardiovascular and skeletal symptoms and minor ocular symptoms [104]. Attias et al reported the cardiovascular symptoms of patients with MFS caused by TGFBR2 mutations were similar in age and incidence of aortic dilatation to those of patients with FBN1 mutations, and their therapeutic effects were similar. Therefore, the severity of MFS cannot be only attributed to the presence of TGFBR2 mutations [105].
TGFBR1
TGFBR1, located on chromosome 9q22.33, consists of 9 exons and encodes the TGFBR1 protein. Mutations of TGFBR1 may also be found in patients with MFS [106]. Lucarini et al suggested that the 6Ala allele of TGFBR1 may be considered as a low penetrance allele in patients with MFS [107]. However, Somers et al demonstrated that 5 MFS patients with the TFGBR1*6A allele did not present phenotypic differences when compared with 21 MFS patients without a TFGBR1*6A allele, although this may partly be due to the small sample size of the study [108].
TGFBRs is also considered a causative gene of LDS.Certain clinical manifestations of LDS and MFS are similar, including cardiovascular symptoms, scoliosis and craniofacial features. Hence, it may be difficult to evaluate the genetic role of TGFBRs in MFS [109]. Stheneur et al demonstrated that the detection rate of TGFBR1/2 mutant genes was 6.2% and 4.8% in classic MFS, and 6.2% and 4.6% in incomplete MFS out of 457 patients with MFS or related disorders [110]. De Cario et al found 10 common polymorphisms of TGFBR2 and 6 of TGFBR1 in 75 patients with MFS. These polymorphisms were correlated with the severity of cardiovascular manifestations in MFS [111].
LTBP genes
LTBP1-4 are extracellular glycoproteins with a similar structure to fibrin. The complex of TGF-β1 and latency-related protein (LAP) is associated with LTBP through two disulfide bonds formed between the third 8-Cys domain of LTBP1, 3 and 4 and LAP, but not LTBP2 [112]. FBN1 microfibrils are responsible for the association between LTBP3 and 4 with the matrix, and the association between LTBP1 and matrix depends on a fibronectin network [113]. The first, second and fourth 8-Cys domains of LTBP1 can independently bind to the matrix, and the N-terminal fragments of LTBP1 more readily bind to the ECM [114]. The C-terminal LTBP1 fragment can form a bipartite interaction with a four-domain FBN1 fragment (EGF2-EGF3-Hyb1-cbEGF1), and LTBP1 connects with FBN1 through two independent epitopes, and have contacts with the ECM network through a flexible pivot [115].
LTBP1 is located on chromosome 2p22.3 and has 38 exons. Quiñones-Pérez et al found a deletion of LTBP1 in 3 patients with MFS in one family, and all patients had thoracic aortic aneurysm (TAA) and certain features of MFS, but they did not meet the criteria for MFS [116]. Sticchi et al reported 5 mutations, including p.Asn542Ser and p.Lys2460Arg of FBN1, p.Val1739Met of NOTCH1, p.Arg1330Gln of LTBP1, and p.Arg423Trp of TGFBR3 in an MFS patient with bicuspid aortic valve and aortic symptoms. These findings showed that the mutation of LTBP1 may participate in regulating the vascular phenotype [117].
The LTBP2 gene is located on 14q24.3 and consists of 36 exons. LTBP2 binds to the matrix through FBN and can negatively regulate the elastic fiber assembly through binding to fibulin-5. LTBP2 also regulates the activity of TGF-β signaling [118]. LTBP2 is essential for the formation of microfibril bundles in the ciliary zonules [119]. The mutation of LTBP2 is related to EL and MFS [120, 121]. LTBP2 mutations may contribute to the systemic phenotype of syndromes related to the abnormality of genes that are associated with the TGF-β pathway [122]. Ramona et al demonstrated that c.1642C>T (p.Arg548*) of LTBP2 may contribute to ocular manifestations, MVP and pectus excavatum of MFS, but is not a causative gene [121].
LTBP3 is mapped to 11q13.1, and is composed of 29 exons. In the FBN1mgR/mgR: LTBP3-/- mouse model, the lifespan of mice is prolonged compared with FBN1mgR/mgR. In mouse models with the absence of FBN3, it was observed that there was a decrease of activation of Smad2/3 and Erk1/2, reduction of disruption of aortic elastic fibers, and improvement of the circumferential mechanical properties of the thoracic aorta, whereas spinal deformities remained or were exacerbated, which may partly affect the overall aortic phenotype [123, 124]. The improper localization of the FBN3/TGF-β complex may promote aortic disease. Bertoli-Avella et al reported that elastic fiber fragmentation, and collagen and proteoglycan deposition were increased in the aortic wall tissues of patients with p.Asp263His of LTBP3. They showed that the pathological manifestations in patients with this gene mutation included severe cardiovascular symptoms that were highly similar to MFS symptoms [125].
SKI
SKI is located on chromosome 1p36.33-p36.32 and contains 9 exons. This gene encodes the nuclear protooncogene protein homolog of avian sarcoma viral oncogene. It inhibits the phosphorylation of R-Smad through activated type I receptors, thereby restraining TGF-β-Smad signal transduction [126]. SKI is associated with SGS, a systemic connective tissue disease that primarily manifests as cardiovascular, skeletal or craniofacial manifestations [127]. Arnaud et al found that three different variants of the SKI gene affected the same amino acid (Thr180) in 6 patients with MFS or MFS-like/ Marfanoid syndrome and 3 patients with marfanoid habitus with learning disabilities. Additionally, 6 of the patients developed TAA. However, none of the patients had aortic dissection, and no aortic dilation (age-related) was found [128].
The correlation of genotype/phenotype in MFS
The mutational heterogeneity of FBN1 may cause age-related penetrance and diversification in clinical manifestations of MFS [129]. Different mutations cause the sequestering of mutant protein in the endoplasmic reticulum then exerting haploinsufficiency (HI) efforts, or promoting secretion of mutant protein in ECM and exerting dominant-negative (DN) efforts. Pathogenic mechanisms and clinical manifestations of MFS may be related to the positional effects of mutations in FBN1, and dominance of the mutant alleles [130]. In the DN model, FBN1 mutant alleles interfere with the function of the protein encoded by the wild-type allele, thereby causing the appearance of MFS [131]. And the HI model refers to the failure of microfibrillar assembly ascribed to HI of normal FBN1 rather than the mutant protein [132]. About one-third mutations of FBN1 result in HI effects including nonsense, splicing or out-of-frame mutations, and other two-third mutations lead to DN effects including missense, splicing and in-frame mutations [133]. DN Mutations of FBN1 affects stability of microfibers on various conditions rather than the microfibril assembly, and therefore different pathological manifestations appear [134]. Conceptually, HI mutations might have a consistent phenotype [135].
