Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(15):2362-2372. doi:10.7150/ijms.47356 This issue Cite
Research Paper
Potential mechanism of RRM2 for promoting Cervical Cancer based on weighted gene co-expression network analysis
Jingtao Wang, Yuexiong Yi, Yurou Chen, Yao Xiong, Wei Zhang ![]()
Department of Obstetrics and Gynecology, Zhongnan Hospital of Wuhan University, Wuhan, Hubei 430071, P.R. China.
Received 2020-4-23; Accepted 2020-8-20; Published 2020-8-29
Abstract
Cervical cancer is the most common gynecologic malignant tumor, with a high incidence in 50-55-year-olds. This study aims to investigate the potential molecular mechanism of RRM2 for promoting the development of cervical cancer based on The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO). RRM2 was found to be significant upregulated in cervical tissue (P<0.05) by extracting the expression of RRM2 from TCGA, GSE63514, GSE7410, GSE7803 and GSE9750. Survival analysis indicated that the overall survival was significantly worse in the patients with high-expression of RRM2 (P<0.05). The top 1000 positively/negatively correlated genes with RRM2 by Pearson Correlation test were extracted. The gene co-expression network by Weighted Gene Co-Expression Network Analysis (WGCNA) with these genes and the clinical characteristics (lymphocyte infiltration, monocyte infiltration, necrosis, neutrophil infiltration, the number of normal/stromal/tumor cells and the number of tumor nuclei) was constructed. By screening the hub nodes from the co-expression network, results suggested that RRM2 may co-express with relevant genes to regulate the number of stromal/tumor cells and the process of lymphocyte infiltration to promote the progression of cervical cancer. RRM2 is likely to become a novel potential diagnostic and prognostic biomarker of cervical cancer and provide evidence to support the study of mechanisms for cervical cancer.
Keywords: Bioinformatics, Cervical cancer, Ribonucleotide reductase M2, Stromal cells, Tumor cells, Lymphocyte infiltration
Introduction
Cervical Cancer is one of the leading causes of cancer-related deaths in women, with an estimated 570,000 new cases and 311,000 deaths in 2018 worldwide [1]. About nine out of ten (87%) deaths related to cervical cancer occurred in developing counties [2]. The main risk factor for cervical cancer is human papillomavirus (HPV) infection, especially subtypes 16 (mostly associated with squamous cell carcinoma) and 18 (mostly associated with adenocarcinoma). Other predisposing factors include low socioeconomic status, early sexual activity, multiple sexual partners, immunosuppression and smoking [3]. Although most patients can be treated at an early stage, poor prognosis can still be identified in patients with metastasis or recurrence [4, 5]. Due to the high incidence and mortality of cervical cancer, uncovering the mechanisms of pathogenesis and identifying novel biomarkers is urgently needed.
The Ribonucleotide reductase subunit M2 (RRM2) gene is located on chromosome 2p25, a region with no copy-number variations (CNVs) or other structural alterations in human cervical cancer samples [6]. RRM2, as a component of ribonucleotide reductase (RNR), plays vital roles in many critical cellular processes such as cell proliferation, invasiveness, migration, angiogenesis and senescence [7]. It is frequently overexpressed and serves as an oncogene in various malignancies, including adrenocortical cancer [8], lung cancer [9], nasopharyngeal cancer [10], gastric cancer [11] and neuroblastoma [12], as well as being a predictor of poor prognosis [13-17]. Our previous study had indicated that RRM2 plays a critical role in the diagnosis and treatment of cervical cancer [18]. In addition, upregulation of RRM2 was found to lead to promotion of angiogenesis, whereas its downregulation significantly increased apoptosis and promoted cell cycle arrest in cervical cancer [6, 19]. However, the mechanisms showing how RRM2 exerted its active role in the progression of cervical cancer were still unclear.
In addition, our previous research has also shown that higher expression of RRM2 is associated with a significantly poorer overall survival [18]. However, relatively little is known about the role of RRM2 in cervical cancer and even less is known of the mechanisms responsible for its effects. Based on current outstanding research, we paid special attention and interest to the potential mechanism of RRM2 in cervical cancer. Therefore, for the main purpose of revealing the potential molecular mechanism of RRM2 for promoting the development of cervical cancer, we carried out our investigation based on The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO). Interestingly, we found that RRM2 may exert its promoted role in cervical cancer by reducing the number of stromal cells, increasing the number of tumor cells and promoting lymphocyte infiltration. Furthermore, our secondary objective was to determine whether RRM2 could be used as a biomarker for the prognosis of cervical cancer. In addition, the relationship between RRM2 and other biomarkers may provide more evidence for the underlying mechanisms of cervical cancer progression. The workflow of the present study is shown in Figure 1.
