Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(8):1848-1856. doi:10.7150/ijms.55866 This issue Cite
Research Paper
Bitter Melon Extract Yields Multiple Effects on Intestinal Epithelial Cells and Likely Contributes to Anti-diabetic Functions
Chi-I Chang1*, Shi-Yie Cheng2*, Annisa Oktafianti Nurlatifah1,3, Wei-Wen Sung1, Jing-Hong Tu1, Lin-Lee Lee4 ![]() , Hsueh-Ling Cheng1
, Hsueh-Ling Cheng1 ![]()
1. Department of Biological Science and Technology, National Pingtung University of Science and Technology, Pingtung 91201, Taiwan.
2. Department of Life Sciences, National University of Kaohsiung, Kaohsiung 811, Taiwan.
3. Department of Agroindustrial Biotechnology, Brawijaya University, Jalan, Veteran Malang 65145, Indonesia.
4. Department of English, National Kaohsiung Normal University, Kaohsiung 80201, Taiwan.
* These authors contributed to this work equally.
Received 2020-11-13; Accepted 2021-1-4; Published 2021-2-24
Abstract

The intestines have been recognized as important tissues for metabolic regulation, including glycemic control, but their vital role in promoting the anti-diabetic effects of bitter melon, the fruit of Momordica charantia L, has seldom been characterized, nor acknowledged. Evidence suggests that bitter melon constituents can have substantial interactions with the intestinal epithelial cells before circulating to other tissues. We therefore characterized the effects of bitter melon extract (BME) on intestinal epithelial cells. BME was found to contain substantial amounts of carbohydrates, proteins, and triterpenoids. TNF-α induced insulin resistance in an enterocyte cell line of IEC-18 cells, and BME promoted glucose utilization of the insulin-resistant cells. Further analysis suggested that the increased glucose consumption was a result of the combined effects of insulin sensitizing and insulin substitution functions of BME. The functions of insulin substitution were likely generated due to the activation of AMP-activated protein kinase. Meanwhile, BME acted as a glucagon-like peptide 1 (GLP-1) secretagogue on enteroendocrine cells, which may be mediated by the activation of bitter-taste receptors. Therefore, BME possesses insulin sensitizing, insulin substitution, and GLP-1 secretagogue functions upon intestinal cells. These effects of BME on intestinal cells likely play a significant part in the anti-diabetic action of bitter melon.
Keywords: AMP-activated protein kinase, bitter-taste receptor, glucagon-like peptide 1, diabetes, intestine, Momordica charantia.
Introduction
Diabetes, a chronic disorder characterized by hyperglycemia, is classified into several types, among which type 1 and type 2 diabetes are the most prevalent. Type 1 diabetes, caused by low or no insulin secretion from impaired pancreatic β cells, likely results from autoimmune problems. Type 2 diabetes is initiated by insulin resistance, suggesting insulin-sensitive tissues cannot respond to insulin stimulation properly, and usually resulting in progressive β-cell dysfunction and insufficient insulin secretion [1, 2].
The whole plant of Momordica charantia L. has been reported to have anti-diabetic functions [3-7], whereas bitter melon (also known as bitter gourds or balsam pears), i.e., the fruit of M. charantia L., are the most commonly consumed parts of the plant as food or herbal medicine. Therefore, we attempted to analyze the lesser-known effects of bitter melon on the intestines regarding glycemic control, in view of the role of the intestines in metabolic regulation including the glycemia, but noting the scarcity of reference to their role in the anti-diabetic effects of bitter melon.
The mechanisms underlying the hypoglycemic effect of bitter melon have been widely studied and explored in insulin-sensitive tissues, but mostly in the liver, the skeletal muscles and the adipose tissues [4, 5, 8, 9]. However, a previous study discovered that triterpenoids purified from bitter melon were transported across an intestinal epithelial cell monolayer in vitro, and small amounts of these triterpenoids stayed inside the epithelial cells [10]. Thus, bitter melon' natural compounds theoretically can have substantial interactions with the intestinal epithelial cells prior to working on other tissues. Moreover, the concentrations of bitter melon compounds encountered by the intestinal epithelial cells in situ during nutrient absorption from the lumen are likely to be higher than those encountered by other organs through circulation. However, other than the inhibition of intestinal α-amylase and α-glucosidase, and the suppression of intestinal glucose absorption from the digestive lumen [11, 12], the effects of bitter melon components on intestinal cells were rarely characterized.
Intestinal cells are also sensitive to insulin and can develop insulin resistance in obese or inflammatory conditions [13-16]. Additionally, the intestines play important roles in metabolism regulation, including glycemic control, since they secrete several hormones to regulate metabolism while being stimulated by food components or metabolic changes [17]. Meanwhile, the intestines occupy a substantial mass of the human body and carry out intensive physiological activities and as such are important vectors in blood glucose utilization, contributing to the reduction of glycemia [13, 18]. Consequently, the responses of intestinal epithelial cells to bitter melon constituents should be carefully re-examined.
The epithelial layer of the intestines is composed of several cell types, including columnar enterocytes in the majority, and enteroendocrine cells that secrete several hormones upon stimulation. The enterocytes were reported to be insulin sensitive [13-15, 19], whereas the enteroendocrine cells were demonstrated to contain bitter-taste receptors (TAS2Rs), a group of G protein-coupled receptors classified as taste receptor 2 family [20]. The activation of TAS2Rs by bitter substances provokes secretion of several hormones from enteroendocrine cells, including glucagon-like peptide 1 (GLP-1) [21]. GLP-1, a notable target for developing anti-diabetic medicines, regulates the glycemic level through several mechanisms, such as the control of insulin secretion [21-23]. The activation of TAS2Rs on enteroendocrine cells triggers the dissociation of β and γ subunits from the α subunit of the coupled trimeric G proteins. The Gβγ complex activates phospholipase C β2 (PLCβ2), which catalyzes the production of inositol-1,4,5-triphosphate (IP3) and diacylglycerol from phosphatidylinositol-4,5-diphosphate in the cell membrane. IP3 activates IP3 receptors in the membrane of the endoplasmic reticulum, resulting in the release of Ca2+ from the endoplasmic reticulum, and thus an increase in cytosolic [Ca2+]. Subsequently, through a not completely clarified mechanism, the increased cytosolic [Ca2+] leads to the secretion of GLP-1 from the enteroendocrine cells [24-26].