Some correlations of genotype/phenotype have been discussed. The widely accepted correlation is that large proportions of MFS children carry mutations located in exons 24-32 and in-frame mutations of FBN1, particularly in neonatal patients [48]. And patients with mutations in exons 43-65 show the same high frequency of major cardiovascular phenotype compared with exons 24-32 [136]. Patients with mutations in exon 1-21 present with a high incidence of EL, and patients with mutations in exons 23-32 are more likely to develop aortic root dilatation [137]. The more severe musculoskeletal and skin phenotype are observed in patients with an FBN1 premature termination codon when compared with those in an in-frame mutation of FBN1 [46, 138]. MFS patients with truncating/splicing mutations have a higher proportion of aortic events and a younger median age, compared with MFS patients with missense mutations [90, 139]. Mutations that alter the structure of the cbEGF module of FBN1 can affect the binding between FBN1 and calcium. Since FBN1 is more likely to be sensitive to proteolytic enzymes. Degradation of mutant fibers may occur during secretion or assembly in ECM [140]. Patients with a missense mutation of FBN1 involving a cysteine residue have a high risk of EL, and a mutation eliminating a cysteine shows a higher presence of aortic dilation and MVP than those with creating a cysteine [138]. Moreover, Franken et al reported that MFS patients with HI variants had 2.5-fold increased risk for the combination of cardiovascular death and 1.6-fold increased risk for aortic complication compared with patients with DN variants after follow-up 8.2 years in 357 MFS patients [141].
Willis et al reported a 24-year-old patient with a de novo mutant c.3037G>A (p.G1013R) located in exon 25 of FBN1. The patient presented the cardiac involvement in early age and relative longevity. Even the mutant location is in exon 24-32 which is associated with neonatal MFS, the mutant G1013R of FBN1 may be associated with a specific phenotype in the mutational hot spot [142]. Whiteman et al reported two mutations in EGF domains of FBN1 (C1117Y, C1129Y) may exert HI effect due to the protein product without being released from the endoplasmic reticulum [143]. The classification of mutant types would influence the correlation of genotype-phenotype, assignment of the HI versus DN effects in MFS should base on the protein products and the effect of a specific mutation would be identified through experimental protein work [133, 135].
In addition, not only the diversity of FBN1 mutations of genotype result in clinical heterogeneity, but also the effect of risk alleles in modifier genes and epigenetics are involved in thoracic aortic involvement [71, 144]. Aubart et al found a second pathogenic event in aneurysm-related genes in nine MFS patients with disease-causing mutations of FBN1 [71]. They also identified two modifier regions containing ECE1 and PRKG1 respectively and a region closing to a cluster of MMPs by cross-mapping of genome-wide strategies in 1070 MFS patients with pathologic FBN1 mutations, these regions were involved in the ECM regulation or SMCs relaxation hence contributing to aortic phenotype of MFS [71]. Wu et al reported two MFS cases had two pathologic mutations of FBN1 (c.A3142G/ c.G1622A; c.G1220A/c.C8080T), and the mutations in PKD1 and FBN1 were both found in 27 MFS patients without kidney disease [145]. Furthermore, Arnaud et al found 4 probands carried missense homozygous and 5 probands with compound heterozygotes FBN1 mutations in 2500 French MFS patients. All the patients were lack of the aggravated clinical manifestations [146]. This report was contrary to previous reports, which demonstrated that homozygous and compound heterozygous cases presented with more severe symptoms compared with patients with only one mutation. These reports demonstrated that the diversification in clinical manifestations might result from a complex molecular mechanism.
Epigenetic Regulation of Clinical Manifestations in MFS
Epigenetic regulation refers to the acquisition of new stable heritable traits that are not related to changes in the DNA sequence [147]. Mechanisms of epigenetic regulation include DNA methylation, or modifications of DNA-associated histones and non-coding RNA [147]. For MFS, molecular mechanisms of intrafamilial heterogeneity in the clinical severity remain unknown [148]. It has been shown that epigenetics serves an essential role in regulation of aortic aneurysms in multiple studies. Arai et al found a negative correlation between DNA methylation on the WT allele within the FBN1 CpG island shore and functional FBN1 mRNA levels in human induced pluripotent stem cells, indicating that epigenetic regulation may affect FBN1 expression in MFS [149]. The increased activity of the methyltransferase EZH2 repressed SM22α, thus promoting aortic disorder in a FBN1C1039G/+ mouse model [150]. Gomez et al demonstrated that an increase in histone H3 acetylation and methylation in the medial layer of TAA were associated with overexpression of Smad2 signaling, specifically in SMCs over several passages in all types of TAA (caused by FBN1 or TGFBR2 mutations). This finding infers that heritability, the cell specificity and epigenetics may all contribute to the pathology of MFS [144]. In a mouse model of FBN1mgΔloxPneo, 47.4% variation of the F2 skeletal phenotype could be explained by the potential modifier genes on chromosomes 3 and 6 participating in the TGF-β signaling pathway or other connective tissue diseases, whereas the modifier genes on chromosomes 4 and 13 were responsible for 40.7% variation of the F2 vascular phenotype [151].
Abnormalities of TGF-β Pathway in MFS
FBN1 regulates the concentration of activated TGF-β in the matrix through binding with the large latent complex, which consists of LAP, LTBP and TGF-β [152]. Abnormalities of FBN1 lead to an imbalance in activation and signal transduction of TGF-β. Active TGF-β binds to the extracellular domain of the type I and type II receptor complex, and conformational changes of the intracellular serine/threonine domain of receptors promote phosphorylation and activation of type I receptors, thus resulting in activation of the SMAD and non-SMAD pathways (ERK, JNK and p38/MAPK) [153, 154] (Figure 1).
The transduction of TGF-β pathway signaling. A: the normal fibrillin-1 as a structural component of extracellular matrix microfibrils participates in the matrix sequestering of TGF-β. B: mutant fibrillin-1 loss interactions with LTBP, hence increasing the concentration of TGF-β in the extracellular matrix and upregulating the TGF-β pathway signal.