Workflow of present study for constructing gene co-expression network. TCGA, the Cancer Genome Atlas; GO, Gene Ontology; WGCNA, Weighted Gene Co-expression Network Analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes.
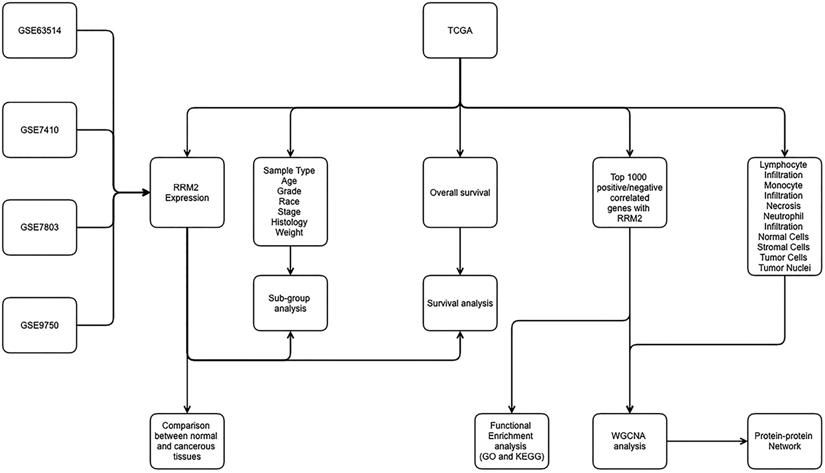
Materials and Methods
Raw data
The Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) is a public repository and distribution center for high-throughput microarray and sequence-based data [20]. The mRNA profiles of GSE63514, GSE7410, GSE7803 and GSE9750 were downloaded from the GEO. The characteristics of these four microarrays are shown in Table 1. In addition, the mRNA expression datasets for cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), which contain 304 cervical cancer samples with 3 normal samples, were also downloaded from The Cancer Genome Atlas (TCGA) using cBioPortal (http://www.cbioportal.org/) [21, 22].
Basic characteristics of the four microarray datasets from GEO
| Profile | RNA type | Platform | Organism | Sample size (T/N) | Published Year | Country |
|---|---|---|---|---|---|---|
| GSE63514 | mRNA | GPL570 | Homo sapiens | 28/24 | 2015 | USA |
| GSE7410 | mRNA | GPL1708 | Homo sapiens | 40/5 | 2008 | Netherlands |
| GSE7803 | mRNA | GPL96 | Homo sapiens | 3/10 | 2007 | USA |
| GSE9750 | mRNA | GPL96 | Homo sapiens | 42/24 | 2008 | USA |
T: Tumor; N: Normal;
GEO: Gene Expression Omnibus.
Expression distribution of RRM2 in cervical cancer tissues and patients' characteristics
The expression of RRM2 was extracted from TCGA, GSE63514, GSE7410, GSE7803 and GSE9750 to identify its regulation between cervical cancer and normal tissue. If several probes were mapped to a same gene, the mean value was utilized as the final expression of this gene [23]. Furthermore, the relationship between RRM2 expression and patients' characteristics was investigated through the UALCAN database, which contains TCGA level 3 RNA-seq and clinical data from 33 cancer types [24]. The comparison for each clinical characteristic between each classification in RRM2 expression was performed by student-t test. Specifically, the patients' characteristics included age, tumor grade, ethnic group, stages, histological type and weight. P<0.05 was considered as significant difference. The classification of body weight was defined as follow: (1) normal weight: patients with BMI ranging from 18 to 24; (2) extreme weight: BMI ranging from 25 to 29; (3) obese: BMI ranging from 30 to 39; (4) extreme obesity: BMI equal or above 40 [24].
