Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(12):2561-2569. doi:10.7150/ijms.56630 This issue Cite
Research Paper
SARS-CoV-2 infection activates a subset of intrinsic pathways to inhibit type I interferons in vitro and in vivo
Weisheng Luo1,2, Lianzhou Huang1,2, Xiaohui Wang1,2, Yuying Ma1,2, Ji Xiao1,2, Xiaowei Song1,2, Ping Liu1,2, Yifei Wang1,2 ![]() , Yiliang Wang1,2
, Yiliang Wang1,2 ![]() , Zhe Ren1,2
, Zhe Ren1,2 ![]()
1. Guangzhou Jinan Biomedicine Research and Development Center, Institute of Biomedicine, College of Life Science and Technology, Jinan University, Guangzhou 510632, PR China.
2. Key Laboratory of Virology of Guangzhou, Jinan University, Guangzhou 510632, PR China.
Received 2020-12-3; Accepted 2021-4-12; Published 2021-4-28
Abstract

SARS-CoV-2 infection poses a global challenge to human health. Upon viral infection, host cells initiate the innate antiviral response, which primarily involves type I interferons (I-IFNs), to enable rapid elimination of the invading virus. Previous studies revealed that SARS-CoV-2 infection limits the expression of I-IFNs in vitro and in vivo, but the underlying mechanism remains incompletely elucidated. In the present study, we performed data mining and longitudinal data analysis using SARS-CoV-2-infected normal human bronchial epithelial (NHBE) cells and ferrets, and the results confirmed the strong inhibitory effect of SARS-CoV-2 on the induction of I-IFNs. Moreover, we identified genes that are negatively correlated with IFNB1 expression in vitro and in vivo based on Pearson correlation analysis. We found that SARS-CoV-2 activates numerous intrinsic pathways, such as the circadian rhythm, phosphatidylinositol signaling system, peroxisome, and TNF signaling pathways, to inhibit I-IFNs. These intrinsic inhibitory pathways jointly facilitate the successful immune evasion of SARS-CoV-2. Our study elucidates the underlying mechanism by which SARS-CoV-2 evades the host innate antiviral response in vitro and in vivo, providing theoretical evidence for targeting these immune evasion-associated pathways to combat SARS-CoV-2 infection.
Keywords: SARS-CoV-2, innate antiviral responses, type I IFNs, immune evasion
Introduction
SARS-CoV-2 is an emerging pathogen that poses a considerable threat to global human health, and a comprehensive understanding of the SARS-CoV-2-host interaction is urgently required [1]. The host innate immune response is the first line of defense against viral infection [2-4]. Upon the recognition of viral components by pattern recognition receptors (PRRs), host cells initiate a concerted innate antiviral response that primarily involves type I interferon (I-IFN) production and the subsequent upregulation of IFN-stimulated genes (ISGs), which encode a subset of antiviral proteins. Importantly, I-IFNs show impressive antiviral activity against SARS-CoV-2 [4-6]. However, several viral proteins have acquired the capability to evade the innate antiviral response via numerous mechanisms [7]. In general, viral proteins suppress innate antiviral responses in two ways: by activating intrinsic immunosuppressive pathways or by directly targeting innate antiviral signaling components to inhibit signal transduction. For example, SARS-CoV, which is closely related to SARS-CoV-2, encodes numerous proteins that antagonize the induction of I-IFNs and the effector functions of downstream ISGs [8]. The balance between the innate antiviral immune response and viral immune evasion is crucial to viral pathogenesis. Indeed, previous studies have reported that SARS-CoV-2 infection induces the expression of a very small subset of ISGs but virtually no I-IFNs [4, 9-13], suggesting the impressive immune evasion activity of SARS-CoV-2. Several studies have revealed that SARS-CoV-2 counteracts the host innate antiviral response [7, 13]. A SARS-CoV-2 protein interaction network revealed that NSP13 of SARS-CoV-2 may interact with TBK1 [14], although the function of this interaction in SARS-CoV-2 immune evasion remains unknown. Thus, the specific and precise mechanism by which SARS-CoV-2 antagonizes I-IFN signaling needs further exploration.
In the present study, we performed data mining and longitudinal data analysis of SARS-CoV-2-infected normal human bronchial epithelial (NHBE) cells and ferrets, and the results confirmed the strong ability of SARS-CoV-2 to inhibit the induction of I-IFNs in vitro and in vivo. We further analyzed genes that are negatively correlated with IFNB1 in SARS-CoV-2-infected NHBE cells and ferrets. Our study reveals the mechanism by which SARS-CoV-2 evades the host innate antiviral response and thus provides a potential therapeutic target for COVID-19.