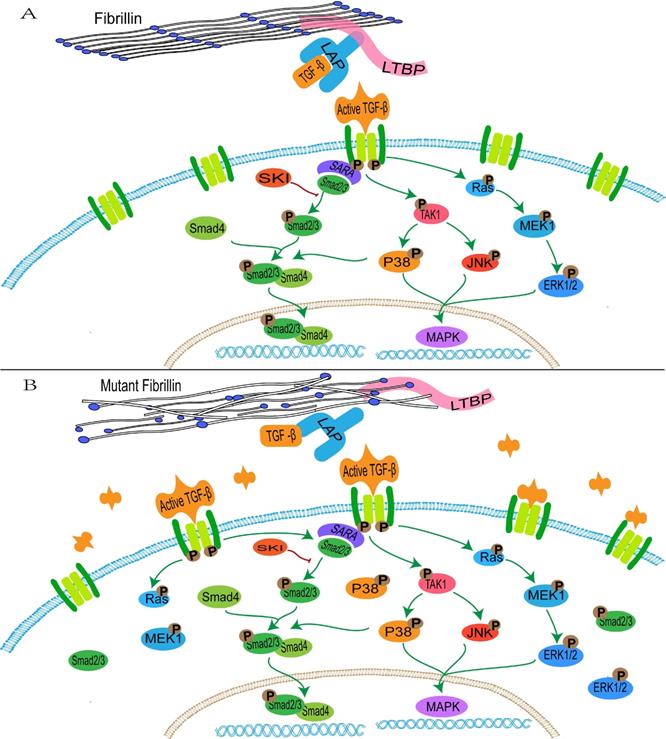
The activated TGFBR1 transmits signals through promoting the phosphorylation of a C-terminal Ser-Ser-X-Ser (SSXS) motif of SMAD2/3. SMAD4 forms a complex with active SMAD2/3 through the two phosphorylated serine residues at the C-terminal of SMAD2/3, and then SMAD4 is transferred to the nucleus [155]. The N-terminal NH1 structures of SMAD3 and SMAD4 can bind to specific DNA sequences in the nucleus, whereas SMAD2 does not directly bind to DNA sequences [155]. In addition, interactions between SMAD2/3 and the TGFBR1 is mediated by a Smad anchor for receptor activation (SARA), which interacts with SMAD2/3 through the Smad-binding domain and contacts with a TGFBR1 via a C-terminal region [156].
It is initially hypothesized that upregulation of the TGF-β pathway promotes the occurrence of TAA in patients with MFS. In the FBN1C1039G/+ mouse model, upregulation of Smad2, ERK1/2, MEK1 and p38 are observed in aortic tissues [157, 158], and the Smad4 haploinsufficiency promotes activation of JNK1, which leads to aortic diseases [159]. The selective inhibition of ERK1/2 reduces the pathological aortic root growth in an FBN1C1039G/+ mouse model [159]. Conversely, in SMCs of patients with MFS, ERK activation drives the overexpression of Notch3, which may serve a protective role in aortic aneurysms through remodeling of tissues [160]. The complex TGF-β signaling induces a mixed synthetic-contractile phenotype in SMCs, hence altering the normal physiological structure of the aorta [160]. In addition, abnormal TGF-β signaling affects contractile protein and collagen 1 in vascular SMCs, thereby increasing cell stiffness and leading to aortic rigidity [161]. TGF-β1 regulates the mitochondrial dynamics through the down-regulation of p-AMPK and induces the increase of reactive oxidative species (ROS) in the mitochondria in vascular progenitor cells of patients with MFS, excessive generation of ROS promotes the phosphorylation and translocation of p65-NF-κB from the cytoplasm to the nucleus in the vascular SMCs of patients with MFS, thus inducing cell senescence [162, 163].
It is worth noting that aortopathies have been observed in the absence of TGF-β signaling by deleting the TGFBR2 in a young MFS mouse model (MFS-TBRII-/- mice) [164]. And this opposite conclusion is confirmed by other studies. Hu et al found that postnatal aortic homeostasis required physiologic TGF-β signaling, and disrupting SMCs TGF-β due to the loss of TGFBR2 in postnatal MFS mouse model caused significant alterations of gene expression and severe aortic lesion including dilation, dissection, elastolysis, and etc. [165]. Cook et al reported that the role of TGF-β was protective in the early stage of TAA formation and pathogenic in later stage in Fbn1mgR/mgR mouse model [166].
Franken et al reported that angiotensin II (AngII) can directly induce Smad2 activation through AngII receptor-1(AT1r) and then increase TGF-β levels in an MFS mouse model, hence AngII may be the primary cause of aortic disease rather than TGF-β signaling, which may instead serve a role in secondary disease progression and appears to be a marker of aortic disease [167]. Cook et al put forward an opposite result that due to the stage-specific dimorphic effects of TGF-β, TGF-β and AT1r were responsible for TAA progression through p-Erk1/2 and p-Smad2/3 signaling, respectively[166]. Moreover, systemic abrogated TGF-β activity in a C57BL/6 mouse model treated with anti-TGF-β antibodies promotes the formation of Ang II-induced abdominal aortic aneurysms (AAAs) [168]. The contradictory results between MFS and AAAs may be explained by the difference in pathological characteristics. Increased TGF-β signaling promotes non-inflammatory excessive accumulation of SMCs and ECM in MFS, and other AAAs are related to the thinned arterial walls, loss of SMCs and ECM, and vascular inflammation [168].
Additionally, androgens enhance Erk/Smad activation, as elastic fiber fragmentation and matrix metalloproteinases 2 (MMP2) expression are higher in males compared with females in MFS mice [13, 169]. MMP2 cleaves matrix proteins in medial SMCs of MFS and can regulate the levels of TGF-β and Erk1/2 phosphorylation [170].
Conclusion
MFS is a complex disease involving multiple systems. The clinical manifestations of MFS are varied and overlap with other diseases. The clinical symptoms of MFS are gradually aggravated with age, and EOMS cases always have serious clinical outcomes. Diagnosis of MFS relies on family history and multi-system scores. Due to the clinical heterogeneity and diversification of mutations, it is difficult for doctors to diagnose mild cases or prenatal patients [171]. However, with the advancements in genetic testing, this problem is expected to be solved.
The majority of the FBN1 mutations in the MFS family affect a single amino acid and lead to abnormalities in protein function. Pathogenic mechanisms of FBN1 include the HI and DN model. But neither model is sufficient to explain the heterogeneity of clinical manifestations. The epigenetic regulation only explains part of the clinical differences. In addition, other genes associated with related disease have been discovered over the years, such as TGFBRs, LTBP2, LTBP3 and SKI. Nevertheless, mutations of these genes are also detected in other diseases, and symptoms caused by them may not be as severe as those caused by FBN1 mutations. Further in vitro/in vivo studies are required to identify the pathogenesis of these genes, the effects of their interactions and the genotype-phenotype correlations.
Abnormalities of the TGF-β pathway signaling is another important pathological mechanism by which aneurysms develop in patients with MFS. It is specifically observed in the SMCs of the vascular medial layer. The TGF-β pathway can regulate a number of processes in several types of cells, including endothelial cells, SMCs and pericytes, amongst others, and its mechanisms may be associated with the induction of proliferation, apoptosis, migration, adhesion, ECM protein production and cytoskeletal organization [172]. The up- and down-regulation of TGF-β pathway signaling are both related to MFS, canonical and non-canonical signaling exert antagonistic effects and co-adjust the mechanism of development of aneurysms in MFS. A better understanding of the TGF-β pathway signaling may assist in determining the exact molecular mechanism underlying the development of MFS. At present, the role of TGF-β pathway signaling abnormalities in MFS requires further elucidation.
