Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(12):2466-2479. doi:10.7150/ijms.54239 This issue Cite
Research Paper
Immune checkpoint molecules are regulated by transforming growth factor (TGF)-β1-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma
Ritu Shrestha1,2, Kim R. Bridle1,2, Darrell H. G. Crawford1,2, Aparna Jayachandran1,2,3 ![]()
1. Faculty of Medicine, The University of Queensland, Brisbane, Queensland, Australia.
2. Gallipoli Medical Research Institute, Greenslopes Private Hospital, Brisbane, Queensland, Australia.
3. Fiona Elsey Cancer Research Institute, Ballarat, Victoria, Australia.
Received 2020-10-8; Accepted 2021-3-30; Published 2021-4-22
Abstract

Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer with a high mortality rate. Epithelial-to-mesenchymal transition (EMT) confers cancer cells with immune evasive ability by modulating the expression of immune checkpoints in many cancers. Thus, the aim of our study is to examine the interplay between EMT and immune checkpoint molecules in HCC. A reversible EMT model was utilised with transforming growth factor (TGF)-β1 as an EMT inducer for HCC cell lines Hep3B and PLC/PRF/5. HCC cells were treated with TGF-β1 for 72 h and the EMT status and immune checkpoint expression were examined. In addition, the migratory ability of HCC cells were examined using wound healing and transwell migration assays in the reversible EMT model. siRNA-mediated knockdown of immune checkpoint molecule, B7-H3, was further utilised to validate the association between TGF-β1-mediated EMT and immune checkpoint expression in HCC. In addition, a web-based platform, SurvExpress, was utilised to evaluate the association between expression of TGF-β1 in combination with immune checkpoint molecules and overall survival in HCC patients. We observed induction of EMT upon treatment of HCC cells with TGF-β1 revealed by reduced expression of epithelial markers along with increased expression of mesenchymal markers. Withdrawal of TGF-β1 reversed the process of EMT with elevated expression of epithelial markers and reduced expression of mesenchymal markers. TGF-β1 treatment elevated the migratory potential of HCC cells which was reversed following reversal assay. Notably, during TGF-β1-induced EMT, there was upregulation of immune checkpoint molecules PD-L1 and B7-H3. However, the reversal of EMT decreased the expression of PD-L1 and B7-H3. In addition, TGF-β1 driven EMT was reversed following knockdown of B7-H3 in both HCC cells further validating the interplay between TGF-β1-mediated EMT and immune checkpoint expression in HCC. Furthermore, the coordinate expression of TGF-β1 with PD-L1 (p=0.01487) and B7-H3 (p=0.009687) was correlated with poor overall survival in 422 HCC patients. Our study has demonstrated a close association between TGF-β1-mediated EMT and regulation of immune checkpoints in HCC.
Keywords: immune checkpoint molecules, epithelial-to-mesenchymal transition, transforming growth factor-β1, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) constitutes the majority of primary liver cancer, which is one of the leading causes of cancer-related deaths and the sixth most common cancer based on incidence [1, 2]. HCC is a rapidly growing second most lethal tumour with rate of mortality almost similar to the rate of incidence [1-4]. HCC is frequently diagnosed in cirrhotic patients with higher risk in patients with underlying liver disease resulting mainly from hepatitis B or C virus infection [2].
Sorafenib and Lenvatinib are the Food and Drug Administration (FDA) approved first-line drugs for the treatment of advanced HCC. Regorafenib, Ramucirab and Cabozantinib are the second-line therapies for HCC patients previously treated with Sorafenib. In addition, immunotherapeutic approaches have been explored in recent years as an alternative second-line treatment modality in HCC. Cancer immunotherapy based on immune checkpoint inhibition has transformed oncology treatment in several cancers including HCC [5-7]. Immune checkpoint inhibitors (ICIs) blocking programmed cell death protein-1 (PD-1) such as Nivolumab and Pembrolizumab have been approved for treatment of HCC. Recently, the FDA has approved the combination of Nivolumab with another checkpoint inhibitor, Ipilimumab that targets cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), for treatment of patients with advanced HCC previously treated with Sorafenib [8]. In addition, the FDA has also approved the combination of Atezolizumab, ICI against programmed cell death protein ligand -1 (PD-L1), and Bevacizumab as first-line treatment for HCC patients based on the response rates in the IMbrave150 trial [9]. Studies have demonstrated that immune checkpoint molecules are often overexpressed in HCC patients with poor prognosis [10, 11]. However, the precise mechanism associated with regulation of immune checkpoint molecules in HCC is still unknown.
The process of epithelial-to-mesenchymal transition (EMT) is a key step in cancer metastasis and invasion that allows phenotypic conversion of cells to gain migratory and invasive features [12-14]. EMT is a major contributor to disease progression and drug resistance in HCC [13, 15]. In addition, the reverse process of mesenchymal-to-epithelial transition (MET) allows tumour recurrence post migration and invasion that is facilitated through the process of EMT [16, 17]. In recent years, EMT has been closely associated with expression and regulation of immune checkpoint molecules in several cancers including lung cancer [18-22], breast cancer [23-26], oesophageal cancer [27], gastric cancer [27-29], salivary adenoid cystic carcinoma [30], pancreatic cancer [31, 32] and head and neck cancer [33]. Previously, we reported an association between the process of EMT and the expression of several immune checkpoint molecules including PD-L1 in HCC [11, 34]. Notably, in HCC patients a close association between EMT and immune checkpoint molecules prognosticated a poor survival [11, 34].
We have previously shown that cytokine, tumour necrosis factor (TNF)-α, regulates expression of immune checkpoint molecules in HCC [34]. In this study, we have used another cytokine, transforming growth factor (TGF)-β1, which is a potent inducer of EMT in HCC cells [35-37]. Our study utilises a reversible EMT model to examine the association between TGF-β1-driven EMT and regulation of immune checkpoint molecules.
Materials and methods
Cell culture and reagents
Human HCC cell line PLC/PRF/5 was purchased from CellBank Australia (85061113) and Hep3B cells were sourced from Prof. V. Nathan Subramaniam, The Queensland University of Technology, Australia. Both cell lines were STR profiled and mycoplasma-tested using the MycoAlert test (Abm, Canada) and cultured as previously described [34]. The cytokine TGF-β1 was purchased from Peprotech, Australia.
EMT reversal assay
An in vitro reversible EMT model was utilised to evaluate the interaction between immune checkpoint molecule expression and EMT as previously described [34]. Briefly, the HCC cells were treated with TGF-β1 for three days and reversal was induced by growing the cells in a TGF-β1 free culture medium for another three days to reverse EMT effects or induce MET. Photographs were taken for cells under all three conditions (Control, EMT and MET) using an inverted microscope equipped with a digital camera (Olympus DP21, Japan).
Wound healing assay
Cells were seeded into a 24 well plate and allowed to reach confluency. A scratch or wound was made using a sterile pipette tip and washed with 1XPBS. 500 µl fresh culture medium was added into the wells and the photographs were taken at the indicated time points under an inverted microscope equipped with a digital camera (Olympus DP21, Japan). For better contrast, images have been enhanced by false colour background using Fiji plug-in for Image J software version 1.53c.
Transwell migration assay
Transwell chambers (8 µm pore size, Corning, Australia) were utilised for the transwell migration assay. 1X105 cells were seeded into the upper chamber in a serum free DMEM culture medium. 500 µl of DMEM culture medium with 10% FBS was added into the lower chamber as the chemoattractant. After 24 h incubation at 37 °C, cells were fixed with 4% paraformaldehyde (Fisher Scientific, Australia) for 15 min. The fixed cells were then stained with 0.1% Crystal Violet (Sigma-Aldrich). The cells on the upper chamber were removed and the migrated cells on the lower chamber were photographed under an inverted microscope equipped with a digital camera (Olympus DP21, Japan).