Results
Analysis of data from SARS-CoV-2-infected NHBE cells demonstrated that SARS-CoV-2 infection inhibits the expression of I-IFNs in NHBE cells
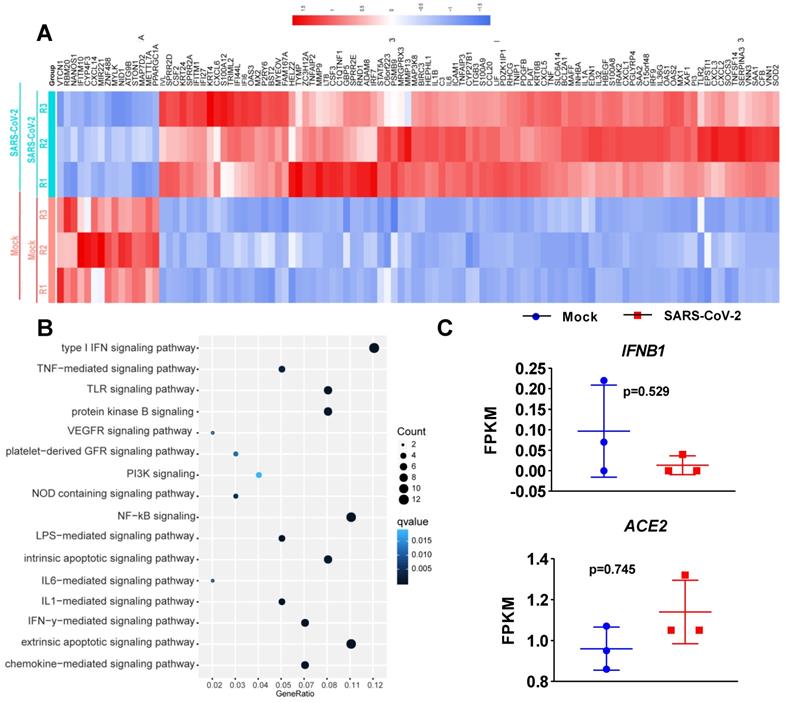
To determine the transcriptional response of NHBE cells to SARS-CoV-2, we performed differential expression analysis comparing SARS-CoV-2-infected cells to corresponding mock-infected cells based on previous transcriptome datasets [4]. NHBE cells were infected with SARS-CoV-2 at an MOI of 2 for 24 h, and polyA RNA was subsequently enriched for subsequent RNA sequencing (RNA-seq) analysis [4]. Our analysis of public transcriptome datasets identified 106 differentially expressed protein-coding genes (DEGs) in SARS-CoV-2-infected NHBE cells: 15 downregulated genes and 91 upregulated genes compared to their expression in control cells (Figure 1A and Table S1). To further understand changes in the expression profile of NHBE cells in response to SARS-CoV-2 infection, gene enrichment analysis of these DEGs was performed. The analysis revealed strong enrichment of these genes in chemotactic and inflammatory response signaling (Figure 1B). Specifically, upon SARS-CoV-2 infection, NHBE cells mount a chemokine-mediated signaling response characterized by the induction of CCL20, CXCL6, CXCL1, CXCL5, CXCL3, CXCL2, and EDN1 (Figure 1B and Table S2). Consistent with previous publications [15-17], SARS-CoV-2 infection was also found to activate interleukin-6 (IL-6)-mediated signaling, which contributes to cytokine release syndrome in severe COVID-19, with representative genes of this pathway including SOCS3 and IL6 (Figure 1B and Table S2). SARS-CoV-2 infection also activated genes downstream of IκB kinase/NF-κB signaling, including ZC3H12A, S100A12, BIRC3, BST2, IL1B, IL36G, IRAK2, TLR2, TNIP1, TNF, and TNFAIP3 (Figure 1B and Table S2). However, the infection of NHBE cells with SARS-CoV-2 failed to induce the expression of I-IFNs, including IFNB1 and a series of IFN genes, as supported by the quantitative results for all protein-coding transcripts (Figure 1C and Table S1). Notably, although a significant increase in IFNB1 was not observed in SARS-CoV-2-infected NHBE cells, ISGs were induced by SARS-CoV-2 infection (Figure 2). The ISGs induced by SARS-CoV-2 infection included IFI6, XAF1, IFITM1, IRF7, OAS1, OAS3, OAS2, IRF9, IFI27, XAF1, BST2, MX2, and MX1 (Figure 2). In contrast, the expression of several other ISGs, primarily ISG15, ISG20, OASL, and CXCL10, was not induced by SARS-CoV-2 (Figure 2). Notably, the expression of ACE2, a receptor for SARS-CoV-2 entry and recently identified ISG [18], was also slightly higher in SARS-CoV-2-infected NHBE cells than in control cells (Figure 2). These results indicate that SARS-CoV-2 exhibits the strong ability to inhibit IFNB1 expression to enable its successful immune escape.