Overall survival analysis of RRM2 in cervical cancer
The prognostic value of RRM2 in overall survival (OS) was assessed using the CESC data in TCGA. Due to the variability in the data, we separated the patients into RRM2 high and low groups according to the best cut-off value confirmed by the pROC package based on the RRM2 expression and the survival status [25]. The Cox proportional hazard model was used to compare the OS status by hazard ratio (HR) between high and low expressions of RRM2 [26]. The P value obtained from a log-rank test was used to indicate statistical significance of the survival correlation between groups. Additionally, P<0.05 was considered to indicate a statistically significant difference [27].
Co-expression analysis with RRM2
To identify the potential gene co-expressed with RRM2, the LinkedOmics database was applied to extract the top 1000 significant positively/negatively correlated genes with RRM2 [28]. The correlation between RRM2 and each other gene was examined using the Pearson correlation test one by one. P<0.05 was considered to be a significant correlation.
Functional and pathway enrichment analysis
Gene ontology (GO) and pathway analysis was carried out on the top 1000 positively/negatively co-expressed genes with RRM2 using the DAVID database (Database for Annotation, Visualization and Integrated Discovery; https://david.ncifcrf.gov/) [29]. The significantly enriched biological items for biological process (BP), cellular components (CC), and molecular functions (MF), as well as pathways, were identified with P<0.05. The significant GO items and pathways were visualized using the ggpubr package [30].
Weighted co-expression network construction and module selection
Weighted Gene Co-Expression Network Analysis (WGCNA) is a systems biology method for constructing relationship patterns that uses the soft threshold method to provide a more extensive and accurate correlation between transcripts compared to more general methods such as Pearson's correlation coefficient [31].
For constructing a co-expression module, we adopted the WGCNA package to analyze the relationship between the genes co-expressed with RRM2 and clinical characteristics, including lymphocyte infiltration, monocyte infiltration, necrosis, neutrophil infiltration, the number of normal/stromal/tumor cells and the number of tumor nuclei. The hclust function was utilized to perform sample cluster analysis on the expression data of each acquired gene in the samples and remove outlier samples. The pickSoftThreshold function was used to select the soft threshold. The soft threshold is a criterion based on approximate scale-free topology. The scale-free topological fitting exponential curve tends to be flat after reaching a higher value. We used the one-step method to construct the co-expression network and the blockwiseModules to identify co-expression modules. The minimum number of genes was set to 30 and the plotDendroAndColors function was applied to draw a dendrogram and to color each module for visualization.
For identifying the association between gene modules and clinical characteristics, we used gene significance (GS), defined as the correlation between genes and characteristics, to quantify the association between individual genes and the characteristics of interest. For each module, the quantitative value of module membership (MM) represented the degree of correlation between the modules and gene expression. The modules that are significant correlated with clinical characteristics by using GS and MM (P<0.05).
Protein-protein interaction network analysis
After identifying the significant modules associated with clinical characteristics, the genes in each module were extracted to construct a protein-protein interaction network (PPI) and the Cytoscape software was used for visualization. To detect the hub genes that related to RRM2, the degree, closeness and betweenness value of each node in the PPI was calculated using the Centiscape plugin [32, 33]. The nodes with degree, closeness and betweenness scores higher than the mean value were considered hub nodes.
Results
Expression distribution of RRM2 in cervical cancer tissues and patients' characteristics
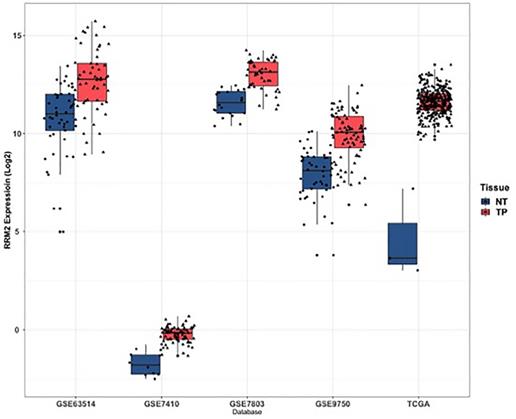
As shown in Figure 2, RRM2 was significantly upregulated in cervical cancer tissues when compared to normal tissues in TCGA, GSE63514, GSE7410, GSE7803 and GSE9750 (P<0.05). In terms of patients' characteristics, similar results were obtained with the expression of RRM2 significantly higher in cervical cancer patients than healthy people with respect to age, tumor grade, ethnic group, stages, histological type and weight (Figure S1; P<0.05). Specifically, the expression of RRM2 in extreme weight was higher than extreme obesity (Figure S1F; P=0.0224). However, no significant difference was detected in cervical cancer patients with respect to age, tumor grade, ethnic group, stages, or histological type (Figure S1A-S1E).