RNA extraction and cDNA synthesis
RNA extraction was performed with Isolate II Bioline RNA synthesis kit (Bioline, Australia) as per the manufacturer's protocol as described previously [38]. Nanodrop 2000c (Thermofisher, Australia) was utilised to confirm the purity and concentration of RNA. Bioline SensiFAST cDNA synthesis kit (Bioline, Australia) was used to reverse transcribe 1 µg RNA into cDNA.
Quantitative reverse transcription-PCR (qRT-PCR)
qRT-PCR was performed using Lo-ROX SYBR Green (Bioline, Australia) on a ViiA7 Applied Biosystems Real-Time PCR system as described previously [38]. Data was analysed using the 2ΔΔCt method where ActB was assigned as the housekeeping gene. In this 2ΔΔCt method, target gene expression was normalized to ActB expression and data are presented as copies of target gene per 10,000 copies of ActB. The sequence for the primers used for qRT-PCR is detailed in previous study [34]. The primer sequence not mentioned is listed as follows: Snai1 forward: GCTGCAGGACTCTAATCCAGA; Snai1 reverse: ATCTCCGGAGGTGGGATG.
Protein isolation and western blot analysis
The protein isolation and western blot analysis were performed as previously described [34]. Briefly, cells cultured and treated in 6 well plates were lysed using RIPA buffer (Thermofisher, Australia) with Complete (Roche, Australia) and phosSTOP (Roche, Australia) protease and phosphatase inhibitors at 4oC to isolate the total protein. A Pierce BCA protein assay kit (Thermofisher, Australia) was used to measure the total protein extracted. 10 µg of protein was used for separation by electrophoresis (SDS-PAGE) in a polyacrylamide gel in the presence of sodium dodecyl sulphate (SDS) and transferred to a polyvinylidene difluoride film (PVDF) membrane. After blocking with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T), the membranes were incubated with primary antibodies at 4 °C overnight. The protein was detected using an enhanced chemiluminescence reagent, SuperSignal West Femto Maximum Sensitivity Substrate (Thermofisher, Australia) following incubation with HRP-conjugated secondary antibodies. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the housekeeping control. Quant LAS 500 was used to capture images and quantified with Image Studio Lite Ver 5.2 software. Antibodies used are listed in previous study [34] and β-Actin (Cat. No. 4967s, Cell Signaling) at dilution of 1:2000.
Immunofluorescence
Immunofluorescence staining was performed as previously described [34]. Briefly, the cells were cultured in 8 well tissue culture treated chamber slides. The cells were then washed with PBS, fixed with 4% paraformaldehyde (Fisher scientific, Australia) for 15 min and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, Australia). 5% FBS was used as blocking buffer and then the cells were incubated overnight with primary antibodies at 4 °C. The cells were then washed with PBS, incubated with secondary antibodies at room temperature followed by 10 min incubation with 4',6-diamidino-2-phenylindole (DAPI) (Thermofisher Scientific, Australia). The slides were then mounted in ProLong Diamond (Thermofisher Scientific, Australia) for observation with Nikon C2 system and captured and analysed with NIS-Elements software (Nikon, Australia). Antibodies used are listed in previous study [34].
B7-H3 knockdown
Cells were transfected at 50% confluency for transient siRNA transfection using a control siRNA (4390843) (Thermofisher, Australia), GAPDH siRNA (4390849) and two different silencer select siRNAs targeting B7-H3 (s37290 and s37288) (Thermofisher, Australia) at 10 nM final concentration with Lipofectamine RNAiMAX (Invitrogen, Australia) according to manufacturer's protocol. Cells were incubated with siRNA complex for 72 hours and then collected for further experiments.
SurvExpress bioinformatics tool
HCC patient datasets were subjected to survival analyses with SurvExpress tool, as previously described [11]. Briefly, the HCC patient database, TCGA-Liver-Cancer with 422 patients was analysed [11, 39-41]. Using cox regression analyses, we evaluated the coordinate gene expression of TGF-β1 and immune checkpoint molecules to explore their association with the survival of HCC patients [11]. The survival times was assessed with Kaplan-Meier curves [11].
Statistical analysis
Experimental data are presented as mean with standard error of mean (SEM). Prism software version 8.00 (GraphPad Software Inc) was used for the statistical analyses. One-way analysis of variance (ANOVA) followed by Dunnet's multiple comparisons test was used for analysis of dose concentration and time course experiments. Comparisons of were performed using In addition, ANOVA followed by Sidak's multiple comparisons test was used for comparisons of TGF-β1-induced EMT and MET following reversal assay. Gene expression differences between control and TGF-β1 treated cells were analysed using Student's t-test. Statistical significance was set at P<0.05. Error bars indicate standard error of the mean (SEM).
A log-rank test was used for testing the P value of survival curves for analysis of survival with SurvExpress. Deviance residuals were applied for the correlation coefficient [11, 42]. Cox model was applied to estimate the hazard ratio (HR) between the groups [11].
Results
Human HCC cells undergo EMT with TGF-β1 treatment
To examine the induction of EMT by TGF-β1, Hep3B and PLC/PRF/5 cells were treated with various doses of TGF-β1 at different time points. The induction of EMT was evaluated by decreased expression of epithelial markers (E-cadherin and Occludin) and increased expression of mesenchymal markers (N-cadherin, Vimentin, Snai1 and Fibronectin). We observed an optimal induction of EMT in Hep3B cells at the concentration of 10 ng/ml of TGF-β1 (Supplementary Fig. S1A) and thus we utilised 10 ng/ml of TGF-β1 for EMT induction in Hep3B cells throughout this study. In addition, we observed robust EMT changes at 72 h post treatment of Hep3B cells with 10 ng/ml of TGF-β1 with the maximum decrease in the expression of epithelial markers E-cadherin and Occludin and the maximum increase in the expression of majority of mesenchymal markers N-cadherin, Vimentin and Snai1 (Supplementary Fig. S1B) and selected the time point of 72 h for TGF-β1 treatment as optimum for this study. Similarly, we also demonstrated that TGF-β1 induces optimal EMT in PLC/PRF/5 cells treated for 72 h with a concentration of 10 ng/ml of TGF-β1 (Supplementary Fig. S2A). In addition, time course experiment reported optimal EMT induction with 10 ng/ml of TGF-β1 treatment in PLC/PRF/5 cells for 72 h (Supplementary Fig. S2B). The induction of EMT in both Hep3B and PLC/PRF/5 cells treated with TGF-β1 was further validated by western blot analysis (Supplementary Fig. S3A and B). Both cell lines demonstrated an upregulation of epithelial markers and downregulation of mesenchymal markers at protein level consistent with EMT changes.