The constituents of bitter melon have been demonstrated to exert effects on insulin-sensitive cells [4, 8], and bitterness is a characteristic taste of this vegetable. Thus, we analyzed the responses of enterocytes and enteroendocrine cells to bitter melon extract (BME) in order to evaluate the effects of bitter melon on the intestines.
Materials and methods
Reagents
Antibodies for total or phosphorylated Akt (Ser 473) and total or phosphorylated AMPK α subunit (Thr 172) were purchased from Cell Signaling Technology (Beverley, MA, USA); horseradish peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Fetal bovine serum (FBS) was acquired from Invitrogen (Carlsbad, CA, USA). Protease inhibitor cocktail and phosphatase inhibitor cocktail were from Calbiochem (Merck Millipore, Darmstadt, Germany). Cell Culture Lysis Reagent was from Promega (Madison, WI, USA). Folin-Ciocalteau reagent was procured from Fluka Biochemica (Buchs, Switzerland), whereas methanol and ethanol were purchased from Merck (Darmstadt, Germany). Cell culture media, bovine insulin solution, dimethylsulfoxide (DMSO), mouse TNF-α, rosiglitazone, denatonium benzoate, Fluo-4 AM, Compound C, gallein, U73122, and other chemicals were ordered from Sigma Chemical Company (St. Louis, MO, USA).
Preparation and chemical composition analysis of bitter melon extract
Bitter melon (the fruit of M. charantia L.) were collected from a local farm in Pingtung County, Taiwan, in July 2018. A voucher specimen (MC) has been identified by Prof. Sheng-Zehn Yang, Curator of Herbarium, National Pingtung University of Science and Technology and deposited in the Department of Life Science, National University of Kaohsiung. The sample was oven-dried (53o C) for one week. The dried specimen (1.0 kg, dry weight) was chopped into small pieces and extracted three times with ethanol (3 × 1.5 L, 2 days each) in a percolator at room temperature. After filtration and solvent evaporation, the combined ethanol extracts were concentrated in vacuo (under 35o C) to afford a resulting light yellow gum (35.0 g), i.e., BME. Part of BME was dissolved in DMSO, and subjected to protein concentration assay by using Bradford reagent (Bio-Rad, Hercules, CA, USA), as well as to total carbohydrate assay by using the phenol-sulfuric acid method [27].
The total phenolic content of BME was determined using the Folin-Ciocalteu assay with some modifications [28]. Briefly, 25 μL of BME solutions (50-1000 μg/mL in methanol) were mixed with 100 μL of 20% Na2CO3. After 2 min, 50 μL of 20% Folin-Ciocalteau reagent was subsequently added to the mixture and then incubated for 30 min at room temperature. The absorbance of the reaction mixtures was measured at 750 nm using a microplate spectrophotometer (Multiskan GO, Thermo Fischer Scientific, Vantaa, Finland). Gallic acid (2-200 μg/mL in methanol) was used as a standard to plot the calibration curve. The total phenolic contents of BME was expressed as mg gallic acid equivalents (GAE) per gram dry sample (mg GAE/g dry sample).
Total flavonoid content of BME was determined following the method described by Barreira et al. with some modifications [29]. Briefly, 25 μL aliquots of BME solutions (125-1000 μg/mL in methanol) were mixed with 152.5μL of deionized water followed by an addition of 7.5 μL of 5% NaNO2. The mixture was allowed to stand at room temperature for 6 min and then 15 μL of a 10% AlCl3·6H2O solution was added into the mixture. After a further 5 min incubation at room temperature, 50 μL of 1M NaOH was added to the mixture. The mixture was then incubated for 15 min at room temperature. The absorbance at 500 nm was read using a microplate spectrophotometer. Quercetin (4-400 μg/mL in methanol) was used as a standard to draw the calibration curve. The total flavonoid content was expressed as mg quercetin equivalents (QE) per gram dry sample (mg QE/g dry sample).
The total triterpenoid content of BME was determined by the method of Fan and He [30] with a slight modification. Briefly, 100 μL of BME (10 mg/mL in methanol) was mixed with 150 μL of 5% (w/v) vanillin-glacial acetic acid solution and 500 μL perchloric acid solution. The mixture was incubated at 80°C for 30 min. Subsequently, the solution was cooled in an ice-water bath to the ambient temperature and diluted to 3 mL. Absorbance at 548 nm was measured using a microplate spectrophotometer and compared to standard ursolic acid calibration curve (25-500 μg/mL in methanol). Results were expressed as mg ursolic acid equivalents (UAE) per gram dry sample (mg UAE/g dry sample).
Cell culture and cytotoxicity assays
NCI-H716 cells and IEC-18 cells were purchased from Bioresource Collection and Research Center (Hsinchu, Taiwan), and cultured at 37° C in a humidified incubator supplied with 5% CO2 using Dulbecco's modified Eagle's medium and RPMI 1640 medium, respectively, containing 10% FBS.
Cells were seeded in 96-well plates in a density of 1 x 104 cells/well (IEC-18) or 1 x 106 cells/well (NCI-H716), and treated in triplicate with the desired concentration of BME, or an equal volume of DMSO (solvent; as the control) in a serum-free medium for 6 h. Subsequently, a quick cell proliferation assay kit (BioVision, Milpitas, CA, USA) was added per the supplier's instructions, and the plates were incubated for 2 h at 37° C. Absorbance at 450 nm and 650 nm was measured afterwards. The relative survival rate versus the control was determined, and the mean ± standard error (SE) was calculated.