Expression of RRM2 in cervical cancer and normal tissues. RRM2 was significantly higher in cervical cancer tissues than in normal tissues (p < 0.05). NT, normal tissue; TP: primary tumor.
Overall survival analysis of RRM2 in cervical cancer
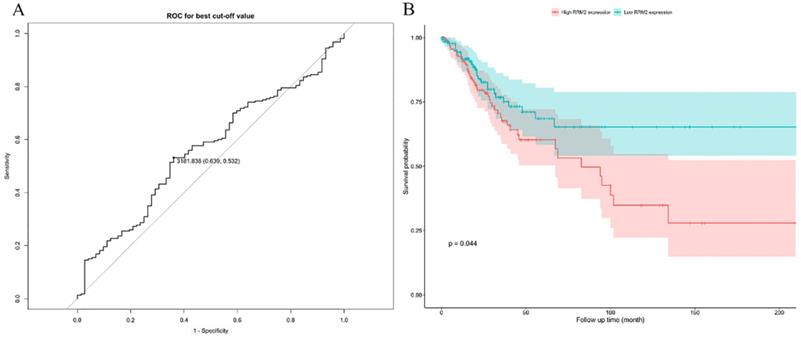
A log-rank test was applied to assess the OS in patients between high and low expressions of RRM2. The optimal cutoff value is 3181.838 (Figure 3A). The results indicated that cervical cancer patients with high expression showed a significantly poorer OS (HR: 1.621; 95% CI: 0.03834-0.9929; P=0.044) (Figure 3B).
The prognostic value of differently expressed RRM2 in cervical cancer patients in survival analysis. (A) The optimal cutoff value is 3181.838. (B) The cervical cancer patients with high expression revealed a significantly poorer OS (HR: 1.621, 95%CI: 0.03834-0.9929, P value: 0.044). HR, hazard ratio; CI, confidence interval; OS, overall survival.
Co-expression analysis with RRM2
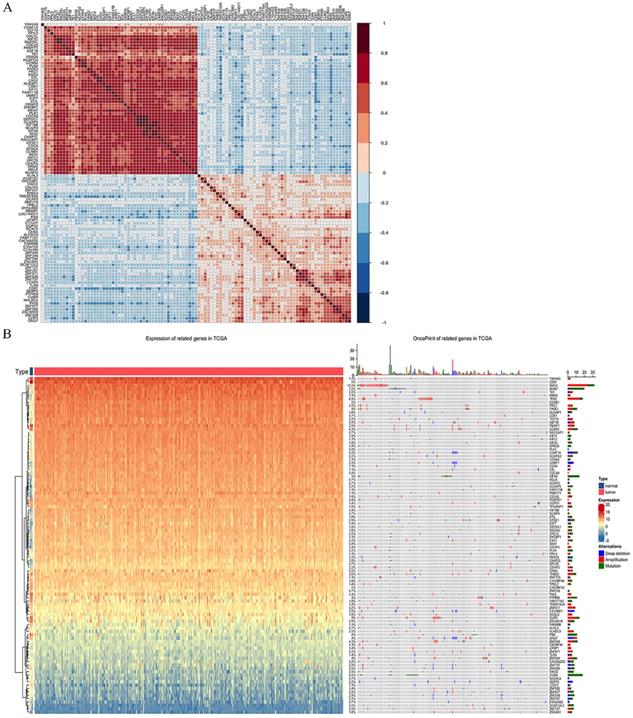
By extracting the top 1000 significant positively/negatively correlated genes with RRM2, a total of 2000 potential genes were acquired. Table 2 and 3 shows the top ten significant positively/negatively correlated genes. Of which, TCF19 (R = 0.539, P = 2.51*10-24), CDCA8 (R = 0.518, P = 2.53*10-22) and CENPO (R = 0.508, P = 4.63*10-21) were the top three positively correlated genes with RRM2, whereas PCMTD1 (R = -0.374, P = 1.69*10-11), TP53INP1 (R = -0.362, P = 7.19*10-11) and ZNF582 (R = -0.358, P = 1.32*10-10) represented the top three negatively correlated genes. Figure 4A shows the correlation matrix for the top 50 significant positively/negatively correlated genes, and Figure 4B shows their expression and oncoprint in TCGA.