TGF-β1 induces upregulation of immune checkpoint molecules
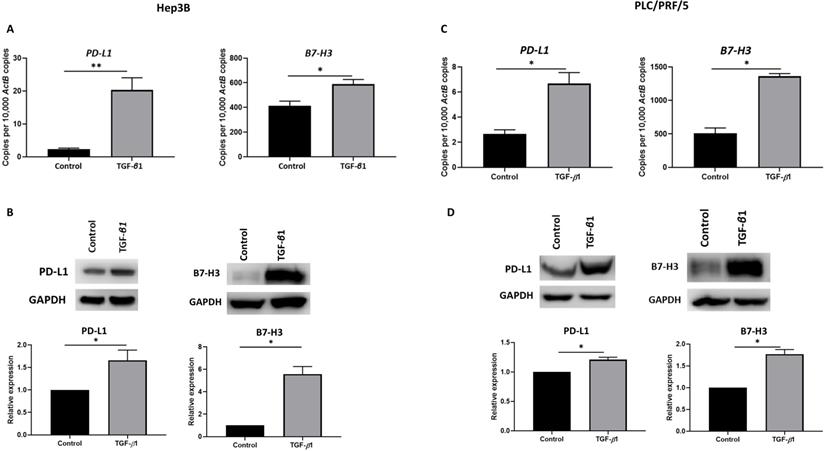
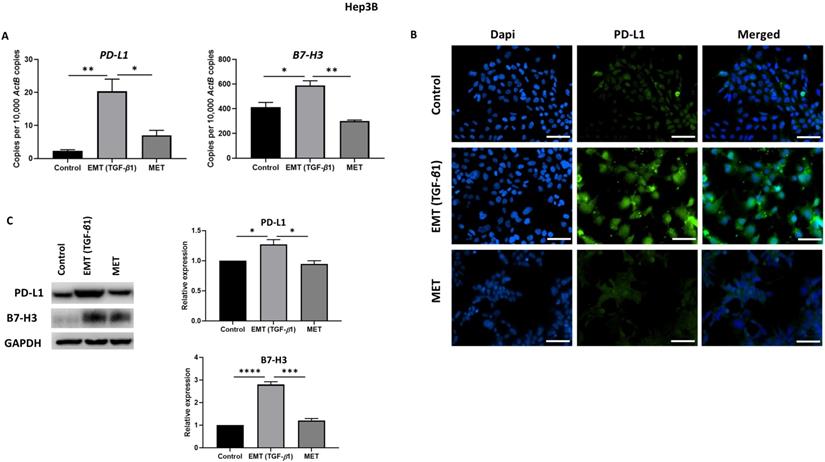
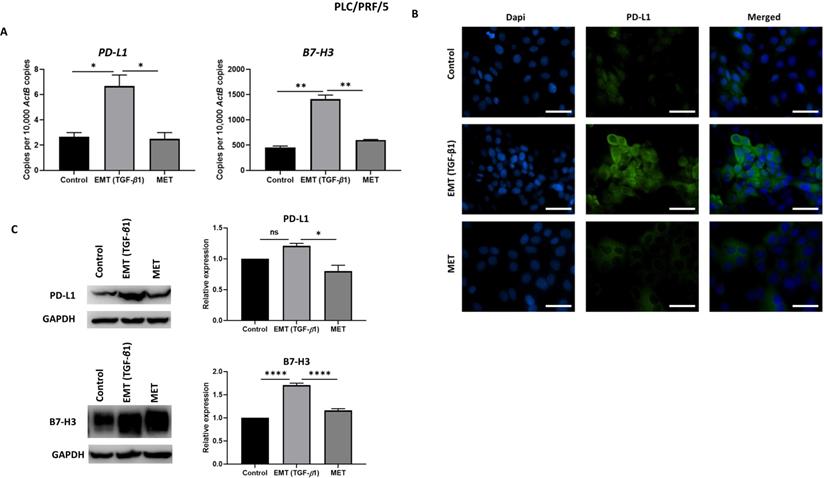
Previously, we have shown that immune checkpoint modulators, PD-L1 and B7-H3 prognosticates a poor outcome in HCC patients [11]. To determine the effect of TGF-β1-mediated EMT on expression of these immune checkpoint molecules, we treated Hep3B and PLC/PRF/5 cells with 10 ng/ml of TGF-β1 for 72 h. We observed the upregulation of these immune checkpoint molecules in both Hep3B (Fig. 1A-B) and PLC/PRF/5 (Fig. 1C-D) cells as demonstrated by qRT-PCR and western blot analysis.
EMT induced by TGF-β1 modulates expression of immune checkpoint molecules in HCC cells. (A) qRT-PCR revealed upregulation of PD-L1 and B7-H3 upon treatment with 10 ng/ml of TGF-β1 for 72 h in Hep3B cells. (B) Western blot analysis revealed upregulated expression of PD-L1 and B7-H3 upon treatment with 10 ng/ml of TGF-β1 for 72 h in Hep3B cells. (C) qRT-PCR revealed upregulation of PD-L1 and B7-H3 upon treatment with 10 ng/ml of TGF-β1 for 72 h in PLC/PRF/5 cells. (D) Western blot analysis revealed upregulated expression of PD-L1 and B7-H3 upon treatment with10 ng/ml of TGF-β1 for 72 h in PLC/PRF/5 cells. (n=3, *p<0.05, **p<0.01). GAPDH was used as loading control.
Abrogation of migratory ability of HCC cells upon reversal of TGF-β1-induced EMT
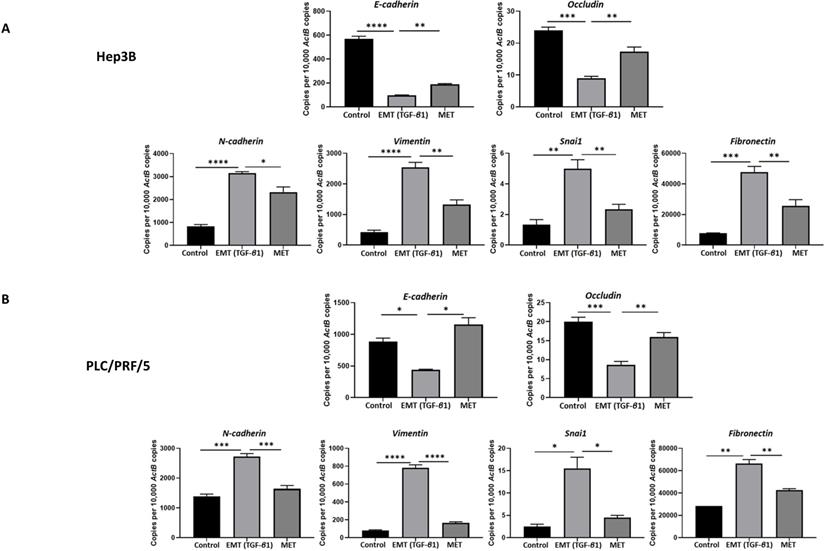
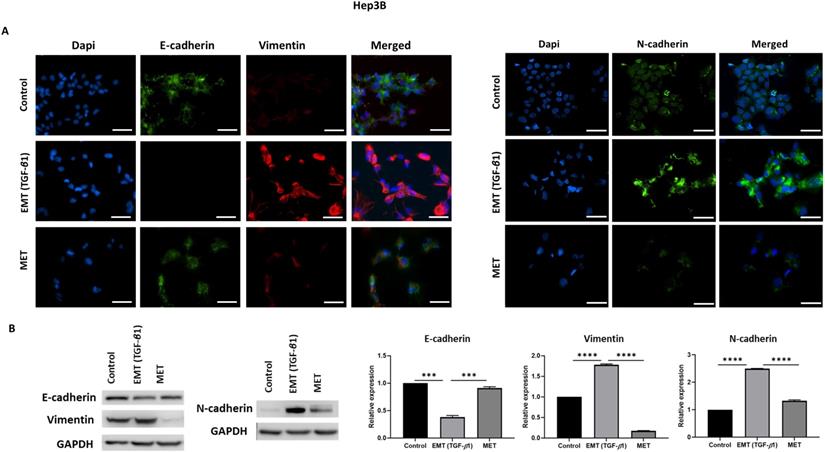
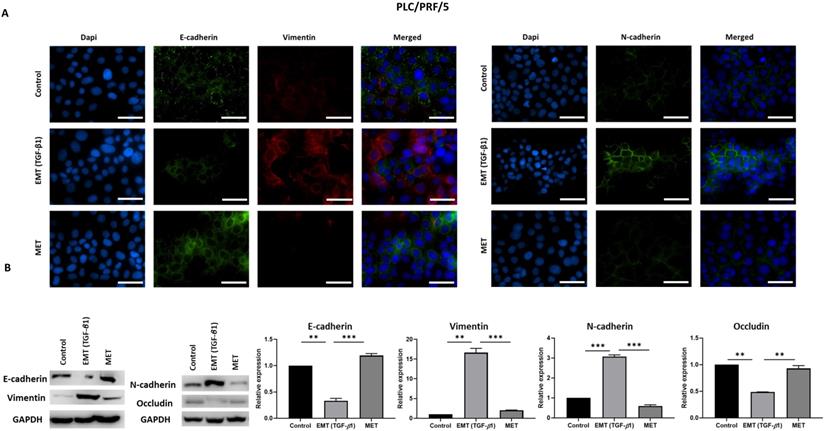
We utilised an in vitro EMT reversal model to induce EMT in a reversible manner in HCC cells. The qRT-PCR results demonstrated the occurrence of EMT post TGF-β1 treatment consistent with the reduced expression of epithelial markers (E-cadherin and Occludin) and concomitant increased expression of mesenchymal markers (N-cadherin, Vimentin, Snai1 and Fibronectin). The removal of TGF-β1 induced the cells to undergo an MET consistent with upregulation of epithelial markers (E-cadherin and Occludin) and downregulation of mesenchymal markers (N-cadherin, Vimentin, Snai1 and Fibronectin) in Hep3B (Fig. 2A) and PLC/PRF/5 (Fig. 2B) cells. The reversible process of EMT and MET was further confirmed by immunofluorescence and western blot analysis in Hep3B (Fig. 3A-B) and PLC/PRF/5 (Fig. 4A-B) cells.