Glucose uptake assays
Glucose uptake assays in normal IEC-18 cells were performed as previously described [19]. For assays in insulin-resistant cells, IEC-18 cells were seeded equally in 96-well plates, cultured overnight, and incubated in a serum-free medium containing the desired concentration of TNF-α for 5 h to induce insulin resistance in the cells. The medium was removed, replaced with 50 μL fresh, serum-free medium containing 100 nM insulin, the desired concentration of BME, or an equal volume of the solvent (control), and incubated at 37° C for 5 h. Compound C was added 30 min earlier than the addition of BME when it was required. Each treatment was performed in triplicate. At 0 and 5 h after the treatment, 5 μL of the medium was withdrawn, mixed with 200 μL of a glucose assay kit (Glucose GOD FS, Diagnostic Systems, Holzheim, Germany), and reacted at 37° C for 10 min. Absorbance at 500 nm was detected in order to analyze the glucose concentration in the medium. The cell number in each well was subsequently analyzed by using the quick cell proliferation assay kit as described above. Glucose consumption by each well was determined based on the OD500 difference between 0 h and 5 h, and was normalized by cell number. Relative glucose uptake versus the control was then calculated.
Western blot analysis
Cells were seeded in 35- or 60-mm plates, cultured until approximately 90% confluence, washed with phosphate-buffered saline (PBS; pH 7.4), and incubated in a serum-free medium containing 20 ng/mL of TNF-α for 5 h. After being washed twice with PBS, the cells were incubated in a serum-free medium containing 100 nM insulin, a desired concentration of BME, 50 μM rosiglitazone, or an equal volume of the solvent for 1 h. The cells were then washed twice with PBS, submerged in a lysis buffer (Cell Culture Lysis Reagent containing a protease inhibitor cocktail, and a phosphatase inhibitor cocktail), and scraped off the plate on ice. The resulting suspension was centrifuged at 12,000 × g for 10 min at 4°C. The supernatant was collected and analyzed for protein concentration using Bradford reagent. Equal amounts of proteins were sampled from different treatments, and subjected to electrophoresis and western blotting as described previously [31, 32]. Immunoreactive bands were developed on the PVDF membrane using Supersignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Rockford, IL, USA) and detected using an imaging system (UVP BioSpectrum, UVP, LLC, Upland, CA, USA). The band intensities were determined using the supplied software.
GLP-1 assays
NCI-H716 cells were seeded equally in 24-well plates precoated with Matrigel (Corning, Corning, NY, USA), cultured as described previously [19], and treated for 1 h with 200 μg/mL BME, 10 mM denatonium benzoate (dissolved in 4% DMSO), or the respective solvent in PBS containing 0.9 mM CaCl2. In cultures treated with an inhibitor, the inhibitor was added 30 min earlier than BME. GLP-1 concentration in the medium was then determined by using a GLP-1 ELISA kit (RayBiotech, Peachtree Corners, GA, USA).
Calcium fluorescent imaging assay
NCI-H716 cells were seeded in clear-bottom 96-well black plates (Corning) precoated with Matrigel. Cells were then incubated with serum-free medium containing 4 μM Fluo-4 AM for 1 h. After being washed 3 times with PBS, the medium was replaced with Dulbecco's phosphate-buffered saline containing 2 mM calcium (DPBS). Cells were incubated in DPBS for 30 min at 37°C, washed 3 times with DPBS, and incubated in DPBS again. The plate was then placed under a fluorescent microscope (IX73, Olympus, Tokyo, Japan) equipped with an imaging system (DP80, Olympus) to start recording fluorescent imaging. BME, denatonium benzoate, or the solvent, was added to the saline, and the real-time intracellular fluorescence fluctuations were recorded by the imaging system and quantified with the supplied software (Dimension, Olympus). For experiments treated with an inhibitor, the inhibitor was added during the 30-min incubation with DPBS, and was present during fluorescence recording. The fold of fluorescence increment relative to the background was calculated as ΔF/F = (Fn - F0)/F0, in which F0 is the relative fluorescence unit obtained at time = 0 sec; Fn is the relative fluorescence unit recorded at time = n sec.
Statistical analysis
Data were analyzed using one-way analysis of variance (ANOVA) followed by Scheffe's post hoc test. Significance was considered when p < 0.05 and F > 3.5546.
Results
Bitter melon extract promoting the glucose utilization of insulin-resistant enterocytes
The protein, carbohydrate, phenolic compound, flavonoid, and triterpenoid content of BME were analyzed. As a result, BME contains 5.46 ± 0.07 % (w/w) of total proteins and 17.20 ± 0.85% (w/w) of total carbohydrates. Total phenolic content, total flavonoids, and total triterpenoids in BME are 2.31 ± 0.11 mg GAE/g, 0.95 ± 0.021 mg QE/g, and 17.24 ± 0.91 mg UAE/g, respectively. Therefore, BME contains substantial amounts of carbohydrates, proteins, and triterpenoids.
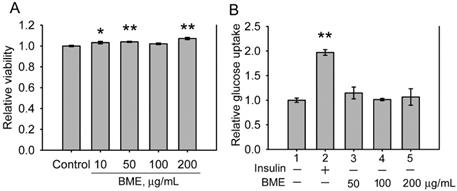
The cytotoxicity of BME to IEC-18 cells, an insulin-sensitive rat enterocyte cell line [19], was examined. Figure 1A displays that BME at 10-200 μg/mL is not toxic to IEC-18 cells. Therefore, the effect of BME on the glucose consumption of IEC-18 cells was characterized. Figure 1B reveals that insulin increased the glucose utilization of IEC-18 cells as reported previously [19]. BME at 50-200 μg/mL did not show an obvious effect on promoting the glucose uptake of the cells, suggesting that BME does not possess an insulin-like effect to stimulate glucose intake by the enterocytes. Hence, BME does not affect the glucose consumption of normal enterocytes.