With the development of next-generation sequencing technologies, WES is being widely used to detect genes related to MFS. In addition, research methods based on the single-cell analysis, such as scRNA-seq, have also been used for analysis of the expression of genes in various tissues, and explaining the differing degrees of involvement of each system. Along with whole gene technology and bioinformatics, the combined application of high throughput sequencing technologies and 'big data' analysis will be helpful in further analyzing the entire genome and the better understanding of the complex traits of patients with MFS. This may highlight novel strategies for prevention, early diagnosis and treatment/management of MFS.
Acknowledgements
This work was supported by grants from the Department of Science and Technology of Sichuan Province (grant no. 2020YFS0435), the Health Care of Sichuan Provincial cadres (grant no. 2020-227), Science & Technology Bureau of Chengdu (grant no. YF05-00198-SN and YF05-00060-SN), and the Sichuan Academy Medical Science & Sichuan Provincial People Hospital (grant no. 2018LY01).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vanem TT, Geiran OR, Krohg-Sorensen K, Roe C, Paus B, Rand-Hendriksen S. Survival, causes of death, and cardiovascular events in patients with Marfan syndrome. Mol Genet Genomic Med. 2018;6:1114-23
2. Gentilini D, Oliveri A, Fazia T, Pini A, Marelli S, Bernardinelli L. et al. NGS analysis in Marfan syndrome spectrum: Combination of rare and common genetic variants to improve genotype-phenotype correlation analysis. PLoS One. 2019;14:e0222506
3. Veiga-Fernandez A, Joigneau Prieto L, Alvarez T, Ruiz Y, Perez R, Gamez F. et al. Perinatal diagnosis and management of early-onset Marfan syndrome: case report and systematic review. J Matern Fetal Neonatal Med. 2020;33:2493-504
4. Ho NC, Tran JR, Bektas A. Marfan's syndrome. Lancet. 2005;366:1978-81
5. Groth KA, Hove H, Kyhl K, Folkestad L, Gaustadnes M, Vejlstrup N. et al. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet J Rare Dis. 2015;10:153
6. Groth KA, Stochholm K, Hove H, Andersen NH, Gravholt CH. Causes of Mortality in the Marfan Syndrome(from a Nationwide Register Study). Am J Cardiol. 2018;122:1231-5
7. Miyahara S, Okita Y. Overview of current surgical strategies for aortic disease in patients with Marfan syndrome. Surg Today. 2016;46:1006-18
8. Coelho SG, Almeida AG. Marfan syndrome revisited: From genetics to the clinic. Rev Port Cardiol. 2020;39:215-26
9. Fleck T, Czerny M, Wolner E, Grabenwoger M. Interventional treatment methods in patients with Marfan Syndrome. Images Paediatr Cardiol. 2004;6:1-11
10. de Vries BB, Pals G, Odink R, Hamel BC. Homozygosity for a FBN1 missense mutation: clinical and molecular evidence for recessive Marfan syndrome. Eur J Hum Genet. 2007;15:930-5
11. Khan AO, Bolz HJ, Bergmann C. Results of fibrillin-1 gene analysis in children from inbred families with lens subluxation. J AAPOS. 2014;18:134-9
12. Verstraeten A, Alaerts M, Van Laer L, Loeys B. Marfan Syndrome and Related Disorders: 25 Years of Gene Discovery. Hum Mutat. 2016;37:524-31
13. Anderson NK, Juzwiak EE, Dietz HC. A seX(X/Y) Article on Marfan Syndrome. J Am Heart Assoc. 2020;9:e018814
14. Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB. et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476-85
15. Salik I, Rawla P. Marfan Syndrome. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2020
16. Speed TJ, Mathur VA, Hand M, Christensen B, Sponseller PD, Williams KA. et al. Characterization of pain, disability, and psychological burden in Marfan syndrome. Am J Med Genet A. 2017;173:315-23
17. Judge DP, Dietz HC. Marfan's syndrome. Lancet. 2005;366:1965-76
18. Rybczynski M, Mir TS, Sheikhzadeh S, Bernhardt AM, Schad C, Treede H. et al. Frequency and age-related course of mitral valve dysfunction in the Marfan syndrome. Am J Cardiol. 2010;106:1048-53
19. van Karnebeek CD, Naeff MS, Mulder BJ, Hennekam RC, Offringa M. Natural history of cardiovascular manifestations in Marfan syndrome. Arch Dis Child. 2001;84:129-37
20. Ramirez F, Caescu C, Wondimu E, Galatioto J. Marfan syndrome; A connective tissue disease at the crossroads of mechanotransduction, TGFbeta signaling and cell stemness. Matrix Biol. 2018;71-72:82-9
21. Sisk HE, Zahka KG, Pyeritz RE. The Marfan syndrome in early childhood: analysis of 15 patients diagnosed at less than 4 years of age. Am J Cardiol. 1983;52:353-8
22. Dormand H, Mohiaddin RH. Cardiovascular magnetic resonance in Marfan syndrome. J Cardiovasc Magn Reson. 2013;15:33
23. Hascoet S, Edouard T, Plaisancie J, Arnoult F, Milleron O, Stheneur C. et al. Incidence of cardiovascular events and risk markers in a prospective study of children diagnosed with Marfan syndrome. Arch Cardiovasc Dis. 2020;113:40-9
24. Detaint D, Faivre L, Collod-Beroud G, Child AH, Loeys BL, Binquet C. et al. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur Heart J. 2010;31:2223-9
25. Wozniak-Mielczarek L, Sabiniewicz R, Drezek-Nojowicz M, Nowak R, Gilis-Malinowska N, Mielczarek M. et al. Differences in Cardiovascular Manifestation of Marfan Syndrome Between Children and Adults. Pediatr Cardiol. 2019;40:393-403
26. Isselbacher EM, Bonaca MP, Di Eusanio M, Froehlich J, Bassone E, Sechtem U. et al. Recurrent Aortic Dissection: Observations From the International Registry of Aortic Dissection. Circulation. 2016;134:1013-24
27. den Hartog AW, Franken R, Zwinderman AH, Timmermans J, Scholte AJ, van den Berg MP. et al. The risk for type B aortic dissection in Marfan syndrome. J Am Coll Cardiol. 2015;65:246-54
28. de Souza RB, Farinha-Arcieri LE, Catroxo MHB, Martins A, Tedesco RC, Alonso LG. et al. Association of thoracic spine deformity and cardiovascular disease in a mouse model for Marfan syndrome. PLoS One. 2019;14:e0224581
29. Jondeau G, Detaint D, Tubach F, Arnoult F, Milleron O, Raoux F. et al. Aortic event rate in the Marfan population: a cohort study. Circulation. 2012;125:226-32
30. Baumgartner H, De Backer J, Babu-Narayan SV, Budts W, Chessa M, Diller GP. et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur Heart J. 2021;42:563-645
31. Welby JP, Kim ST, Carr CM, Lehman VT, Rydberg CH, Wald JT. et al. Carotid Artery Tortuosity Is Associated with Connective Tissue Diseases. AJNR Am J Neuroradiol. 2019;40:1738-43
32. Franken R, El Morabit A, de Waard V, Timmermans J, Scholte AJ, van den Berg MP. et al. Increased aortic tortuosity indicates a more severe aortic phenotype in adults with Marfan syndrome. Int J Cardiol. 2015;194:7-12