EMT mediated by TGF-β1 in HCC cells is reversible. qRT-PCR revealed upregulation of E-cadherin and Occludin and downregulation of N-cadherin, Vimentin, Snai1 and Fibronectin following EMT reversal assay in (A) Hep3B and (B) PLC/PRF/5 cells (n=3, *p<0.05, **p<0.01, ***p<0.005, ****p<0.001).
TGF-β1 induces reversible EMT in Hep3B cells. (A) Fluorescence microscopy demonstrated lower expression of E-cadherin and higher expression of Vimentin and N-cadherin during EMT whereas higher expression of E-cadherin and lower expression of Vimentin and N-cadherin was observed following MET (scale bar = 200 µm, magnification 20X). (B) Western blot analysis revealed decreased expression of E-cadherin and increased expression of Vimentin and N-cadherin during EMT and upregulation of E-cadherin and downregulation of Vimentin and N-cadherin during reversal or MET (n=3, ***p<0.005, ****p<0.001). GAPDH was used as loading control.
TGF-β1-mediated EMT is reversible in PLC/PRF/5 cells. (A) Fluorescence microscopy revealed decreased expression of E-cadherin and increased expression of Vimentin and N-cadherin during EMT whereas increased expression of E-cadherin and decreased expression of Vimentin and N-cadherin was observed following MET (scale bar = 200 µm, magnification 20X). (B) Western blot analysis revealed downregulation of E-cadherin and Occludin and upregulation of Vimentin and N-cadherin during EMT and upregulation of E-cadherin and downregulation of Vimentin and N-cadherin during MET. (n=3, **p<0.01, ***p<0.005). GAPDH was used as loading control.
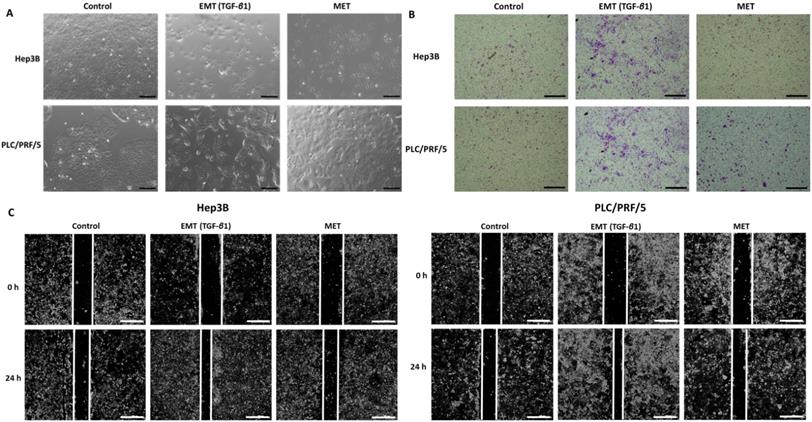
As the process of EMT is involved in tumour metastasis, the migratory ability of HCC cells were compared upon treatment with TGF-β1 and reversal assay using wound healing and transwell migration assay along with observation of morphology changes. We observed morphological changes in both Hep3B and PLC/PRF/5 during the process of EMT and MET (Fig. 5A). During EMT, cells appear to be fibroblast-like and elongated and during MET, they display a cobblestone morphology similar to the untreated control cells. Furthermore, we observed that TGF-β1-induced EMT enhances the migratory ability of Hep3B and PLC/PRF/5 cells whereas the reversal assay reduces the motility both Hep3B and PLC/PRF/5 cells as demonstrated by transwell migration assay (Fig. 5B) and wound healing assay (Fig. 5C). The EMT marker changes together with the functional changes observed in HCC cells in the reversible EMT model makes this assay a robust in vitro tool to investigate the changes associated with the EMT status.
TGF-β1-mediated EMT modulates migratory ability of HCC cells. (A) Morphology changes in Hep3B and PLC/PRF/5 cells during EMT and MET (scale bar = 500 µm, magnification 10X). (B) Transwell migration assay revealed increased motility of Hep3B and PLC/PRF/5 cells upon TGF-β1-induced EMT and decreased motility following MET (scale bar = 500 µm, magnification 10X). (C) The wound healing assay demonstrated that the migratory ability is increased upon EMT and decreased following MET in Hep3B and PLC/PRF/5 cells (scale bar = 500 µm, magnification 10X).
Reversal of EMT reverses expression of immune checkpoint molecules
To examine whether the expression of immune checkpoint molecules were altered with EMT status, we utilised the reversible EMT assay. We observed downregulation of PD-L1 and B7-H3 following EMT reversal assay as demonstrated by qRT-PCR, immunofluorescence staining and western blot analysis in Hep3B (Fig. 6A-C) and PLC/PRF/5 (Fig. 7A-C) cells. The changes in EMT status affects the expression of immune checkpoint molecules suggesting a role for TGF-β1 in the regulation of immune checkpoint molecules PD-L1 and B7-H3.
Expression of immune checkpoint molecules is reversed following reversal of EMT in Hep3B cells. (A) qRT-PCR, (B) fluorescence microscopy (scale bar = 200 µm, magnification 20X) and (C) western blot analysis demonstrated upregulation of PD-L1 and B7-H3 upon induction of EMT by TGF-β1 and downregulation of PD-L1 and B7-H3 upon reversal of MET. (n=3, * p<0.05, **p<0.01, ***p<0.005, ****p<0.001). GAPDH was used as loading control.
Expression of immune checkpoint molecules is reversed following reversal of EMT in PLC/PRF/5 cells. (A) qRT-PCR, (B) fluorescence microscopy (scale bar = 200 µm, magnification 20X) and (C) western blot analysis demonstrated upregulation of PD-L1 and B7-H3 upon induction of EMT by TGF-β1 and downregulation of PD-L1 and B7-H3 upon reversal of MET. (n=3, *p<0.05, **p<0.01, ****p<0.001). GAPDH was used as loading control.
Knockdown of B7-H3 can reverse TGF-β1-induced EMT in HCC cells
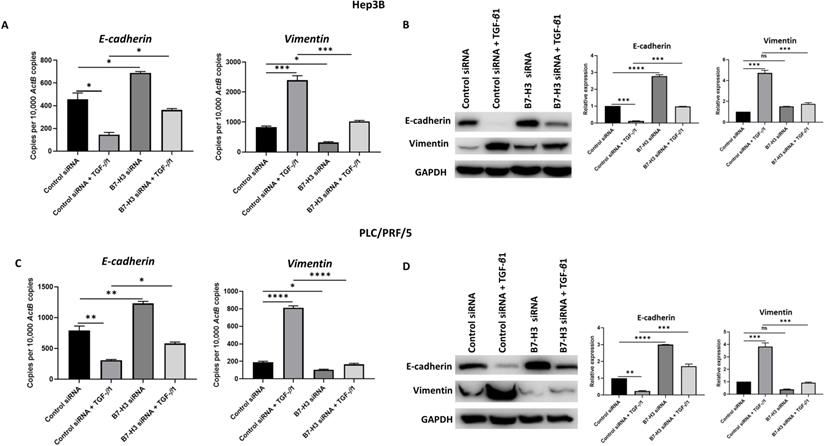
To further validate the association between TGF-β1-induced EMT and expression of immune checkpoint molecules such as B7-H3, we utilised two different siRNAs to silence the expression of B7-H3. The efficacy of B7-H3 knockdown was confirmed by qRT-PCR and western blot analysis in both Hep3B (Supplementary Fig. S4A-B) and PLC/PRF/5 (Supplementary Fig. S4C-D) cells. Next, we examined the effects of siRNA-mediated silencing of B7-H3 on expression of EMT markers to validate the aforementioned association between EMT and immune checkpoint expression. Interestingly, we observed reversal of TGF-β1-mediated EMT following B7-H3 knockdown evident by upregulation of epithelial marker E-cadherin and downregulation of mesenchymal marker Vimentin as revealed by qRT-PCR and western blot analysis in both Hep3B (Fig. 8A-B) and PLC/PRF/5 (Fig. 8C-D) cells.