The effects of BME on normal IEC-18 cells. A, cytotoxicity assays. IEC-18 cells were treated with the solvent (control), 10, 50, 100 or 200 μg/mL BME for 6 h and subjected to a cell proliferation assay. B, glucose uptake assays in IEC-18 cells treated for 5 h with 100 nM insulin, 50, 100, or 200 μg/mL EPSs as indicated underneath the histogram. Relative glucose uptake versus Group 1 was determined. Both experiments were performed twice independently, each in triplicate. The data are the mean ± SE (N = 6). *P < 0.05 and **P < 0.005 versus the control (A) or Group 1 (B).
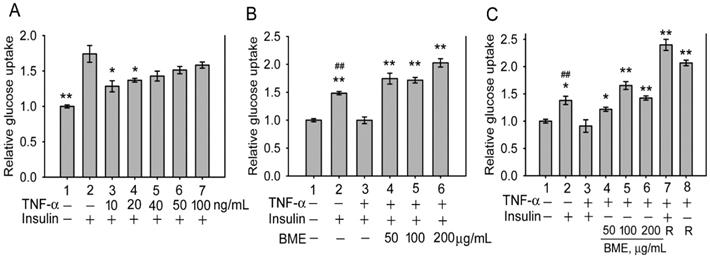
Subsequently, the effect of BME on insulin-resistant cells was analyzed. Inflammatory cytokines such as TNF-α are critical factors causing insulin resistance of tissues [33-35]. Figure 2A shows that pretreatment by TNF-α at 10 and 20 ng/mL effectively decreased insulin-promoted glucose uptake by IEC-18 cells (Groups 3 and 4 versus Group 2), suggesting the occurrence of insulin resistance in these cells, whereas higher concentrations of TNF-α did not show a higher efficacy (Groups 5-7). Thus, cells were pretreated with 20 ng/mL TNF-α, followed by insulin and BME co-stimulation. Figure 2B demonstrates that 50, 100, and 200 μg/mL BME all effectively enhanced the glucose uptake of TNF-α-pretreated cells (Groups 4-6) compared to the insulin-resistant control (Group 3), indicating that BME can promote the glucose consumption of insulin-resistant enterocytes. Subsequently, TNF-α-pretreated cells were stimulated by BME alone, without insulin. As shown in Figure 2C, BME at 50, 100, and 200 μg/mL (Groups 4, 5, 6) still significantly elevated the glucose uptake of the cells compared to the control (Group 3). Meanwhile, as a positive control, rosiglitazone, a thiazolidinedione-type medicine for treating diabetes, also promoted the glucose consumption of the cells in the presence or absence of insulin (Groups 7 and 8). Figure 2C reveals that BME alone, without insulin, can still enhance the glucose uptake of insulin-resistant cells. We define this function as insulin substitute herein. Therefore, the increased glucose consumption by the co-treatment of BME and insulin in Figure 2B might be irrelevant to the presence of insulin. Put differently, BME might not work as an insulin sensitizer. To clarify this issue, the effect of BME on Akt, an effector in the insulin-signaling pathway, was assayed. Figures 3A and 3B show that TNF-α decreased insulin-induced Akt activation (Lane 3 versus Lane 2), whereas the addition of 100 and 200 μg/mL BME recovered the elevation of Akt phosphorylation in the presence of insulin (Figure 3A, Lanes 4, 5), similar to the effect of rosiglitazone (Figure 3A, Lane 6). However, this effect of BME or rosiglitazone on Akt activation was not obvious in the absence of insulin (Figure 3B, Lanes 4, 5, 6). Therefore, BME alone could not activate Akt, yet it recovered insulin-promoted Akt activation in TNF-α-treated cells, indicating a function of insulin sensitizer. Together, Figures 2, 3A, and 3B suggest that BME was bi-functional, acting as an insulin sensitizer and an insulin substitute. Hence, the increased glucose uptake observed in insulin-resistant cells co-treated by BME and insulin (Figure 2B Groups 4-6) could be a result of the combined effects of insulin sensitizing and insulin substitution functions of BME. Meanwhile, data in Figure 3B also reveal that the insulin-substitute function of BME was not mediated by the activation of Akt, suggesting that it depends on other mechanisms to confer the insulin-substitute function.
Glucose uptake assays in insulin-resistant IEC-18 cells. A, cells were pretreated with 10, 20, 40, 50 or 100 ng/mL TNF-α, followed by stimulation with 100 nM insulin. The experiment was performed in triplicate. B and C, cells were pretreated with 20 ng/mL TNF-α, followed by stimulation with 50, 100 or 200 μg/mL BME in the presence (B) or absence (C) of 100 nM insulin. In C, 50 μM rosiglitazone (R) was also tested as a positive control with (Group 7) or without (Group 8) insulin. Experiments were performed twice independently, each in triplicate. Relative glucose uptake versus Group 1 is shown. The data are the mean ± SE (N = 3 in A; N = 6 in B and C). *P < 0.05 and **P < 0.005 versus Group 2 (A) or Group 3 (B and C); ##P < 0.005 versus Group 1 (B and C).
The effects of BME on Akt and AMPK. A, B, and C, western blot analysis of the levels of phosphorylated Akt, total Akt (A, B), and phosphorylated AMPK, total AMPK (C). IEC-18 cells were pretreated with 20 ng/mL TNF-α, followed by stimulation with 100 or 200 μg/mL BME, or 50 μM rosiglitazone (R) in the presence or absence of 100 nM insulin, as indicated underneath the blots. In C, the concentration of BME (indicated as B) was 200 μg/mL. Experiments were performed three times independently. The result from one of the experiments is shown. D, glucose uptake assays in IEC-18 cells. Cells were pretreated with 20 ng/mL TNF-α, followed by the treatment as indicated underneath the histogram. Experiments were performed in triplicate. The data are the mean ± SE (N = 3). *P < 0.05 and **P < 0.005 between the indicated groups.