33. Morris SA, Orbach DB, Geva T, Singh MN, Gauvreau K, Lacro RV. Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation. 2011;124:388-96
34. Agg B, Szilveszter B, Daradics N, Benke K, Stengl R, Kolossvary M. et al. Increased visceral arterial tortuosity in Marfan syndrome. Orphanet J Rare Dis. 2020;15:91
35. Gehle P, Goergen B, Pilger D, Ruokonen P, Robinson PN, Salchow DJ. Biometric and structural ocular manifestations of Marfan syndrome. PLoS One. 2017;12:e0183370
36. Vanhonsebrouck E, Consejo A, Coucke PJ, Leroy BP, Kreps EO. The corneoscleral shape in Marfan syndrome. Acta Ophthalmol. 2020
37. Wang Y, Lian Z, Zhou Y, Li X, Wu J, Zhang X. et al. Differential diagnosis of Marfan syndrome based on ocular biologic parameters. Ann Transl Med. 2020;8:1354
38. Sandvik GF, Vanem TT, Rand-Hendriksen S, Cholidis S, Saethre M, Drolsum L. Ten-year reinvestigation of ocular manifestations in Marfan syndrome. Clin Exp Ophthalmol. 2019;47:212-8
39. Salchow DJ, Gehle P. Ocular manifestations of Marfan syndrome in children and adolescents. Eur J Ophthalmol. 2019;29:38-43
40. Giampietro PF, Raggio C, Davis JG. Marfan syndrome: orthopedic and genetic review. Curr Opin Pediatr. 2002;14:35-41
41. Sponseller PD, Erkula G, Skolasky RL, Venuti KD, Dietz HC 3rd. Improving clinical recognition of Marfan syndrome. J Bone Joint Surg Am. 2010;92:1868-75
42. Shirley ED, Sponseller PD. Marfan syndrome. J Am Acad Orthop Surg. 2009;17:572-81
43. Bitterman AD, Sponseller PD. Marfan Syndrome: A Clinical Update. J Am Acad Orthop Surg. 2017;25:603-9
44. De Maio F, Fichera A, De Luna V, Mancini F, Caterini R. Orthopaedic Aspects of Marfan Syndrome: The Experience of a Referral Center for Diagnosis of Rare Diseases. Adv Orthop. 2016;2016:8275391
45. Stheneur C, Tubach F, Jouneaux M, Roy C, Benoist G, Chevallier B. et al. Study of phenotype evolution during childhood in Marfan syndrome to improve clinical recognition. Genet Med. 2014;16:246-50
46. Haine E, Salles JP, Khau Van Kien P, Conte-Auriol F, Gennero I, Plancke A. et al. Muscle and Bone Impairment in Children With Marfan Syndrome: Correlation With Age and FBN1 Genotype. J Bone Miner Res. 2015;30:1369-76
47. Sheikhzadeh S, Kade C, Keyser B, Stuhrmann M, Arslan-Kirchner M, Rybczynski M. et al. Analysis of phenotype and genotype information for the diagnosis of Marfan syndrome. Clin Genet. 2012;82:240-7
48. Faivre L, Masurel-Paulet A, Collod-Beroud G, Callewaert BL, Child AH, Stheneur C. et al. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics. 2009;123:391-8
49. Teare MD, Santibanez Koref MF. Linkage analysis and the study of Mendelian disease in the era of whole exome and genome sequencing. Brief Funct Genomics. 2014;13:378-83
50. Tsipouras P, Del Mastro R, Sarfarazi M, Lee B, Vitale E, Child AH. et al. Genetic linkage of the Marfan syndrome, ectopia lentis, and congenital contractural arachnodactyly to the fibrillin genes on chromosomes 15 and 5. The International Marfan Syndrome Collaborative Study. N Engl J Med. 1992;326:905-9
51. Collod G, Babron MC, Jondeau G, Coulon M, Weissenbach J, Dubourg O. et al. A second locus for Marfan syndrome maps to chromosome 3p24.2-p25. Nat Genet. 1994;8:264-8
52. Bailey-Wilson JE, Wilson AF. Linkage analysis in the next-generation sequencing era. Hum Hered. 2011;72:228-36
53. Nataraj AJ, Olivos-Glander I, Kusukawa N, Highsmith WE Jr. Single-strand conformation polymorphism and heteroduplex analysis for gel-based mutation detection. Electrophoresis. 1999;20:1177-85
54. Singh KK, Elligsen D, Liersch R, Schubert S, Pabst B, Arslan-Kirchner M. et al. Multi-exon out of frame deletion of the FBN1 gene leading to a severe juvenile onset cardiovascular phenotype in Marfan syndrome. J Mol Cell Cardiol. 2007;42:352-6
55. Wang B, Hu D, Xia J, Li Q, Yang J, Lu G. FBN1 mutation in Chinese patients with Marfan syndrome and its gene diagnosis using haplotype linkage analysis. Chin Med J (Engl). 2003;116:1043-6
56. Rommel K, Karck M, Haverich A, Schmidtke J, Arslan-Kirchner M. Mutation screening of the fibrillin-1 (FBN1) gene in 76 unrelated patients with Marfan syndrome or Marfanoid features leads to the identification of 11 novel and three previously reported mutations. Hum Mutat. 2002;20:406-7
57. Korkko J, Annunen S, Pihlajamaa T, Prockop DJ, Ala-Kokko L. Conformation sensitive gel electrophoresis for simple and accurate detection of mutations: comparison with denaturing gradient gel electrophoresis and nucleotide sequencing. Proc Natl Acad Sci U S A. 1998;95:1681-5
58. Hill M. Conformation sensitive gel electrophoresis. Methods Mol Biol. 2011;688:7-16
59. Korkko J, Kaitila I, Lonnqvist L, Peltonen L, Ala-Kokko L. Sensitivity of conformation sensitive gel electrophoresis in detecting mutations in Marfan syndrome and related conditions. J Med Genet. 2002;39:34-41
60. Loeys B, De Backer J, Van Acker P, Wettinck K, Pals G, Nuytinck L. et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat. 2004;24:140-6
61. Uyeda T, Takahashi T, Eto S, Sato T, Xu G, Kanezaki R. et al. Three novel mutations of the fibrillin-1 gene and ten single nucleotide polymorphisms of the fibrillin-3 gene in Marfan syndrome patients. J Hum Genet. 2004;49:404-7
62. Song SQ, Zhao BJ, Li S, Zhang JQ, Wang H, Jia CW. et al. [Mutation analysis and prenatal diagnosis of FBN1 gene mutations for four patients with Marfan syndrome]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2013;30:534-8
63. Howarth R, Yearwood C, Harvey JF. Application of dHPLC for mutation detection of the fibrillin-1 gene for the diagnosis of Marfan syndrome in a National Health Service Laboratory. Genet Test. 2007;11:146-52
64. Huang X, Wu Y, Chen F, Huang Y, Ma X, Chen T. [Two novel mutations in fibrillin-1 gene of Marfan syndrome]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2004;21:562-5
65. Kosaki K, Udaka T, Okuyama T. DHPLC in clinical molecular diagnostic services. Mol Genet Metab. 2005;86:117-23
66. Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870-9
67. Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59:5-15
68. Jelin AC, Vora N. Whole Exome Sequencing: Applications in Prenatal Genetics. Obstet Gynecol Clin North Am. 2018;45:69-81
69. Majewski J, Schwartzentruber J, Lalonde E, Montpetit A, Jabado N. What can exome sequencing do for you? J Med Genet. 2011;48:580-9
70. LaDuca H, Farwell KD, Vuong H, Lu HM, Mu W, Shahmirzadi L. et al. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PLoS One. 2017;12:e0170843