Knockdown of B7-H3 can reverse TGF-β1-induced EMT in HCC cells. siRNA-mediated silencing of B7-H3 expression in Hep3B cells resulted in reversal of TGF-β1-induced EMT as demonstrated by (A) qRT-PCR and (B) western blot analysis. Knockdown of B7-H3 expression in PLC/PRF/5 cells caused reversal of TGF-β1-induced EMT as demonstrated by (C) qRT-PCR and (D) western blot analysis. GAPDH was used as loading control. (n=3, *p<0.05, **p<0.01, ***p<0.005, ****p<0.001).
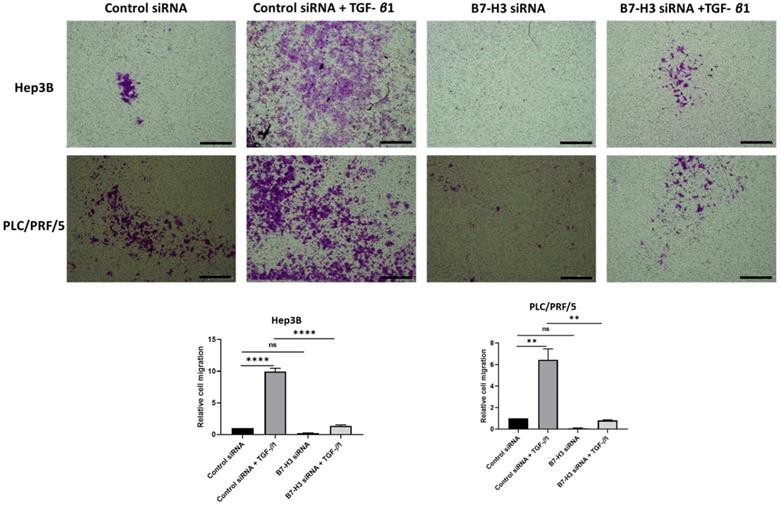
This relationship between TGF-β1-mediated EMT and expression of immune checkpoint molecules was further validated at functional level with transwell migration. We observed reduced migration of cells following siRNA-mediated knockdown of B7-H3 (Fig. 9). This further justified our hypothesis on the significant association between TGF-β1-mediated EMT and immune checkpoint molecule expression in HCC.
Silencing of B7-H3 reduces motility of HCC cells. Transwell migration revealed that number of cells migrating reduced significantly following B7-H3 knockdown in both Hep3B and PLC/PRF/5 cells (scale bar = 500 µm, magnification 10X) (n=3, **p<0.01, ****p<0.001).
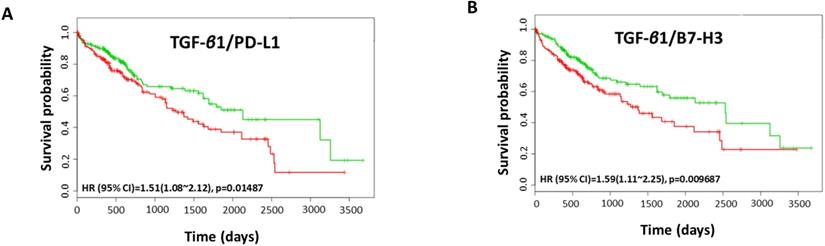
Coordinate expression of TGF-β1 and immune checkpoint molecules in HCC patients
We utilised TCGA-Liver-Cancer HCC database with 422 HCC patients within Survexpress to assess the association between coordinate expression of TGF-β1 and immune checkpoint molecules with overall survival in HCC patients. We observed that the coordinate expression of TGF-β1 in combination with PD-L1 resulted in a significantly poorer overall survival in 422 patients (HR: 1.51, CI: 1.08~2.12, Log-Rank Equal Curves p=0.01487) (Fig. 10A). Similarly, the combination of TGF-β1 with B7-H3 showed significant poorer overall survival (HR: 1.59, CI: 1.11~2.25, Log-Rank Equal Curves p=0.009687) (Fig. 10B). This data suggests that a significant correlation of TGF-β1 with PD-L1 and B7-H3 in HCC patients prognosticates a poor outcome.
Coordinate expression of TGF-β1 and immune checkpoint molecules in HCC patients. Kaplan-Meier survival curves demonstrating the (A) gene expression of TGF-β1/PD-L1 and overall survival, and (B) gene expression of TGF-β1/B7-H3 and overall survival in HCC patient samples. Red curve represents high-risk group, while green curve represents low-risk group [11]. Markers (+) represent censoring samples. X-axis represents the study time in days. Y-axis represents survival probability.
Discussion
In this study, we explored the relationship between TGF-β1-induced EMT and expression of immune checkpoint molecules in human HCC cells based on a reversible model of EMT. We observed induction of EMT and upregulation of PD-L1 and B7-H3 in both Hep3B and PLC/PRF/5 cells along with enhanced motility upon treatment with TGF-β1. The close association of EMT and immune checkpoints in HCC was further confirmed by reversibility of the expression of immune checkpoint molecules during EMT and MET. In addition, we also observed that HCC patients with coordinate expression of TGF-β1 and immune checkpoint molecules PD-L1 or B7-H3 had poor overall survival.
TGF-β signalling plays a key role in the process of EMT and carcinogenesis in several cancers [43, 44]. In hepatocarcinogenesis, TGF-β signalling has been implicated in all the stages of disease progression, from inflammation to cirrhosis to carcinoma [45, 46]. TGF-β is a widely studied EMT inducer that regulates EMT-transcription factors (EMT-TFs) [46, 47]. Several in vitro studies have utilised TGF-β for inducing EMT in HCC [37, 48-50]. In addition, studies have demonstrated reversible induction of EMT with cytokine TNF-α [34] or combination of TGF-β1 and TNF-α [18]. The present study utilised TGF-β1 as a reversible EMT inducer in human HCC cells, Hep3B and PLC/PRF/5. Our study is the first to explore the association of TGF-β1-induced EMT and expression of immune checkpoint molecules in HCC.
Immunotherapeutic approaches based on ICIs have revolutionized the cancer treatment scenario by modulating the patient's immune system to combat the tumour [51, 52]. Several ICI drugs targeting CTLA-4 or PD-1/PD-L1 interaction have been approved by the FDA for various cancer types including melanoma, lung cancer, renal cell carcinoma, breast cancer, head and neck cancer, colorectal cancer, urothelial cancer and HCC [18-33]. Despite the approval of few ICIs and several ongoing clinical trials, ICIs as a monotherapy have failed to show promising result as first-line treatment in HCC compared to Sorafenib [53, 54]. In addition, the response of HCC patients to ICIs may be affected by several molecular features including genetic alterations, variation in inflammatory infiltrates, microsatellite instability (MSI) and expression of alternative immune checkpoint molecules [53, 55, 56]. Thus, there is a need for identification of robust predictive biomarkers along with combined ICI therapeutic approaches for enhanced clinical outcome in HCC patients [53].
Tissue-agnostic biomarkers such as tumour mutation burden (TMB) and MSI have been examined for cancer immunotherapy [57-59]. TMB has been utilised as a predictive biomarker in cancer immunotherapy in several studies [11, 60-62]. Our previous study demonstrated significant association of TMB with poor clinical outcomes in HCC patients [11]. Recently, FDA has approved Pembrolizumab, an anti-PD-1 ICI, for the treatment of tumour mutational burden-high (TMB-H) cancers based on KEYNOTE-158 study [63]. This approval of Pembrolizumab for TMB-H cancers is subsequent to previous FDA approval of the drug in MSI-high (MSI-H) or mismatch repair deficient (dMMR) cancers [64]. Thus, genetic and immunogenic features of tumour should be considered with use of tissue-agnostic biomarkers before the selection of ICIs for cancer immunotherapy.