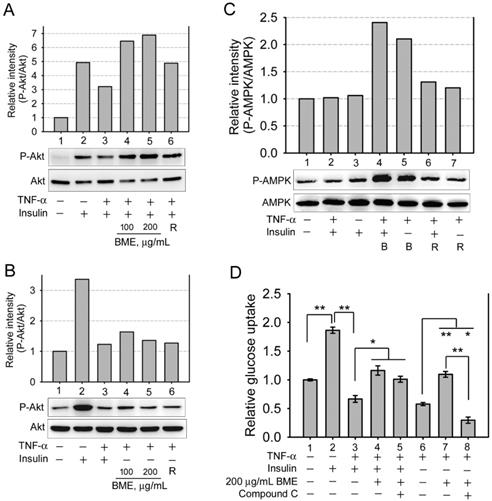
Several natural compounds isolated from bitter melon were demonstrated to activate AMP-activated protein kinase (AMPK) [4, 8], which can increase glucose consumption of cells without insulin and without Akt activation. Therefore, whether BME activated AMPK in the enterocyte cell line was analyzed. Figure 3C manifests that 200 μg/mL BME obviously activated AMPK in the presence or absence of insulin in TNF-α-treated IEC-18 cells (Lanes 4 and 5), and 50 μM rosiglitazone showed a weaker effect (Lanes 6 and 7). Meanwhile, insulin did not obviously activate AMPK in this cell line (Lane 3), meaning that AMPK is probably not involved in insulin-mediated hypoglycemic function in this cell line. Moreover, Figure 3D shows that compound C, an AMPK inhibitor, did not significantly inhibit BME-promoted glucose uptake of insulin-resistant cells when insulin was present (Group 5 versus Group 4). This further supports that BME has a function of insulin sensitizer, which recovers the function of insulin, yet the function of insulin does not depend on AMPK in this cell line. Therefore, AMPK inhibitor did not obviously interfere with the glucose uptake of this group. On the contrary, compound C effectively suppressed BME-promoted glucose uptake of TNF-α-pretreated cells when insulin was absent (Figure 3D, Group 8 versus Group 7), suggesting that AMPK mediates the insulin substitute function of BME in the absence of insulin.
Bitter melon extract stimulating GLP-1 secretion from the enteroendocrine cells
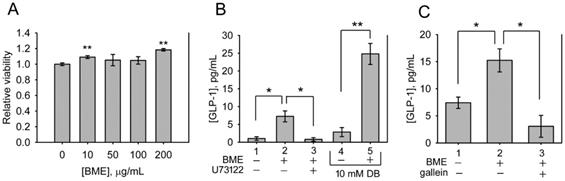
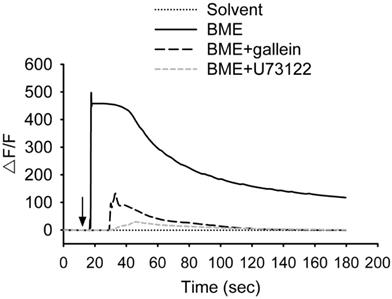
The bitterness of bitter melon indicates that the constituents of bitter melon activate TAS2Rs. Therefore, whether BME promotes the secretion of GLP-1 from enteroendocrine cells was analyzed. NCI-H716 cells, a human enteroendocrine cell line, were treated with 10-200 μg/mL BME to test cytotoxicity. Figure 4A shows that BME is not toxic to NCI-H716 cells. Subsequently, the secretion of GLP-1 from NCI-H716 was found to be enhanced while being treated with BME, as revealed in Figures 4B and 4C (Group 2 versus Group 1). Denatonium benzoate, a bitter compound demonstrated to elevate GLP-1 secretion, also increased GLP-1 secretion from the cells (Figure 4B, Group 5 versus Group 4). To evaluate whether the activation of TAS2R contributes to BME-induced GLP-1 secretion, the inhibitor of PLCβ2 (U73122) or Gβγ complex (gallein) was added. Consequently, U73122 (Figure 4B, Group 3) and gallein (Figure 4C, Group 3) both effectively suppressed BME-induced elevation of GLP-1 secretion. Furthermore, Figure 5 demonstrates that BME induced an obvious increase in intracellular [Ca2+], a biomarker of the activation of the TAS2R-signaling pathway. When cells were pre-treated with gallein or U73122, the BME-induced calcium response was apparently suppressed. Together, these data suggest that BME can effectively enhance GLP-1 secretion from enteroendocrine cells, which is likely mediated by the activation of the TAS2R-signaling pathway.
BME promoted the secretion of GLP-1 from NCI-H716 cells. A, cytotoxicity assays. NCI-H716 cells were treated with the solvent (0; control), 10, 50, 100 or 200 μg/mL BME for 6 h and subjected to a cell proliferation assay. Experiments were performed twice independently, each in triplicate. The data are the mean ± SE (N = 6). B and C, GLP-1 secretion assays. NCI-H716 cells were treated for 1 h with 200 μg/mL BME, 10 μM U73122, 10 μM gallein, 10 mM denatonium benzoate (DB), or the respective solvent, as indicated underneath the histogram. The culture medium was subjected to GLP-1 concentration assay by using an ELISA kit. Experiments were performed in quadruplicate (B) or in triplicate (C). The data are the mean ± SE. *P < 0.05 and **P < 0.005 versus the control (A) or between the indicated groups (B and C).