71. Aubart M, Gazal S, Arnaud P, Benarroch L, Gross MS, Buratti J. et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur J Hum Genet. 2018;26:1759-72
72. Hamilton A, Tetreault M, Dyment DA, Zou R, Kernohan K, Geraghty MT. et al. Concordance between whole-exome sequencing and clinical Sanger sequencing: implications for patient care. Mol Genet Genomic Med. 2016;4:504-12
73. Ku CS, Cooper DN, Polychronakos C, Naidoo N, Wu M, Soong R. Exome sequencing: dual role as a discovery and diagnostic tool. Ann Neurol. 2012;71:5-14
74. Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G. Clinical sequencing: From raw data to diagnosis with lifetime value. Clin Genet. 2018;93:508-19
75. Pipis M, Rossor AM, Laura M, Reilly MM. Next-generation sequencing in Charcot-Marie-Tooth disease: opportunities and challenges. Nat Rev Neurol. 2019;15:644-56
76. Yohe S, Thyagarajan B. Review of Clinical Next-Generation Sequencing. Arch Pathol Lab Med. 2017;141:1544-57
77. Benke K, Agg B, Meienberg J, Kopps AM, Fattorini N, Stengl R. et al. Hungarian Marfan family with large FBN1 deletion calls attention to copy number variation detection in the current NGS era. J Thorac Dis. 2018;10:2456-60
78. Yuan P, Xia J, Ou S, Liu P, Du T, Zheng L. et al. A whole-genome sequencing-based novel preimplantation genetic testing method for de novo mutations combined with chromosomal balanced translocations. J Assist Reprod Genet. 2020;37:2525-33
79. Bewicke-Copley F, Arjun Kumar E, Palladino G, Korfi K, Wang J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput Struct Biotechnol J. 2019;17:1348-59
80. Ballester LY, Luthra R, Kanagal-Shamanna R, Singh RR. Advances in clinical next-generation sequencing: target enrichment and sequencing technologies. Expert Rev Mol Diagn. 2016;16:357-72
81. Li J, Lu C, Wu W, Liu Y, Wang R, Si N. et al. Application of next-generation sequencing to screen for pathogenic mutations in 123 unrelated Chinese patients with Marfan syndrome or a related disease. Sci China Life Sci. 2019;62:1630-7
82. Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T. et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS One. 2011;6:e29500
83. Stuppia L, Antonucci I, Palka G, Gatta V. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int J Mol Sci. 2012;13:3245-76
84. Varga RE, Mumtaz R, Jahic A, Rudenskaya GE, Sanchez-Ferrero E, Auer-Grumbach M. et al. MLPA-based evidence for sequence gain: pitfalls in confirmation and necessity for exclusion of false positives. Anal Biochem. 2012;421:799-801
85. Yang H, Ma Y, Luo M, Zhao K, Zhang Y, Zhu G. et al. Identification of gross deletions in FBN1 gene by MLPA. Hum Genomics. 2018;12:46
86. Furtado LV, Wooderchak-Donahue W, Rope AF, Yetman AT, Lewis T, Plant P. et al. Characterization of large genomic deletions in the FBN1 gene using multiplex ligation-dependent probe amplification. BMC Med Genet. 2011;12:119
87. Li J, Wu W, Lu C, Liu Y, Wang R, Si N. et al. Gross deletions in FBN1 results in variable phenotypes of Marfan syndrome. Clin Chim Acta. 2017;474:54-9
88. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. 2018;50:96
89. Pedroza AJ, Tashima Y, Shad R, Cheng P, Wirka R, Churovich S. et al. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2020;40:2195-211
90. Becerra-Munoz VM, Gomez-Doblas JJ, Porras-Martin C, Such-Martinez M, Crespo-Leiro MG, Barriales-Villa R. et al. The importance of genotype-phenotype correlation in the clinical management of Marfan syndrome. Orphanet J Rare Dis. 2018;13:16
91. Biery NJ, Eldadah ZA, Moore CS, Stetten G, Spencer F, Dietz HC. Revised genomic organization of FBN1 and significance for regulated gene expression. Genomics. 1999;56:70-7
92. Pereira L, D'Alessio M, Ramirez F, Lynch JR, Sykes B, Pangilinan T. et al. Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum Mol Genet. 1993;2:1762
93. Corson GM, Chalberg SC, Dietz HC, Charbonneau NL, Sakai LY. Fibrillin binds calcium and is coded by cDNAs that reveal a multidomain structure and alternatively spliced exons at the 5' end. Genomics. 1993;17:476-84
94. Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R. et al. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218-22
95. Collod-Beroud G, Le Bourdelles S, Ades L, Ala-Kokko L, Booms P, Boxer M. et al. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat. 2003;22:199-208
96. Groth KA, Von Kodolitsch Y, Kutsche K, Gaustadnes M, Thorsen K, Andersen NH. et al. Evaluating the quality of Marfan genotype-phenotype correlations in existing FBN1 databases. Genet Med. 2017;19:772-7
97. Groth KA, Gaustadnes M, Thorsen K, Ostergaard JR, Jensen UB, Gravholt CH. et al. Difficulties in diagnosing Marfan syndrome using current FBN1 databases. Genet Med. 2016;18:98-102
98. Ramirez F, Pereira L. The fibrillins. Int J Biochem Cell Biol. 1999;31:255-9
99. Robinson PN, Godfrey M. The molecular genetics of Marfan syndrome and related microfibrillopathies. J Med Genet. 2000;37:9-25
100. Gupta PA, Wallis DD, Chin TO, Northrup H, Tran-Fadulu VT, Towbin JA. et al. FBN2 mutation associated with manifestations of Marfan syndrome and congenital contractural arachnodactyly. J Med Genet. 2004;41:e56
101. Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T. et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855-60
102. Disabella E, Grasso M, Marziliano N, Ansaldi S, Lucchelli C, Porcu E. et al. Two novel and one known mutation of the TGFBR2 gene in Marfan syndrome not associated with FBN1 gene defects. Eur J Hum Genet. 2006;14:34-8
103. Lin J, Vora M, Kane NS, Gleason RJ, Padgett RW. Human Marfan and Marfan-like Syndrome associated mutations lead to altered trafficking of the Type II TGFbeta receptor in Caenorhabditis elegans. PLoS One. 2019;14:e0216628
104. Zhang L, Gao LG, Zhang M, Zhou XL. Genotype-phenotype analysis of F-helix mutations at the kinase domain of TGFBR2, including a type 2 Marfan syndrome familial study. Mol Vis. 2012;18:55-63
105. Attias D, Stheneur C, Roy C, Collod-Beroud G, Detaint D, Faivre L. et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation. 2009;120:2541-9
106. Singh KK, Rommel K, Mishra A, Karck M, Haverich A, Schmidtke J. et al. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum Mutat. 2006;27:770-7
107. Lucarini L, Evangelisti L, Attanasio M, Lapini I, Chiarini F, Porciani MC. et al. May TGFBR1 act also as low penetrance allele in Marfan syndrome? Int J Cardiol. 2009;131:281-4
108. Somers AE, Hinton RB, Pilipenko V, Miller E, Ware SM. Analysis of TGFBR1*6A variant in individuals evaluated for Marfan syndrome. Am J Med Genet A. 2016;170:1786-90