The HCC TME includes cancer cells, immune cells [T-cells, B-cells, dendritic cells, myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs)], carcinoma-associated fibroblasts, endothelial cells, hepatic stellate cells, and the extracellular matrix (ECM) [65]. As the TME is a crucial facilitator of HCC progression and resistance to immunotherapies, much of the recent research is focused on the various components of the TME to identify predictive and prognostic biomarkers for ICI treatment in HCC patients [56, 66]. For instance, a study developed a prognostic biomarker for immunotherapy comprising of eight gene risk signature based on hypoxia status in the TME of HCC patient [56]. Another study reported CD38 expression within the TME including tumour and certain immune subsets, such as macrophages as a predictive marker of responsiveness to anti-PD-1/PD-L1 single agent treatment in HCC patients [66]. HCC TME is rich source of soluble factors and cytokines that can regulate the expression of immune checkpoints and can be utilised as prognostic biomarkers [67]. Thus, further investigation of potential biomarkers that predict the efficacy of ICI treatment in HCC patients is of the utmost importance. Furthermore, it is important to examine the mechanism underlying the poor response and therapy resistance to ICI monotherapies to improve therapeutic efficacy in cancer therapy.The process of EMT is known to influence immune evasion of tumour cells contributing to immunosuppression through regulation of immune checkpoint molecules [52, 68, 69]. EMT is closely associated with immune checkpoint-dependent immunosuppression resulting in aggressive and drug resistant tumours [52, 70-72]. As previously mentioned, EMT is known to regulate immune checkpoint molecules, in particular PD-L1, in several cancers. In recent years, studies have explored several aspects of EMT process responsible for regulation of immune checkpoint molecules, tumour microenvironment and prognosis [33, 70, 73, 74].
This study reported that TGF-β1-mediated EMT promotes migratory ability and enhances expression of PD-L1 and B7-H3 in human HCC cells, Hep3B and PLC/PRF/5. In HCC, we have previously reported a significant association of PD-L1 expression with EMT phenotype characterised by lower expression of epithelial marker E-cadherin and higher expression of mesenchymal marker Vimentin in 422 patients [11]. Other studies have reported a close association between EMT and PD-L1 in various cancers. A study by Asgarova et al reported that cytokines TNF-α and TGF-β1 regulates PD-L1 expression in non-small cell lung carcinoma [18]. A study in gastric carcinoma demonstrated that TGF-β1 promotes motility and PD-L1 expression via NF-κB activation [28]. Similarly, another study revealed that TGF-β1-mediated EMT enhances PD-L1 expression in head and neck squamous cell carcinoma [33]. Furthermore, a study in pancreatic carcinoma revealed that TGF-β upregulates the expression of PD-L1 via both Smad 2/3-dependent and independent pathways along with increased migration and invasion in vitro [32]. The study also reported that PD-L1 expression induces Snail transcription factors which results in TGF-β-mediated EMT [32]. Likewise, another interesting study by David et al examined PD-L1 expression through TGF-β-induced EMT by utilizing M7824, a bifunctional fusion protein, inhibiting TGF-β and PD-L1 [19]. Besides EMT associated with TGF-β, the regulation of immune checkpoints are associated with some inflammatory cytokines such as interferon-γ (IFN-γ), TNF-α and interleukin-17 (IL-17) in various cancer types [31, 75]. Studies have reported other mechanisms of EMT that regulates PD-L1 expression such as EGF-induced EGFR activation [30]; ZEB1 and miR-200-mediated EMT [26, 70], Ataxia Telangiectasia Mutated (ATM) through JAK/STAT3 signalling activation [22].
In addition to PD-L1, we have reported TGF-β1-induced EMT mediated upregulation of another immune checkpoint molecule, B7-H3 in HCC cells [34]. We have previously reported upregulation of B7-H3 expression via TNF-α-mediated EMT in HCC cells [34]. B7-H3 is often overexpressed in cancer types characterised by increased proliferation and invasive potential such as liver, bladder, esophageal, breast, cervical, glioma, colorectal and gastric cancer [76]. A study in HCC revealed that B7-H3 promotes metastasis and invasion of HCC cells by undergoing EMT via JAK2/STAT2/Slug signalling pathway and elevated B7-H3 expression is correlated with poor clinical outcomes in HCC patients [77]. B7-H3 has also been reported to induce EMT and cancer stemness in colorectal cancer [78], induce cell invasion and sphere formation in glioma cells [79] and regulate cancer-initiating cells in ovarian cancer [80].
EMT is known to be significantly associated with overexpression of multiple immune checkpoint molecules resulting in immunosuppression [21]. A study demonstrated close link between EMT score and expression of panel of immune checkpoint molecules including PD-1, PD-L1, B7-H3, PD-L2 and others [69, 81]. A study by Lou et al reported strong association between EMT and upregulation of multiple immune checkpoint molecules including PD-1, CTLA-4, PD-L1, B7-H3, PD-L2, BTLA, and TIM-3 in lung adenocarcinoma [82]. Another study in head and neck squamous cell carcinoma also suggested that an EMT gene signature with a total of 82 genes was closely associated with prognosis and expression of immune checkpoint molecules [33]. Similarly Thompson et al suggested that gene signatures reflecting immune infiltration and EMT may be crucial in predicting patient's response to immunotherapy in lung cancer [83].
A few other studies have explored the regulation of immune checkpoint molecules in HCC by cytokines. These studies did not evaluate the EMT status and the role of EMT in modulating immune checkpoint molecules. One of the studies demonstrated that combined blocking of TGF-β and PD-L1 improved the immune response against tumour [84]. It has been reported that TNF-α and IFN-γ enhance the expression of PD-L1 synergistically in HCC cells [85].
Failure of immune checkpoint therapy also results from expression of alternative immune checkpoint molecules such as TIM-3, LAG-3, IDO-1 and VISTA [55]. Brown et al have reported resistance to ICIs (anti-PD-1 and anti-CTLA-4) through upregulation of alternative immune checkpoint IDO-1 in HCC patients [86]. Inhibition of IDO-1 resulted in enhancing the therapeutic efficacy of ICIs suggesting combination approach in HCC [86].
Our study provides valuable insight into regulation of immune checkpoint molecules PD-L1 and B7-H3 through TGF-β1-induced EMT that may result in an aggressive tumour phenotype and therapy resistance. Furthermore, HCC patients showing coordinate expression of TGF-β1 with PD-L1 or B7-H3 have poor overall survival. Hence, it is conceivable that by using combination approach by targeting TGF-β1-induced EMT along with ICIs such as PD-L1 and/or B7-H3 may lead to better clinical outcome in HCC patients. Moreover, this study suggests that EMT markers may be useful biomarkers to predict patient response in ICI immunotherapy as EMT status is closely associated with the expression of immune checkpoint molecules. However, further studies to determine molecular mechanisms involved in regulation of immune checkpoints and its association with EMT are warranted.
Abbreviations
HCC: hepatocellular carcinoma; EMT: epithelial-to-mesenchymal transition; TGF: transforming growth factor; PD-L1: programmed cell death protein ligand-1; FDA: food and drug administration; ICI: immune checkpoint inhibitor; PD-1: programmed cell death protein-1; SDS: sodium dodecyl sulphate; PVDF: polyvinylidene difluoride film; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; DAPI: 4',6-diamidino-2-phenylindole; SEM: standard error of mean; TMB: tumour mutation burden; TME: tumour microenvironment; MSI: microsatellite instability; TMB-H: tumour mutation burden high; MSI-H: microsatellite instability high; dMMR: mismatch repair deficient.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Gallipoli Medical Research Foundation for providing the necessary facilities and support for this study. We acknowledge SurvExpress for providing a platform for biomarker evaluation. We also acknowledge the TCGA Research Network for generating TCGA datasets.