Transcriptional response of primary human bronchial epithelial cells to SARS-CoV-2. A. Heatmap of differentially expressed protein-coding genes in SARS-CoV-2-infected NHBE cells compared with the corresponding mock-infected cells. B. Dot plot visualization of enriched GO terms in SARS-CoV-2-infected NHBE cells. Gene enrichment analysis of the differentially expressed genes between SARS-CoV-2- and mock-infected NHBE cells was performed. The colors of the dots represent the q values for each enriched GO term, and their sizes represent the number of genes enriched in the corresponding signaling pathway. C. Levels of the innate antiviral factors IFNB1, ACE2, IFI6, XAF1, MX1, MX2, IFITM1, IRF7, OAS1, OAS2, OAS3, IRF9, IFI27, ISG15, ISG20, OASL and CXCL10 in SARS-CoV-2- and mock-infected NHBE cells were determined as FPKM values. The p values were calculated with DESeq2 software.
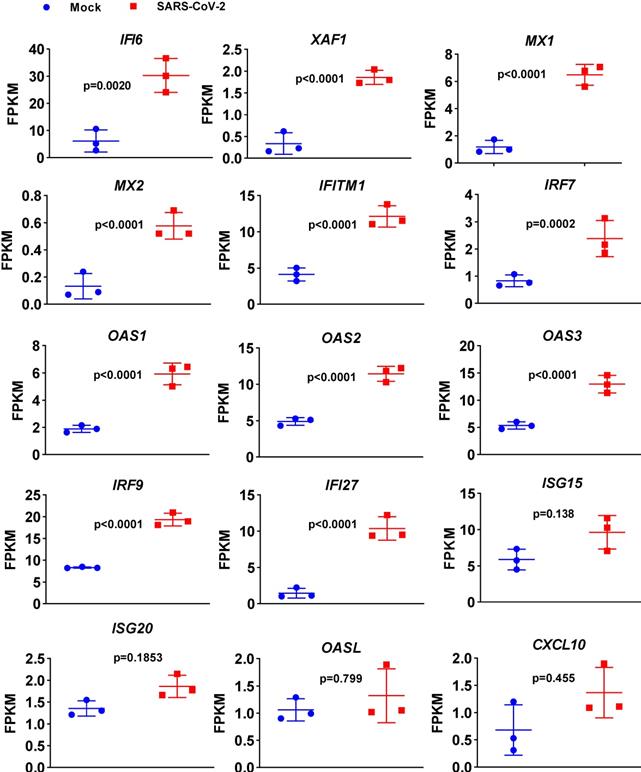
Expression levels of a subset of ISGs in response to SARS-CoV-2 infection in primary human bronchial epithelial cells. Levels of the innate antiviral factors IFI6, XAF1, MX1, MX2, IFITM1, IRF7, OAS1, OAS2, OAS3, IRF9, IFI27, ISG15, ISG20, OASL and CXCL10 in SARS-CoV-2- and mock-infected NHBE cells are presented as FPKM values.