BME induced calcium responses in NCI-H716 cells. Cells were treated with 200 μg/mL BME or the solvent, or pretreated with 10 μM gallein or 10 μM U73122 for 30 min before the addition of 200 μg/mL BME, and subjected to calcium fluorescent imaging assays. The arrow indicates when BME or solvent was added to the medium. Experiments were performed three times independently. The result from one of the experiments is shown.
Discussion
In this study, the extract of bitter melon exhibited three functions upon intestinal cells that are likely associated with its hypoglycemic effect. BME works as an insulin sensitizer and an insulin substitute against insulin-resistant enterocytes, and as a GLP-1 secretagogue on enteroendocrine cells. The insulin sensitizing function recovers insulin-induced activation of Akt. The insulin substitution function is likely due to the activation of AMPK, which mediates an insulin-independent increase of glucose uptake by the cells. The insulin sensitizing and the insulin substitution functions together re-boost glucose utilization by insulin-resistant enterocytes. The GLP-1 secretagogue function of BME may be rendered by the activation of TAS2Rs. These data support that BME has a hypoglycemic function on insulin-resistant or type 2 diabetic cases. Indeed, animal tests and at least some clinical trials have shown anti-diabetic effects of bitter melon on type 2 diabetic models or individuals [12, 36, 37], while the effect of bitter melon on the intestines may be one of the critical mechanisms underlying the anti-diabetic functions of bitter melon in vivo, but was not revealed, nor explored previously. On the other hand, BME did not show an insulin-like activity to increase the glucose consumption of normal enterocytes (Figure 1B). Thus, BME probably does not promote glucose utilization by normal tissues and may not be helpful in type 1 diabetic cases.
Bitter melon was reported to ameliorate insulin resistance [4, 11]. Our data are consistent with this previous finding, owing to that BME exhibited an insulin-sensitizing function on insulin-resistant IEC-18 cells. This activity was revealed by that BME recovered Akt activation only in the presence of insulin in TNF-α-treated cells (Figure 3A and 3B). Moreover, in TNF-α-treated cells, the inhibition of AMPK suppressed the insulin substitute function (Figure 3D, Group 8), but did not inhibit the increased glucose uptake caused by the co-stimulation of BME and insulin (Figure 3D, Group 5). The latter further supports the existence of an insulin sensitizing activity of BME.
The findings that BME functions as an insulin sensitizer and an insulin substitute in insulin-resistant cells are consistent with the results of earlier studies using different cell types or tissues. Previously, bitter melon extract was shown to promote the glucose consumption of an insulin-resistant liver cell line [3, 4]. Natural compounds isolated from the fruit and the stalk of M. charantia L. were reported to work as insulin sensitizers and insulin substitutes in insulin-resistant liver cell line [8]. Bitter melon extract was also reported to improve the insulin sensitivity of skeletal muscle in high-fat-fed rats with signs of insulin resistance [38]. Therefore, there is consistency between results obtained from cells of different tissues. In addition, one study showed that bitter melon natural compounds were transported across an intestinal epithelial cell layer, and some of them were absorbed inside the epithelial cells [10], stating that bitter melon compounds can interact with the intestines in situ, as well as with other tissues through circulation. Overall, our findings and others suggest that apart from being absorbed by the intestines and circulated to other tissues, bitter melon ingredients also interact with intestinal cells in situ, and the interactions are likely part of the mechanisms underlying the hypoglycemic function of bitter melon. The responses of the intestines to bitter melon likely work in concert with those of other tissues to result in the anti-diabetic effect of bitter melon.
In agreement with our findings that BME acts as GLP-1 secretagogue on enteroendocrine cells, bitter melon extract was shown recently to increase serum GLP-1 level in normal and diabetic rats [39]. Several of the reported beneficial effects of bitter melon on glycemic control coincide with the functions of GLP-1. Bitter melon were shown to improve insulin resistance, increase insulin secretion in type 2 diabetic patients, as well as repair damaged β-cells [4, 11, 12, 36]. These functions are consistent with several functions of GLP-1, which promotes glucose-dependent insulin secretion, increases insulin sensitivity of peripheral tissues, enhances insulin synthesis in β-cell, and promotes β-cell survival [40]. Therefore, it is likely that some of the reported functional effects of bitter melon on glycemic control are mediated by GLP-1.
The mechanism underlying the insulin sensitizing effect of BME on insulin-resistant enterocytes was not elucidated in this study. However, insulin resistance is closely associated with inflammation, and type 2 diabetes is regarded as a low-grade, chronic inflammation-related disorder [34]. Thus, the pro-inflammatory factor TNF-α obviously induced insulin resistance in IEC-18 cells. Earlier studies have demonstrated that bitter melon contain ingredients possessing anti-inflammatory activities [32, 41]. Specifically, bitter melon' natural compounds were shown to suppress TNF-α-induced inflammation in a hepatic cell line [32, 42]. Thus, we propose that the insulin sensitizing effect of BME on the TNF-α-treated enterocytes may be a result of its anti-inflammatory function, ameliorating inflammation-induced insulin resistance.
Our data suggest that BME activates the TAS2R-signaling pathway in enteroendocrine cells and leads to GLP-1 secretion. However, which isoforms of TAS2Rs are activated by BME is not clarified. There are 25 members in the human TAS2R family, and one molecule may activate more than one TAS2R isoform [26, 43]. Considering the diversities of molecules contained in BME, it is expected that multiple isoforms of TAS2Rs are activated by BME. Using natural compounds isolated from bitter melon to identify the TAS2R isoforms being activated will provide more insightful information.
Conclusion
In summary, BME contains components with insulin sensitizing, insulin substitution, and GLP-1 secretagogue functions upon intestinal cells. The exact principles of bitter melon possessing these functions deserve to be identified. Meanwhile, these findings suggest that apart from absorbing bitter melon ingredients from the lumen for transporting to other organs through circulation, the intestines may actively participate in the hypoglycemic mechanisms of bitter melon. The interactions of bitter melon compounds with the intestinal epithelial cells in situ are likely a significant part in the hypoglycemic action of bitter melon.