Funding
We acknowledge Gallipoli Medical Research Foundation for funding this study.
Availability of data and materials
The datasets used and/or analysed during the current study are available on reasonable request from the corresponding author.
Author's contributions
Data acquisition, analysis and interpretation were performed by RS and AJ. RS and AJ wrote the manuscript. KB and DC critically revised the manuscript. RS and AJ designed the study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J Hepatol. 2020;72:250-61
2. Villanueva A. Hepatocellular Carcinoma. N Engl J Med. 2019;380:1450-62
3. Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C. et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017;3:1683-91
4. Dal Bo M, De Mattia E, Baboci L, Mezzalira S, Cecchin E, Assaraf YG. et al. New insights into the pharmacological, immunological, and CAR-T-cell approaches in the treatment of hepatocellular carcinoma. Drug Resist Updat. 2020;51:100702
5. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E. et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123-35
6. Hato T, Goyal L, Greten TF, Duda DG, Zhu AX. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology. 2014;60:1776-82
7. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B. et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84
8. Yau T, Kang YK, Kim TY, El-Khoueiry AB, Santoro A, Sangro B. et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020
9. Cheng AL, Qin S, Ikeda M, Galle P, Ducreux M, Zhu A. et al. IMbrave150: Efficacy and safety results from a ph III study evaluating atezolizumab (atezo) 1 bevacizumab (bev) vs sorafenib (Sor) as first treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). Annals of Oncology. 2019 30
10. Li Z, Li N, Li F, Zhou Z, Sang J, Chen Y. et al. Immune checkpoint proteins PD-1 and TIM-3 are both highly expressed in liver tissues and correlate with their gene polymorphisms in patients with HBV-related hepatocellular carcinoma. Medicine (Baltimore). 2016;95:e5749
11. Shrestha R, Prithviraj P, Anaka M, Bridle KR, Crawford DHG, Dhungel B. et al. Monitoring Immune Checkpoint Regulators as Predictive Biomarkers in Hepatocellular Carcinoma. Front Oncol. 2018;8:269
12. Jayachandran A, Dhungel B, Steel JC. Epithelial-to-mesenchymal plasticity of cancer stem cells: therapeutic targets in hepatocellular carcinoma. J Hematol Oncol. 2016;9:74
13. van Zijl F, Zulehner G, Petz M, Schneller D, Kornauth C, Hau M. et al. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5:1169-79
14. Yan L, Xu F, Dai CL. Relationship between epithelial-to-mesenchymal transition and the inflammatory microenvironment of hepatocellular carcinoma. J Exp Clin Cancer Res. 2018;37:203
15. Mir N, Jayachandran A, Dhungel B, Shrestha R, Steel JC. Epithelial-to-Mesenchymal Transition: A Mediator of Sorafenib Resistance in Advanced Hepatocellular Carcinoma. Curr Cancer Drug Targets. 2017;17:698-706
16. Chaffer CL, Thompson EW, Williams ED. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs. 2007;185:7-19
17. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED. et al. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374-83
18. Asgarova A, Asgarov K, Godet Y, Peixoto P, Nadaradjane A, Boyer-Guittaut M. et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology. 2018;7:e1423170
19. David JM, Dominguez C, Palena C. Pharmacological and immunological targeting of tumor mesenchymalization. Pharmacol Ther. 2017;170:212-25
20. Li F, Zhu T, Yue Y, Zhu X, Wang J, Liang L. Preliminary mechanisms of regulating PD-L1 expression in non-small cell lung cancer during the EMT process. Oncol Rep. 2018;40:775-82
21. Manjunath Y, Upparahalli SV, Avella DM, Deroche CB, Kimchi ET, Staveley-O'Carroll KF. et al. PD-L1 Expression with Epithelial Mesenchymal Transition of Circulating Tumor Cells Is Associated with Poor Survival in Curatively Resected Non-Small Cell Lung Cancer. Cancers (Basel). 2019 11
22. Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding Q. et al. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 2019;38:149
23. Alsuliman A, Colak D, Al-Harazi O, Fitwi H, Tulbah A, Al-Tweigeri T. et al. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: significance in claudin-low breast cancer cells. Mol Cancer. 2015;14:149
24. Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL. et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017;77:3982-9
25. Kumar S, Davra V, Obr AE, Geng K, Wood TL, De Lorenzo MS. et al. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. Oncoimmunology. 2017;7:e1376155
26. Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ. et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology. 2017;6:e1263412
27. Chen L, Xiong Y, Li J, Zheng X, Zhou Q, Turner A. et al. PD-L1 Expression Promotes Epithelial to Mesenchymal Transition in Human Esophageal Cancer. Cell Physiol Biochem. 2017;42:2267-80
28. Xu D, Li J, Li RY, Lan T, Xiao C, Gong P. PD-L1 Expression Is Regulated By NF-κB During EMT Signaling In Gastric Carcinoma. Onco Targets Ther. 2019;12:10099-105
29. Xu Z, Gu C, Yao X, Guo W, Wang H, Lin T. et al. CD73 promotes tumor metastasis by modulating RICS/RhoA signaling and EMT in gastric cancer. Cell Death Dis. 2020;11:202
30. Wang Y, Hu J, Ye W, Zhang X, Ju H, Xu D. et al. EGFR activation induced Snail-dependent EMT and myc-dependent PD-L1 in human salivary adenoid cystic carcinoma cells. Cell Cycle. 2018;17:1457-70
31. Imai D, Yoshizumi T, Okano S, Itoh S, Ikegami T, Harada N. et al. IFN-γ Promotes Epithelial-Mesenchymal Transition and the Expression of PD-L1 in Pancreatic Cancer. J Surg Res. 2019;240:115-23
32. Kang JH, Jung MY, Leof EB. B7-1 drives TGF-β stimulated pancreatic carcinoma cell migration and expression of EMT target genes. PLoS One. 2019;14:e0222083
33. Jung AR, Jung CH, Noh JK, Lee YC, Eun YG. Epithelial-mesenchymal transition gene signature is associated with prognosis and tumor microenvironment in head and neck squamous cell carcinoma. Sci Rep. 2020;10:3652
34. Shrestha R, Bridle KR, Crawford DHG, Jayachandran A. TNF-α-mediated epithelial-to-mesenchymal transition regulates expression of immune checkpoint molecules in hepatocellular carcinoma. Mol Med Rep. 2020;21:1849-60
35. Liu L, Li N, Zhang Q, Zhou J, Lin L, He X. Inhibition of ERK1/2 Signaling Impairs the Promoting Effects of TGF-β1 on Hepatocellular Carcinoma Cell Invasion and Epithelial-Mesenchymal Transition. Oncol Res. 2017;25:1607-16
36. Rawal P, Siddiqui H, Hassan M, Choudhary MC, Tripathi DM, Nain V. et al. Endothelial Cell-Derived TGF-β Promotes Epithelial-Mesenchymal Transition via CD133 in HBx-Infected Hepatoma Cells. Front Oncol. 2019;9:308
37. Wang B, Liu T, Wu JC, Luo SZ, Chen R, Lu LG. et al. STAT3 aggravates TGF-β1-induced hepatic epithelial-to-mesenchymal transition and migration. Biomed Pharmacother. 2018;98:214-21
38. Jayachandran A, Shrestha R, Dhungel B, Huang IT, Vasconcelos MYK, Morrison BJ. et al. Murine hepatocellular carcinoma derived stem cells reveal epithelial-to-mesenchymal plasticity. World J Stem Cells. 2017;9:159-68
39. Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY. et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385-92
40. Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A. et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995-2004
41. Tsuchiya M, Parker JS, Kono H, Matsuda M, Fujii H, Rusyn I. Gene expression in nontumoral liver tissue and recurrence-free survival in hepatitis C virus-positive hepatocellular carcinoma. Mol Cancer. 2010;9:74