109. Loeys BL, Dietz HC. Loeys-Dietz Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021.
110. Stheneur C, Collod-Beroud G, Faivre L, Gouya L, Sultan G, Le Parc JM. et al. Identification of 23 TGFBR2 and 6 TGFBR1 gene mutations and genotype-phenotype investigations in 457 patients with Marfan syndrome type I and II, Loeys-Dietz syndrome and related disorders. Hum Mutat. 2008;29:E284-95
111. De Cario R, Sticchi E, Lucarini L, Attanasio M, Nistri S, Marcucci R. et al. Role of TGFBR1 and TGFBR2 genetic variants in Marfan syndrome. J Vasc Surg. 2018;68:225-33 e5
112. Saharinen J, Keski-Oja J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol Biol Cell. 2000;11:2691-704
113. Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Courousse T, Sakai LY. et al. Specificity of latent TGF-beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiol. 2012;227:3828-36
114. Unsold C, Hyytiainen M, Bruckner-Tuderman L, Keski-Oja J. Latent TGF-beta binding protein LTBP-1 contains three potential extracellular matrix interacting domains. J Cell Sci. 2001;114:187-97
115. Robertson IB, Dias HF, Osuch IH, Lowe ED, Jensen SA, Redfield C. et al. The N-Terminal Region of Fibrillin-1 Mediates a Bipartite Interaction with LTBP1. Structure. 2017;25:1208-21 e5
116. Quinones-Perez B, VanNoy GE, Towne MC, Shen Y, Singh MN, Agrawal PB. et al. Three-generation family with novel contiguous gene deletion on chromosome 2p22 associated with thoracic aortic aneurysm syndrome. Am J Med Genet A. 2018;176:560-9
117. Sticchi E, De Cario R, Magi A, Giglio S, Provenzano A, Nistri S. et al. Bicuspid Aortic Valve: Role of Multiple Gene Variants in Influencing the Clinical Phenotype. Biomed Res Int. 2018;2018:8386123
118. Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-beta-binding proteins. Matrix Biol. 2015;47:44-53
119. Inoue T, Ohbayashi T, Fujikawa Y, Yoshida H, Akama TO, Noda K. et al. Latent TGF-beta binding protein-2 is essential for the development of ciliary zonule microfibrils. Hum Mol Genet. 2014;23:5672-82
120. Thomson J, Singh M, Eckersley A, Cain SA, Sherratt MJ, Baldock C. Fibrillin microfibrils and elastic fibre proteins: Functional interactions and extracellular regulation of growth factors. Semin Cell Dev Biol. 2019;89:109-17
121. Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, Yazdani S, Nilforushan N, Fan JB. et al. LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat. 2012;33:1182-7
122. Morlino S, Alesi V, Cali F, Lepri FR, Secinaro A, Grammatico P. et al. LTBP2-related "Marfan-like" phenotype in two Roma/Gypsy subjects with the LTBP2 homozygous p.R299X variant. Am J Med Genet A. 2019;179:104-12
123. Korneva A, Zilberberg L, Rifkin DB, Humphrey JD, Bellini C. Absence of LTBP-3 attenuates the aneurysmal phenotype but not spinal effects on the aorta in Marfan syndrome. Biomech Model Mechanobiol. 2019;18:261-73
124. Zilberberg L, Phoon CK, Robertson I, Dabovic B, Ramirez F, Rifkin DB. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci U S A. 2015;112:14012-7
125. Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JMA, de Graaf BM, van de Beek G. et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65:1324-36
126. Prunier C, Pessah M, Ferrand N, Seo SR, Howe P, Atfi A. The oncoprotein Ski acts as an antagonist of transforming growth factor-beta signaling by suppressing Smad2 phosphorylation. J Biol Chem. 2003;278:26249-57