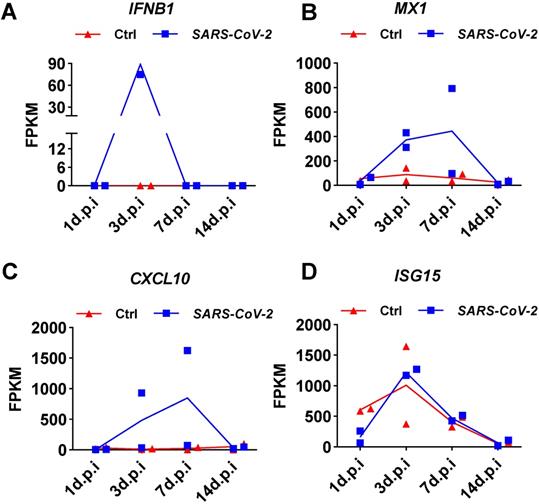
Analysis of longitudinal data from SARS-CoV-2-infected ferrets demonstrated transient induction of IFNB1 by SARS-CoV-2 infection in vivo
To determine whether the weak induction of IFNB1 by SARS-CoV-2 observed in NHBE cells was an artifact of the cell culture conditions, we analyzed public transcriptome data obtained from SARS-CoV-2-infected ferrets. Naive ferrets were intranasally infected with 5 × 104 PFU of the SARS-CoV-2 isolate USA-WA1/2020 [4]. Small pellets consisting of upper respiratory tract cells isolated from nasal washes were subjected to transcriptome analysis. Transient induction of IFNB1 by SARS-CoV-2 infection in ferrets over time was shown by the quantitative results presented as the corresponding fragments per kilobase million (FPKM) values (Figure 3). Specifically, on day 1 after infection, no SARS-CoV-2-mediated induction of IFNB1 was observed (Figure 3A). Significant IFNB1 upregulation was observed at 3 days post-infection (Figure 3A). However, the expression of IFNB1 quickly decreased to the baseline level at 7 days post-infection (Figure 3A). Inconsistent with the observed trend in the IFNB1 level, persistent upregulation of numerous ISGs, including CXCL10, MX1, and ISG15, was observed in response to SARS-CoV-2 infection from 3 to 7 days post-infection (Figure 3B, 3C, and 3D).
Expression changes in IFNB1 and several ISGs in nasal washes samples collected from SARS-CoV-2- and mock-infected control ferrets. A-D. Results of transcriptome analysis indicating the levels of IFNB1 (A) and several ISGs (B, C, and D) in nasal washes samples collected from SARS-CoV-2- and mock-infected control ferrets at indicated time points post-infection. The FPKM values of these genes are presented and reflect their expression levels.
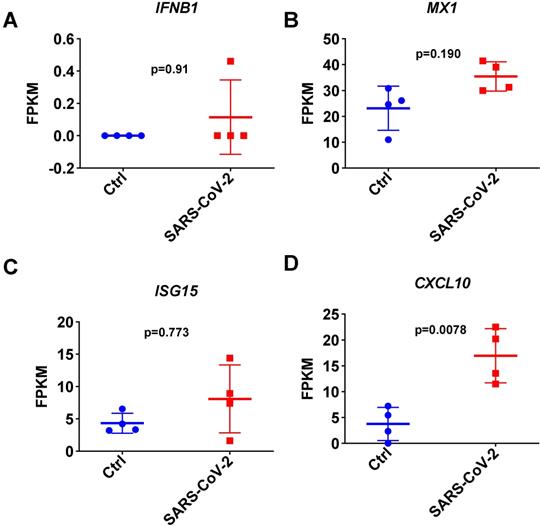
To further investigate the I-IFN response to SARS-CoV-2 in detail, we next focused on analyzing the respiratory tract in SARS-CoV-2-infected ferrets infected in parallel on day 3 using public transcriptome data [4]. Consistent with the results observed with the small cell pellets, the quantitative results of transcriptome data analysis indicated that SARS-CoV-2 infection inhibited the expression of IFNB1 in the respiratory tracts of SARS-CoV-2-infected ferrets on day 3 (Figure 4A). Although SARS-CoV-2 infection induced significant CXCL10 upregulation, most ISGs were not significantly upregulated in the respiratory tract in SARS-CoV-2-infected ferrets (Figure 4B, 4C, and 4D). These results indicated that SARS-CoV-2 infection induces the transient expression of IFNB1 but causes an insufficient response to I-IFNs in the respiratory tract. The upregulation of ISGs observed in the nasal wash samples may be attributable to the small size of the upper respiratory tract cell pellets.
Expression changes in IFNB1 and several ISGs in respiratory tract samples collected from SARS-CoV-2- and mock-infected control ferrets. A-D. Results of transcriptome analysis indicating the levels of IFNB1 (A) and several ISGs (B, C, and D) in respiratory tract samples from SARS-CoV-2- and mock-infected control ferrets on day 3 post-infection. The FPKM values of these genes are presented and reflect their expression levels.