42. Awan FM, Naz A, Obaid A, Ali A, Ahmad J, Anjum S. et al. Identification of Circulating Biomarker Candidates for Hepatocellular Carcinoma (HCC): An Integrated Prioritization Approach. PLoS One. 2015;10:e0138913
43. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
44. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156-72
45. Dooley S, ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res. 2012;347:245-56
46. Fabregat I, Caballero-Díaz D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front Oncol. 2018;8:357
47. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21-45
48. Huang J, Qiu M, Wan L, Wang G, Huang T, Chen Z. et al. TGF-β1 Promotes Hepatocellular Carcinoma Invasion and Metastasis via ERK Pathway-Mediated FGFR4 Expression. Cell Physiol Biochem. 2018;45:1690-9
49. Malfettone A, Soukupova J, Bertran E, Crosas-Molist E, Lastra R, Fernando J. et al. Transforming growth factor-β-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett. 2017;392:39-50
50. Xu Z, Shen MX, Ma DZ, Wang LY, Zha XL. TGF-beta1-promoted epithelial-to-mesenchymal transformation and cell adhesion contribute to TGF-beta1-enhanced cell migration in SMMC-7721 cells. Cell Res. 2003;13:343-50
51. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938-45
52. Soundararajan R, Fradette JJ, Konen JM, Moulder S, Zhang X, Gibbons DL. et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers (Basel). 2019 11
53. Nishida N, Kudo M. Immune Phenotype and Immune Checkpoint Inhibitors for the Treatment of Human Hepatocellular Carcinoma. Cancers (Basel). 2020 12
54. Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J. et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Annals of Oncology. 2019;30:v874-v5
55. Xu F, Jin T, Zhu Y, Dai C. Immune checkpoint therapy in liver cancer. J Exp Clin Cancer Res. 2018;37:110
56. Liu Z, Liu L, Lu T, Wang L, Li Z, Jiao D. et al. Hypoxia Molecular Characterization in Hepatocellular Carcinoma Identifies One Risk Signature and Two Nomograms for Clinical Management. J Oncol. 2021;2021:6664386
57. Ang C, Klempner SJ, Ali SM, Madison R, Ross JS, Severson EA. et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget. 2019;10:4018-25
58. Kim SI, Cassella CR, Byrne KT. Tumor Burden and Immunotherapy: Impact on Immune Infiltration and Therapeutic Outcomes. Front Immunol. 2020;11:629722
59. Sung WWY, Chow JCH, Cho WCS. Tumor mutational burden as a tissue-agnostic biomarker for cancer immunotherapy. Expert Rev Clin Pharmacol. 2020:1-3
60. Kim H, Hong JY, Lee J, Park SH, Park JO, Park YS. et al. Clinical sequencing to assess tumor mutational burden as a useful biomarker to immunotherapy in various solid tumors. Ther Adv Med Oncol. 2021;13:1758835921992992
61. Wong CN, Fessas P, Dominy K, Mauri FA, Kaneko T, Parcq PD. et al. Qualification of tumour mutational burden by targeted next-generation sequencing as a biomarker in hepatocellular carcinoma. Liver Int. 2021;41:192-203
62. Yin L, Zhou L, Xu R. Identification of Tumor Mutation Burden and Immune Infiltrates in Hepatocellular Carcinoma Based on Multi-Omics Analysis. Front Mol Biosci. 2020;7:599142
63. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353-65
64. Prasad V, Kaestner V, Mailankody S. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type-FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. JAMA Oncol. 2018;4:157-8
65. Zhang FP, Huang YP, Luo WX, Deng WY, Liu CQ, Xu LB. et al. Construction of a risk score prognosis model based on hepatocellular carcinoma microenvironment. World J Gastroenterol. 2020;26:134-53
66. Ng HHM, Lee RY, Goh S, Tay ISY, Lim X, Lee B. et al. Immunohistochemical scoring of CD38 in the tumor microenvironment predicts responsiveness to anti-PD-1/PD-L1 immunotherapy in hepatocellular carcinoma. J Immunother Cancer. 2020 8
67. Hilmi M, Neuzillet C, Calderaro J, Lafdil F, Pawlotsky JM, Rousseau B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: current knowledge and future research directions. J Immunother Cancer. 2019;7:333
68. Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009;15:195-206
69. Shrestha R, Bridle KR, Crawford DHG, Jayachandran A. Immune checkpoint blockade therapies for HCC: current status and future implications. Hepatoma Research. 2019. 2019
70. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA. et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241
71. Chouaib S, Janji B, Tittarelli A, Eggermont A, Thiery JP. Tumor plasticity interferes with anti-tumor immunity. Crit Rev Immunol. 2014;34:91-102
72. Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front Oncol. 2018;8:386
73. De Matteis S, Canale M, Verlicchi A, Bronte G, Delmonte A, Crinò L. et al. Advances in Molecular Mechanisms and Immunotherapy Involving the Immune Cell-Promoted Epithelial-to-Mesenchymal Transition in Lung Cancer. J Oncol. 2019;2019:7475364
74. Lequeux A, Noman MZ, Xiao M, Sauvage D, Van Moer K, Viry E. et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019;458:13-20
75. Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L. et al. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett. 2017;184:7-14
76. Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. B7H3 As a Promoter of Metastasis and Promising Therapeutic Target. Front Oncol. 2018;8:264
77. Kang FB, Wang L, Jia HC, Li D, Li HJ, Zhang YG. et al. B7-H3 promotes aggression and invasion of hepatocellular carcinoma by targeting epithelial-to-mesenchymal transition via JAK2/STAT3/Slug signaling pathway. Cancer Cell Int. 2015;15:45
78. Jiang B, Zhang T, Liu F, Sun Z, Shi H, Hua D. et al. The co-stimulatory molecule B7-H3 promotes the epithelial-mesenchymal transition in colorectal cancer. Oncotarget. 2016;7:31755-71
79. Lemke D, Pfenning PN, Sahm F, Klein AC, Kempf T, Warnken U. et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin Cancer Res. 2012;18:105-17
80. Fauci JM, Sabbatino F, Wang Y, Londoño-Joshi AI, Straughn JM, Landen CN. et al. Monoclonal antibody-based immunotherapy of ovarian cancer: targeting ovarian cancer cells with the B7-H3-specific mAb 376.96. Gynecol Oncol. 2014;132:203-10
81. Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN. et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin Cancer Res. 2016;22:609-20
82. Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH. et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin Cancer Res. 2016;22:3630-42
83. Thompson JC, Hwang WT, Davis C, Deshpande C, Jeffries S, Rajpurohit Y. et al. Gene signatures of tumor inflammation and epithelial-to-mesenchymal transition (EMT) predict responses to immune checkpoint blockade in lung cancer with high accuracy. Lung Cancer. 2020;139:1-8
84. Ihling C, Naughton B, Zhang Y, Rolfe PA, Frick-Krieger E, Terracciano LM. et al. Observational Study of PD-L1, TGF-β, and Immune Cell Infiltrates in Hepatocellular Carcinoma. Front Med (Lausanne). 2019;6:15
85. Li N, Wang J, Zhang N, Zhuang M, Zong Z, Zou J. et al. Cross-talk between TNF-α and IFN-γ signaling in induction of B7-H1 expression in hepatocellular carcinoma cells. Cancer Immunol Immunother. 2018;67:271-83
86. Brown ZJ, Yu SJ, Heinrich B, Ma C, Fu Q, Sandhu M. et al. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol Immunother. 2018;67:1305-15
Author contact
![]() Corresponding author: Aparna Jayachandran, PhD, Fiona Elsey Cancer Research Institute, Ballarat Technology Park- Central Suite 23, 106-110 Lydiard St Sth, Ballarat, VIC 3350, Australia. E-mail: aparnaorg.au; Phone +61 4 2424 8058.
Corresponding author: Aparna Jayachandran, PhD, Fiona Elsey Cancer Research Institute, Ballarat Technology Park- Central Suite 23, 106-110 Lydiard St Sth, Ballarat, VIC 3350, Australia. E-mail: aparnaorg.au; Phone +61 4 2424 8058.