Abbreviations
AMPK: AMP-activated protein kinase; BME: bitter melon extract; DB: denatonium benzoate; DPBS: Dulbecco's phosphate-buffered saline containing 2 mM calcium; GAE: gallic acid equivalents; GLP-1: glucagon-like peptide 1; IP3: inositol-1,4,5-triphosphate; PLCβ2: phospholipase C β2; QE: quercetin equivalents; TAS2R: bitter-taste receptor; UAE: ursolic acid equivalents.
Acknowledgements
Funding
This work was supported by the Ministry of Science and Technology of Taiwan (Grant No. MOST 103-2313-B-020-009-MY3 and MOST 106-2320-B-020-001-MY3).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Golden SH, Sapir T. Methods for insulin delivery and glucose monitoring in diabetes: summary of a comparative effectiveness review. J Manag Care Pharm. 2012;18:S1-17
2. Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol. 2009;9:753-62
3. Cheng HL, Huang HK, Chang CI, Tsai CP, Chou CH. A cell-based screening identifies compounds from the stem of Momordica charantia that overcome insulin resistance and activate AMP-activated protein kinase. J Agric Food Chem. 2008;56:6835-43
4. Chang CI, Tseng HI, Liao YW, Yen CH, Chen TM, Lin CC. et al. In vivo and in vitro studies to identify the hypoglycaemic constituents of Momordica charantia wild variant WB24. Food Chemistry. 2011;125:521-8
5. Efird JT, Choi YM, Davies SW, Mehra S, Anderson EJ, Katunga LA. Potential for improved glycemic control with dietary Momordica charantia in patients with insulin resistance and pre-diabetes. Int J Environ Res Public Health. 2014;11:2328-45
6. Tariq A, Sadia S, Fan Y, Ali S, Amber R, Mussarat S. et al. Herbal medicines used to treat diabetes in Southern regions of Pakistan and their pharmacological evidence. J Herb Med. 2020;21:100323
7. Chinsembu KC. Diabetes mellitus and nature's pharmacy of putative antidiabetic plants. J Herb Med. 2019;15:100230
8. Chang CI, Chou CH, Liao MH, Chen TM, Cheng CH, Anggriani R. et al. Bitter melon triterpenes work as insulin sensitizers and insulin substitutes in insulin-resistant cells. J Funct Foods. 2015;13:214-24
9. Popovich DG, Li L, Zhang W. Bitter melon (Momordica charantia) triterpenoid extract reduces preadipocyte viability, lipid accumulation and adiponectin expression in 3T3-L1 cells. Food Chem Toxicol. 2010;48:1619-26
10. Wu SB, Yue GG, To MH, Keller AC, Lau CB, Kennelly EJ. Transport in Caco-2 cell monolayers of antidiabetic cucurbitane triterpenoids from Momordica charantia fruits. Planta Med. 2014;80:907-11
11. Wang S, Li Z, Yang G, Ho CT, Li S. Momordica charantia: a popular health-promoting vegetable with multifunctionality. Food Funct. 2017;8:1749-62
12. Chaturvedi P. Antidiabetic potentials of Momordica charantia: multiple mechanisms behind the effects. J Med Food. 2012;15:101-7
13. Honka H, Makinen J, Hannukainen JC, Tarkia M, Oikonen V, Teras M. et al. Validation of [18F]fluorodeoxyglucose and positron emission tomography (PET) for the measurement of intestinal metabolism in pigs, and evidence of intestinal insulin resistance in patients with morbid obesity. Diabetologia. 2013;56:893-900
14. Hsieh J, Hayashi AA, Webb J, Adeli K. Postprandial dyslipidemia in insulin resistance: Mechanisms and role of intestinal insulin sensitivity. Atherosclerosis Supp. 2008;9:7-13
15. Veilleux A, Grenier E, Marceau P, Carpentier AC, Richard D, Levy E. Intestinal lipid handling. Evidence and implication of insulin signaling abnormalities in human obese subjects. Arterioscler Thromb Vasc Biol. 2014;34:644-53
16. Qin B, Qiu W, Avramoglu RK, Adeli K. Tumor necrosis factor-alpha induces intestinal insulin resistance and stimulates the overproduction of intestinal apolipoprotein B48-containing lipoproteins. Diabetes. 2007;56:450-61
17. Vella A, Camilleri M. The gastrointestinal tract as an integrator of mechanical and hormonal response to nutrient ingestion. Diabetes. 2017;66:2729-37
18. Saeidi N, Meoli L, Nestoridi E, Gupta NK, Kvas S, Kucharczyk J. et al. Reprogramming of intestinal glucose metabolism and glycemic control in rats after gastric bypass. Science. 2013;341:406-10
19. Chen YC, Huang SD, Tu JH, Yu JS, Nurlatifah AO, Chiu WC. et al. Exopolysaccharides of Bacillus amyloliquefaciens modulate glycemic level in mice and promote glucose uptake of cells through the activation of Akt. Int J Biol Macromol. 2020;146:202-11
20. Behrens M, Meyerhof W. Gustatory and extragustatory functions of mammalian taste receptors. Physiol Behav. 2011;105:4-13
21. Pais R, Gribble FM, Reimann F. Stimulation of incretin secreting cells. Ther Adv Endocrinol Metab. 2016;7:24-42
22. Lehrke M, Marx N. New antidiabetic therapies: innovative strategies for an old problem. Current opinion in lipidology. 2012;23:569-75
23. Lee YS, Jun HS. Glucagon-like peptide-1 receptor agonist and glucagon increase glucose-stimulated insulin secretion in beta cells via distinct adenylyl cyclases. Int J Med Sci. 2018;15:603-9