Analysis of SARS-CoV-2-induced genes whose expression is negatively correlated with IFNB1 in vitro and in vivo
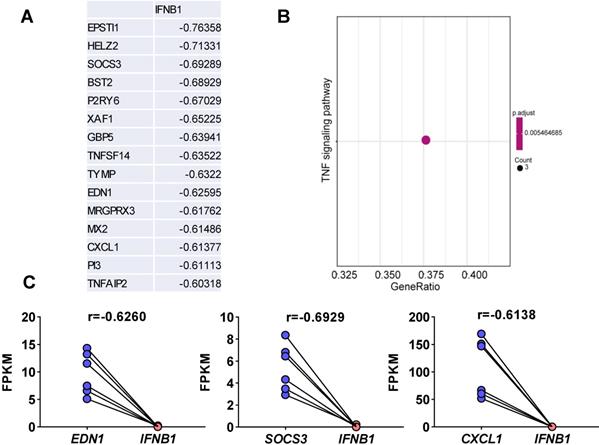
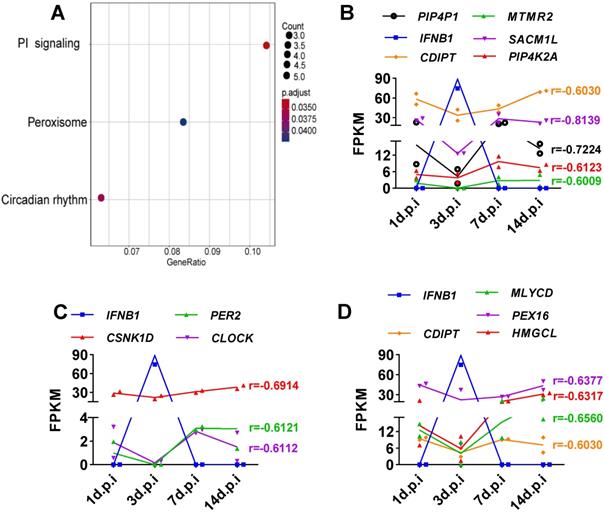
Viruses usually encode numerous type I IFN antagonists to ensure successful evasion of host innate immunity [7, 14]. These antagonists suppress the expression of I-IFNs by activating intrinsic pathways, restricting I-IFN induction, or directly manipulating the crucial factors that activate these I-IFN pathways. To identify the pathway by which SARS-CoV-2-mediated I-IFN inhibition is activated, we analyzed SARS-CoV-2-induced genes that were negatively associated with IFNB1 in vitro and in vivo based on Pearson correlation analysis. We identified 15 genes whose expression was negatively correlated with IFNB1 expression: EPSTI1, HELZ2, SOCS3, BST2, P2RY6, XAF1, GBP5, TNFSF14, TYMP, EDN1, MRGPRX3, MX2, CXCL1, PI3, and TNFAIP2 (Figure 5A and Table S3). The results of KEGG analysis suggested that a number of these correlated genes were enriched in the TNF signaling pathway (Figure 5B); these genes included SOCS3, CXCL1, and EDN1 (Figure 5C). Moreover, we identified genes that were negatively correlated with IFNB1 in our longitudinal study of SARS-CoV-2-infected ferrets via Pearson correlation-based analysis (Table S4). The results of KEGG analysis revealed that the genes that were negatively correlated with IFNB1 in SARS-CoV-2-infected ferrets were primarily enriched in the following pathways: the phosphatidylinositol signaling system (PIP4P1, MTMR2, SACM1L, CDIPT, and PIP4K2A), the circadian rhythm (PER2, CSNK1D, and CLOCK), and peroxisomes (CDIPT, MLYCD, PEX16, and HMGCL) (Figure 6).
Elucidation of SARS-CoV-2-induced genes whose expression was negatively correlated with IFNB1 expression in vitro. A. Pearson correlation coefficient-based analysis of genes whose expression was negatively correlated with IFNB1 expression. The expression of 15 genes was negatively correlated with that of IFNB1: EPSTI1, HELZ2, SOCS3, BST2, P2RY6, XAF1, GBP5, TNFSF14, TYMP, EDN1, MRGPRX3, MX2, CXCL1, PI3, and TNFAIP2. B. The results of KEGG analysis showed that the correlated genes were enriched in the TNF signaling pathway (SOCS3, CXCL1, and EDN1; adjusted p value=0.005464685 and gene count=3). C. The levels of genes enriched in the TNF signaling pathway upon SARS-CoV-2 infection in NHBEs. The FPKM values of the indicated genes were calculated and reflect the expression levels of these genes. The correlation coefficients (r) between IFNB1 and the corresponding genes are also indicated.
Elucidation of SARS-CoV-2-induced genes whose expression was negatively correlated with IFNB1 expression in ferrets with SARS-CoV-2 infection. A. Based on the obtained Pearson correlation coefficients, the results of KEGG analysis revealed that genes that are negatively correlated with IFNB1 were primarily enriched in the phosphatidylinositol (PI) signaling system, circadian rhythm, and peroxisome pathways. B-D. Longitudinal changes in genes enriched in the signaling pathways shown in (A), the PI signaling system(B), circadian rhythm (C), and peroxisome (D) pathways, in SARS-CoV-2-infected ferrets. The levels of these genes are presented as FPKM values. The correlation coefficients (r) between IFNB1 and the corresponding genes are also indicated.