127. Greally MT. Shprintzen-Goldberg Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021.
128. Arnaud P, Racine C, Hanna N, Thevenon J, Alessandri JL, Bonneau D. et al. A new mutational hotspot in the SKI gene in the context of MFS/TAA molecular diagnosis. Hum Genet. 2020;139:461-72
129. Overwater E, Efrat R, Barge-Schaapveld D, Lakeman P, Weiss MM, Maugeri A. et al. Autosomal dominant Marfan syndrome caused by a previously reported recessive FBN1 variant. Mol Genet Genomic Med. 2019;7:e00518
130. Ramachandra CJ, Mehta A, Guo KW, Wong P, Tan JL, Shim W. Molecular pathogenesis of Marfan syndrome. Int J Cardiol. 2015;187:585-91
131. Eldadah ZA, Brenn T, Furthmayr H, Dietz HC. Expression of a mutant human fibrillin allele upon a normal human or murine genetic background recapitulates a Marfan cellular phenotype. J Clin Invest. 1995;95:874-80
132. Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL. et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172-81
133. Loeys B. The search for genotype/phenotype correlation in Marfan syndrome: to be or not to be? Eur Heart J. 2016;37:3291-3
134. Charbonneau NL, Carlson EJ, Tufa S, Sengle G, Manalo EC, Carlberg VM. et al. In vivo studies of mutant fibrillin-1 microfibrils. J Biol Chem. 2010;285:24943-55
135. Landis BJ, Veldtman GR, Ware SM. Genotype-phenotype correlations in Marfan syndrome. Heart. 2017;103:1750-2
136. Gao L, Tian T, Zhou X, Fan L, Wang R, Wu H. Detection of ten novel FBN1 mutations in Chinese patients with typical or incomplete Marfan syndrome and an overview of the genotype-phenotype correlations. Int J Cardiol. 2019;293:186-91
137. Pees C, Michel-Behnke I, Hagl M, Laccone F. Detection of 15 novel mutations in 52 children from 40 families with the Marfan or Loeys-Dietz syndrome and phenotype-genotype correlations. Clin Genet. 2014;86:552-7
138. Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E. et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454-66
139. Baudhuin LM, Kotzer KE, Lagerstedt SA. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet Med. 2015;17:177-87
140. Reinhardt DP, Ono RN, Notbohm H, Muller PK, Bachinger HP, Sakai LY. Mutations in calcium-binding epidermal growth factor modules render fibrillin-1 susceptible to proteolysis. A potential disease-causing mechanism in Marfan syndrome. J Biol Chem. 2000;275:12339-45
141. Franken R, Groenink M, de Waard V, Feenstra HM, Scholte AJ, van den Berg MP. et al. Genotype impacts survival in Marfan syndrome. Eur Heart J. 2016;37:3285-90
142. Willis BR, Lee M, Rethanavelu K, Fung JLF, Wong RMS, Hui P. et al. A case of G1013R FBN1 mutation: A potential genotype-phenotype correlation in severe Marfan syndrome. Am J Med Genet A. 2020;182:1329-35
143. Whiteman P, Handford PA. Defective secretion of recombinant fragments of fibrillin-1: implications of protein misfolding for the pathogenesis of Marfan syndrome and related disorders. Hum Mol Genet. 2003;12:727-37
144. Gomez D, Coyet A, Ollivier V, Jeunemaitre X, Jondeau G, Michel JB. et al. Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms. Cardiovasc Res. 2011;89:446-56
145. Wu Y, Sun H, Wang J, Wang X, Gong M, Han L. et al. Marfan syndrome: whole-exome sequencing reveals de novo mutations, second gene and genotype-phenotype correlations in the Chinese population. Biosci Rep. 2020;40:BSR20203356
146. Arnaud P, Hanna N, Aubart M, Leheup B, Dupuis-Girod S, Naudion S. et al. Homozygous and compound heterozygous mutations in the FBN1 gene: unexpected findings in molecular diagnosis of Marfan syndrome. J Med Genet. 2017;54:100-3
147. Michel JB, Jondeau G, Milewicz DM. From genetics to response to injury: vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc Res. 2018;114:578-89
148. De Backer J, Loeys B, Leroy B, Coucke P, Dietz H, De Paepe A. Utility of molecular analyses in the exploration of extreme intrafamilial variability in the Marfan syndrome. Clin Genet. 2007;72:188-98
149. Arai Y, Umeyama K, Okazaki N, Nakano K, Nishino K, Nagashima H. et al. DNA methylation ambiguity in the Fibrillin-1 (FBN1) CpG island shore possibly involved in Marfan syndrome. Sci Rep. 2020;10:5287
150. Lino Cardenas CL, Kessinger CW, MacDonald C, Jassar AS, Isselbacher EM, Jaffer FA. et al. Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm. JCI Insight. 2018;3:e97493
151. Fernandes GR, Massironi SM, Pereira LV. Identification of Loci Modulating the Cardiovascular and Skeletal Phenotypes of Marfan Syndrome in Mice. Sci Rep. 2016;6:22426
152. Benke K, Agg B, Szilveszter B, Tarr F, Nagy ZB, Polos M. et al. The role of transforming growth factor-beta in Marfan syndrome. Cardiol J. 2013;20:227-34
153. Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790-811
154. Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742-50
155. Rezaei HB, Kamato D, Ansari G, Osman N, Little PJ. Cell biology of Smad2/3 linker region phosphorylation in vascular smooth muscle. Clin Exp Pharmacol Physiol. 2012;39:661-7
156. Rozes-Salvador V, Siri SO, Musri MM, Conde C. New Player in Endosomal Trafficking: Differential Roles of Smad Anchor for Receptor Activation (SARA) Protein. Mol Cell Biol. 2018;38:e00446-18
157. Tae HJ, Petrashevskaya N, Marshall S, Krawczyk M, Talan M. Cardiac remodeling in the mouse model of Marfan syndrome develops into two distinctive phenotypes. Am J Physiol Heart Circ Physiol. 2016;310:H290-9
158. Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB. et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. 2009;284:5630-6
159. Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C. et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358-61
160. Pedroza AJ, Koyano T, Trojan J, Rubin A, Palmon I, Jaatinen K. et al. Divergent effects of canonical and non-canonical TGF-beta signalling on mixed contractile-synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. J Cell Mol Med. 2020;24:2369-83
161. Crosas-Molist E, Meirelles T, Lopez-Luque J, Serra-Peinado C, Selva J, Caja L. et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35:960-72
162. He H, Yu B, Liu Z, Ye G, You W, Hong Y. et al. Vascular progenitor cell senescence in patients with Marfan syndrome. J Cell Mol Med. 2019;23:4139-52
163. You W, Hong Y, He H, Huang X, Tao W, Liang X. et al. TGF-beta mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging (Albany NY). 2019;11:3574-84
164. Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R. et al. Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor beta Signaling. J Am Heart Assoc. 2017;6:e004968
165. Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN. et al. Postnatal Deletion of the Type II Transforming Growth Factor-beta Receptor in Smooth Muscle Cells Causes Severe Aortopathy in Mice. Arterioscler Thromb Vasc Biol. 2015;35:2647-56
166. Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S. et al. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35:911-7
167. Franken R, Radonic T, den Hartog AW, Groenink M, Pals G, van Eijk M. et al. The revised role of TGF-beta in aortic aneurysms in Marfan syndrome. Neth Heart J. 2015;23:116-21
168. Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S. et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. 2010;120:422-32
169. Tashima Y, He H, Cui JZ, Pedroza AJ, Nakamura K, Yokoyama N. et al. Androgens Accentuate TGF-beta Dependent Erk/Smad Activation During Thoracic Aortic Aneurysm Formation in Marfan Syndrome Male Mice. J Am Heart Assoc. 2020;9:e015773
170. Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ Res. 2012;110:e92-e101
171. Wang S, Niu Z, Wang H, Ma M, Zhang W, Fang Wang S. et al. De Novo Paternal FBN1 Mutation Detected in Embryos Before Implantation. Med Sci Monit. 2017;23:3136-46
172. Goumans MJ, Ten Dijke P. TGF-beta Signaling in Control of Cardiovascular Function. Cold Spring Harb Perspect Biol. 2018;10:a022210
Author contact
![]() Corresponding author: Professor Dingding Zhang, Sichuan Provincial Key Laboratory for Genetic Disease, Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and Technology of China, 2006 Xiyuan Road, Gaoxin, Chengdu, Sichuan 611731, P.R. China. E-mail: zhangdingdinguestc.edu.cn.
Corresponding author: Professor Dingding Zhang, Sichuan Provincial Key Laboratory for Genetic Disease, Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and Technology of China, 2006 Xiyuan Road, Gaoxin, Chengdu, Sichuan 611731, P.R. China. E-mail: zhangdingdinguestc.edu.cn.