24. Kim KS, Egan JM, Jang HJ. Denatonium induces secretion of glucagon-like peptide-1 through activation of bitter taste receptor pathways. Diabetologia. 2014;57:2117-25
25. Yu Y, Hao G, Zhang Q, Hua W, Wang M, Zhou W. et al. Berberine induces GLP-1 secretion through activation of bitter taste receptor pathways. Biochem Pharmacol. 2015;97:173-7
26. Jaggupilli A, Howard R, Upadhyaya JD, Bhullar RP, Chelikani P. Bitter taste receptors: Novel insights into the biochemistry and pharmacology. Int J Biochem Cell Biol. 2016;77:184-96
27. DuBois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Colorimetric method for determination of sugars and related substances. Anal Chem. 1956;28:350-6
28. Meda A, Lamien CE, Romito M, Millogo J, Nacoulma OG. Determination of the total phenolic, flavonoid and proline contents in Burkina Fasan honey, as well as their radical scavenging activity. Food Chem. 2005;91:571-7
29. Barreira JCM, Ferreira ICFR, Oliveira MBPP, Pereira JA. Antioxidant activities of the extracts from chestnut flower, leaf, skins and fruit. Food Chem. 2008;107:1106-13
30. Fan JP, He CH. Simultaneous quantification of three major bioactive triterpene acids in the leaves of Diospyros kaki by high-performance liquid chromatography method. J Pharm Biomed Anal. 2006;41:950-6
31. Cheng HL, Chang SM, Cheng YW, Liu HJ, Chen YC. Characterization of the activities of p21Cip1/Waf1 promoter-driven reporter systems during camptothecin-induced senescence-like state of BHK-21 cells. Mol Cell Biochem. 2006;291:29-38
32. Cheng HL, Kuo CY, Liao YW, Lin CC. EMCD, a hypoglycemic triterpene isolated from Momordica charantia wild variant, attenuates TNF-alpha-induced inflammation in FL83B cells in an AMP-activated protein kinase-independent manner. Eur J Pharmacol. 2012;689:241-8
33. Baboota RK, Bishnoi M, Ambalam P, Kondepudi KK, Sarma SM, Boparai RK. et al. Functional food ingredients for the management of obesity and associated co-morbidities - A review. J Funct Foods. 2013;5:997-1012
34. Kolb H, Mandrup-Poulsen T. The global diabetes epidemic as a consequence of lifestyle-induced low-grade inflammation. Diabetologia. 2010;53:10-20
35. Srikanthan K, Feyh A, Visweshwar H, Shapiro JI, Sodhi K. Systematic review of metabolic syndrome biomarkers: a panel for early detection, management, and risk stratification in the West Virginian population. Int J Med Sci. 2016;13:25-38
36. Cortez-Navarrete M, Martinez-Abundis E, Perez-Rubio KG, Gonzalez-Ortiz M, Mendez-Del Villar M. Momordica charantia administration Improves insulin secretion in type 2 diabetes mellitus. J Med Food. 2018;21:672-7
37. Habicht SD, Ludwig C, Yang RY, Krawinkel MB. Momordica charantia and type 2 diabetes: from in vitro to human studies. Curr Diabetes Rev. 2014;10:48-60
38. Sridhar MG, Vinayagamoorthi R, Arul Suyambunathan V, Bobby Z, Selvaraj N. Bitter gourd (Momordica charantia) improves insulin sensitivity by increasing skeletal muscle insulin-stimulated IRS-1 tyrosine phosphorylation in high-fat-fed rats. Br J Nutr. 2008;99:806-12
39. Bhat GA, Khan HA, Alhomida AS, Sharma P, Singh R, Paray BA. GLP-I secretion in healthy and diabetic Wistar rats in response to aqueous extract of Momordica charantia. BMC Complement Altern Med. 2018;18:162
40. Muller TD, Finan B, Bloom SR, D'Alessio D, Drucker DJ, Flatt PR. et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. 2019;30:72-130
41. Bortolotti M, Mercatelli D, Polito L. Momordica charantia, a nutraceutical approach for inflammatory related diseases. Front Pharmacol. 2019;10:486
42. Cheng HL, Yang MH, Anggriani R, Chang CI. Comparison of anti-inflammatory activities of structurally similar triterpenoids isolated from bitter melon. Nat Prod Commun. 2017;12:1847-50
43. Behrens M, Meyerhof W. Bitter taste receptor research comes of age: from characterization to modulation of TAS2Rs. Semin Cell Dev Biol. 2013;24:215-21
Author contact
![]() Corresponding authors: Hsueh-Ling Cheng, Professor, E-mail: hlchengnpust.edu.tw; Fax: 886-8-774-0550; Phone: +886-8-7703202 ext. 518; Address: Room BT212, Biotechnology Hall, National Pingtung University of Science and Technology, No. 1, Shuehfu Rd., Neipu Township, Pingtung 91201, Taiwan; Lin-Lee Lee, Professor, E-mail: linleeleeedu.tw; Phone: +886-7-7172930 ext. 2690; Address: Department of English, National Kaohsiung Normal University, No. 116, Heping 1st Road, Lingya District, Kaohsiung 80201, Taiwan.
Corresponding authors: Hsueh-Ling Cheng, Professor, E-mail: hlchengnpust.edu.tw; Fax: 886-8-774-0550; Phone: +886-8-7703202 ext. 518; Address: Room BT212, Biotechnology Hall, National Pingtung University of Science and Technology, No. 1, Shuehfu Rd., Neipu Township, Pingtung 91201, Taiwan; Lin-Lee Lee, Professor, E-mail: linleeleeedu.tw; Phone: +886-7-7172930 ext. 2690; Address: Department of English, National Kaohsiung Normal University, No. 116, Heping 1st Road, Lingya District, Kaohsiung 80201, Taiwan.