Discussion
Numerous studies have reported that SARS-CoV-2 infection induces a weakened innate antiviral defense response characterized by a low level of I-IFNs and a strong proinflammatory response [4, 7, 15, 18]. I-IFNs are the primary initiators that activate the expression of antiviral genes. Indeed, patients with life-threatening COVID-19 show an inherited defect in the innate antiviral response [19, 20], further implying the importance of I-IFNs in combating SARS-CoV-2 infection. However, the underlying mechanisms by which type I IFNs are inhibited by SARS-CoV-2 remain largely unknown. In general, viruses can produce numerous proteins that target crucial factors involved in I-IFN signaling or activate intrinsic pathways to inhibit I-IFN signaling and achieve immune evasion [8, 21-23]. The activation of intrinsic pathways that inhibit I-IFNs is a crucial negative feedback mechanism that maintains host immune hemostasis to avoid damage due to the excessive production of I-IFNs [22]. In the present study, we focused on elucidating the intrinsic inhibitory pathways activated by SARS-CoV-2 infection in vitro and in vivo.
First, we confirmed that SARS-CoV-2 infection inhibited the expression of IFNB1 in vitro and in vivo by analyzing published in vitro and in vivo transcriptome data, and our findings were consistent with those of previous studies showing that SARS-CoV-2 infection triggers decreased I-IFN production [4, 9-13]. Specifically, SARS-CoV-2 infection induced limited IFNB1 expression. In contrast, SARS-CoV-2 infection inhibited the expression of IFNB1 in NHBE cells. Moreover, although the expression of most ISGs could not be induced by SARS-CoV-2 infection, the expression of several ISGs was induced. Taken together, these findings indicate that SARS-CoV-2 infection induced a weakened innate antiviral response in vitro. Similar to the results observed in vitro, SARS-CoV-2-infected ferrets showed the transient induction of IFNB1, as demonstrated by transcriptome analysis of the respiratory tract and nasal wash samples. Given the suppressed innate antiviral response observed in the context of SARS-CoV-2 infection, we speculated that SARS-CoV-2 exhibits strong immune evasion activity. Thus, we performed Pearson correlation analysis to analyze genes whose expression was negatively correlated with IFNB1 expression in the context of SARS-CoV-2 infection in vitro and in vivo. The in vitro correlation analysis results suggested that the enrichment of primarily the SOCS3, EDN1, and CXCL1 genes was negatively correlated with IFNB1. SOCS3, a component of the TNF signaling pathway, is a factor known to inhibit overactive I-IFN signaling [22]. Given that TNF activates an IRF1-dependent autocrine loop, leading to sustained expression of STAT1-dependent I-IFN response genes [24], the activation of TNF signaling may explain the induction of ISGs but not IFNB1. Indeed, accumulating evidence indicates that anti-TNF therapy shows impressive efficacy against COVID-19 and should accordingly receive priority in COVID-19 treatment trials [25]. In contrast, no studies have addressed the roles of EDN1 and CXCL1 in suppressing I-IFN signaling. The in vivo correlation analysis revealed that SARS-CoV-2 infection significantly activates the circadian rhythm, phosphatidylinositol signaling, and peroxisome signaling pathways, all of which may jointly contribute to suppressing the innate antiviral response to SARS-CoV-2 in vivo. Indeed, the disruption of circadian rhythms has been suggested to lead to the impairment of innate immunity [26-28]. In particular, mice lacking circadian rhythms showed robust activation of the IFN pathway [27], and the circadian clock has been shown to regulate cellular susceptibility to the herpes and influenza viruses [29]. A recent study revealed that systemic Bmal1-knockout (KO) mice had an exacerbated imiquimod-induced ISG response in the skin and isolated epidermis, suggesting an involvement of the circadian clock in the innate antiviral response [27]. Indeed, the importance of the circadian clock in COVID-19 management has been demonstrated, although the associated mechanism remains obscure [30]. Peroxisomes are also signaling platforms for antiviral innate immunity [31], implying that they may be involved in limiting the innate antiviral response to SARS-CoV-2. However, a limitation of our present study is that we performed only transcriptome analysis, and the findings need further experimental confirmation. Despite this limitation, our study provides novel insight into the mechanisms underlying the immune evasion of SARS-CoV-2. An inhibitor targeting inhibition of the intrinsic pathway of I-IFN activated by SARS-CoV-2 is expected to be an ideal therapeutic target for COVID-19 by initiating intrinsic innate antiviral immunity.
Materials and Methods
Acquisition and analysis of high-throughput RNA-seq data
The raw high-throughput RNA-seq data analyzed in the present study were obtained from GEO (GSE147507) and published in a previous study [4]. The method used to analyze the raw data in the present study was similar to that described in our previous study with minor modifications [32]. FastQC software with the default parameters was used to filter the low-quality reads to obtain clean data of high quality. The filtered RNA-seq data were then mapped to the genome of the corresponding species using HISAT2 v2.0.4 [33]. The mapped reads of each sample were assembled with StringTie (v1.3.1) via a reference-based approach [34]. StringTie uses a novel network flow algorithm as well as an optional de novo assembly step to assemble and quantitate full-length transcripts representing multiple splice variants for each gene locus [34]. The FPKM value is an indicator of gene expression. DEGs were identified using the R package DESeq2 v4.0 [35]. An adjusted p value < 0.05 (according to the Benjamini-Hochberg procedure) [36] and an absolute fold change > 2 were considered to indicate significantly differential gene expression.
Pearson correlation analysis
Correlations between IFNB1 and other genes were analyzed by Pearson correlation analysis [37]. In brief, we calculated the correlation coefficients (r, Pearson correlation coefficient; p value ≤ 0.05) between IFNB1 and other genes and used the criterion |r|≥0.6 to identify the correlated genes. Pearson correlation analysis was conducted using the Python package scipy [38].
Pathway enrichment analysis
Genes usually interact to take part in specific biological functions. Pathway-based analysis facilitates further understanding of the biological functions of genes, and KEGG is a major public pathway-related database [39, 40]. In the present study, KEGG enrichment analysis was performed with the R package clusterProfiler v3.0.4 [41]. Pathway enrichment analysis identified metabolic pathways and signal transduction pathways significantly enriched in the DEGs compared to the whole-genome background using the formula presented below. Specifically, N is the number of all genes with KEGG annotations, n is the number of DEGs among the N genes, M is the number of all genes annotated to specific pathways, and m is the number of DEGs among the M genes. The calculated p value was subjected to FDR correction, with FDR ≤ 0.05 as the threshold. Pathways meeting this condition were defined as pathways significantly enriched in DEGs.
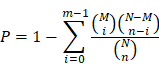
Supplementary Material
Supplementary tables.
Acknowledgements
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81872908 and 81573471), the Science and Technology Program of Guangzhou, China (201604020178), the Key Projects of Biological Industry Science & Technology of Guangzhou China [grant No. 201300000060], and the Science & Technology Plan Program of Guangdong Province, China [grant No. 2012A080204003].
Authors' contributions
Yiliang Wang and Weisheng Luo: conception and design, collection and/or assembly of data, data analysis and interpretation, and manuscript writing; Lianzhou Huang, and Ji Xiao: collection and/or assembly of data and data analysis and interpretation; Fujun Jin, Zhe Ren, and Yifei Wang: conception and design, manuscript writing, and final approval of the manuscript. All authors read and approved the final manuscript. Yiliang Wang and Weisheng Luo contributed equally to this article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang Y, Wang Y, Luo W, Huang L, Xiao J, Li F. et al. A comprehensive investigation of the mRNA and protein level of ACE2, the putative receptor of SARS-CoV-2, in human tissues and blood cells. Int J Med Sci. 2020;17:1522-31
2. Wang YL, Wang Y, Luo WS, Song XW, Huang LZ, Xiao J. et al. Roles of long non-coding RNAs and emerging RNA-binding proteins in innate antiviral responses. Theranostics. 2020;10:9407-24
3. Wang Y, Jia J, Wang Y, Li F, Song X, Qin S. et al. Roles of HSV-1 infection-induced microglial immune responses in CNS diseases: friends or foes? Crit Rev Microbiol. 2019;45:581-94
4. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R. et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036 -+
5. Mantlo E, Bukreyeva N, Maruyama J, Paessler S, Huang C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res. 2020;179:104811
6. Sallard E, Lescure FX, Yazdanpanah Y, Mentre F, Peiffer-Smadja N. Type 1 interferons as a potential treatment against COVID-19. Antiviral Res. 2020;178:104791
7. Wu Y, Ma L, Zhuang Z, Cai S, Zhao Z, Zhou L. et al. Main protease of SARS-CoV-2 serves as a bifunctional molecule in restricting type I interferon antiviral signaling. Signal Transduct Target Ther. 2020;5:221
8. Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol. 2012;2:264-75
9. Trouillet-Assant S, Viel S, Gaymard A, Pons S, Richard JC, Perret M. et al. Type I IFN immunoprofiling in COVID-19 patients. J Allergy Clin Immunol. 2020
10. Chu H, Chan JF, Wang Y, Yuen TT, Chai Y, Hou Y. et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis. 2020
11. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497-506
12. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z. et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe. 2020;27:883-90 e2
13. Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020;16:e1008737
14. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020
15. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. Journal of Clinical Investigation. 2020
16. Wei L, Ming S, Zou B, Wu Y, Hong Z, Li Z. et al. Viral Invasion and Type I Interferon Response Characterize the Immunophenotypes during COVID-19 Infection. Available at SSRN 3555695. 2020
17. Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020;368:473-4
18. Ziegler C, Allon S, Nyquist S, Mbano I, Miao V, Tzouanas C. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020
19. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020 370
20. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020 370
21. Jin S, Cui J. BST2 inhibits type I IFN (interferon) signaling by accelerating MAVS degradation through CALCOCO2-directed autophagy. Autophagy. 2018;14:171-2
22. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36-49
23. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y. et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is further increased by a naturally occurring elongation variant. bioRxiv. 2020. 2020 05.11.088179
24. Yarilina A, Park-Min K-H, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nature immunology. 2008;9:378-87
25. Robinson PC, Richards D, Tanner HL, Feldmann M. Accumulating evidence suggests anti-TNF therapy needs to be given trial priority in COVID-19 treatment. Lancet Rheumatol. 2020;2:E653-E5
26. Sutton CE, Finlay CM, Raverdeau M, Early JO, DeCourcey J, Zaslona Z. et al. Loss of the molecular clock in myeloid cells exacerbates T cell-mediated CNS autoimmune disease. Nat Commun. 2017;8:1923
27. Greenberg EN, Marshall ME, Jin S, Venkatesh S, Dragan M, Tsoi LC. et al. Circadian control of interferon-sensitive gene expression in murine skin. Proc Natl Acad Sci U S A. 2020;117:5761-71
28. Aiello I, Fedele MLM, Roman F, Marpegan L, Caldart C, Chiesa JJ. et al. Circadian disruption promotes tumor-immune microenvironment remodeling favoring tumor cell proliferation. Sci Adv. 2020 6
29. Edgar RS, Stangherlin A, Nagy AD, Nicoll MP, Efstathiou S, O'Neill JS. et al. Cell autonomous regulation of herpes and influenza virus infection by the circadian clock. Proc Natl Acad Sci U S A. 2016;113:10085-90
30. Ray S, Reddy AB. COVID-19 management in light of the circadian clock. Nat Rev Mol Cell Biol. 2020;21:494-5
31. Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B. et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668-81
32. Wang Y, Huang L, Wang Y, Luo W, Li F, Xiao J. et al. Single-cell RNA-sequencing analysis identifies host long noncoding RNA MAMDC2-AS1 as a co-factor for HSV-1 nuclear transport. Int J Biol Sci. 2020;16:1586-603
33. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357-9
34. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650-67
35. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
36. Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological). 1995;57:289-300
37. Benesty J, Chen J, Huang Y, Cohen I. Noise reduction in speech processing: Springer Science & Business Media; 2009
38. Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17:261-72
39. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27-30
40. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139-40
41. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284-7
Author contact
![]() Corresponding authors: Zhe Ren, Biomedicine Research and Development Center, Jinan University, Guangzhou 510632, Guangdong, PR China. E-mail: rzl62000com; Yiliang Wang, Biomedicine Research and Development Center, Jinan University, Guangzhou 510632, Guangdong, PR China. E-mail: wang_yiliangcn; Yifei Wang, Biomedicine Research and Development Center, Jinan University, Guangzhou 510632, Guangdong, PR China. E-mail: twang-yfcom.
Corresponding authors: Zhe Ren, Biomedicine Research and Development Center, Jinan University, Guangzhou 510632, Guangdong, PR China. E-mail: rzl62000com; Yiliang Wang, Biomedicine Research and Development Center, Jinan University, Guangzhou 510632, Guangdong, PR China. E-mail: wang_yiliangcn; Yifei Wang, Biomedicine Research and Development Center, Jinan University, Guangzhou 510632, Guangdong, PR China. E-mail: twang-yfcom.