Top 10 positively correlated genes with RRM2
| Gene | Cor. | P value | Sample size |
|---|---|---|---|
| TCF19 | 0.539 | 2.51e-24 | 304 |
| CDCA8 | 0.519 | 2.53e-22 | 304 |
| CENPO | 0.505 | 4.63e-21 | 304 |
| CCNA2 | 0.496 | 2.68e-20 | 304 |
| KIF2C | 0.493 | 4.81e-20 | 304 |
| KIF4A | 0.491 | 8.28e-20 | 304 |
| RACGAP1 | 0.488 | 1.41e-19 | 304 |
| ORC1L | 0.486 | 1.87e-19 | 304 |
| KIFC1 | 0.481 | 4.77e-19 | 304 |
| MELK | 0.474 | 1.97e-18 | 304 |
Cor.: Correlation,
Top 10 negatively correlated genes with RRM2
| Gene | Cor. | P value | Sample size |
|---|---|---|---|
| PCMTD1 | -0.374 | 1.69e-11 | 304 |
| TP53INP1 | -0.362 | 7.19e-11 | 304 |
| ZNF582 | -0.358 | 1.32e-10 | 304 |
| ZNF737 | -0.356 | 1.55e-10 | 304 |
| TMEM150A | -0.345 | 6.20e-10 | 304 |
| ZNF248 | -0.343 | 7.99e-10 | 304 |
| LOC144571 | -0.339 | 1.31e-09 | 304 |
| FAM171A1 | -0.337 | 1.57e-09 | 304 |
| ZNF626 | -0.335 | 2.05e-09 | 304 |
| RNF170 | -0.334 | 2.39e-09 | 304 |
Cor.: Correlation.
Co-expression analysis with RRM2. (A) Correlations between the top 50 positively/negatively correlated genes. Red represents positive correlation, and blue indicates negative correlation. (B) Expression between normal and cervical cancer tissue and oncoprint of the top 50 positively/negatively correlated genes in TCGA.
Functional and pathway enrichment analysis
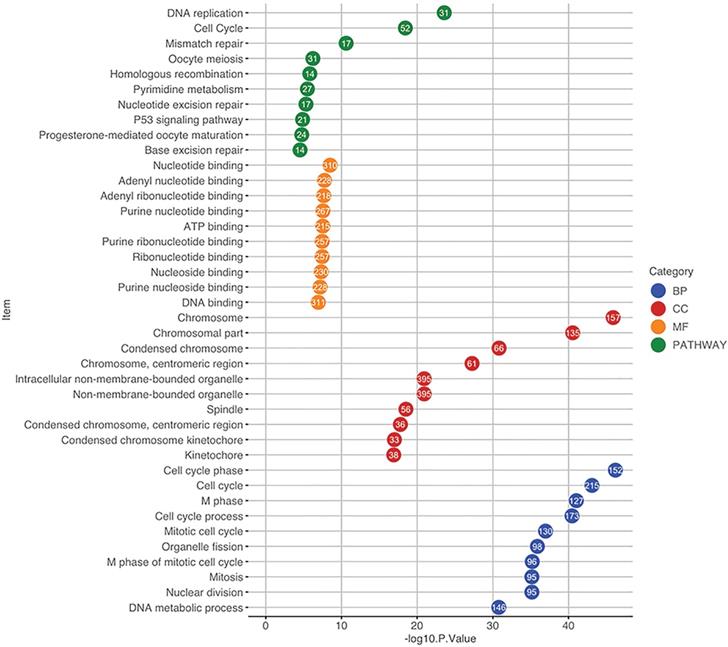
The 2000 significant correlated genes were used for GO and pathway enrichment analysis using the DAVID database. For GO analysis, when considering BP, these genes were mainly enriched in cell cycle, cell cycle phase and M phase. With regards to CC, the top three enriched items were chromosome, chromosomal part and centromeric region. In terms of MF, nucleotide binding, adenyl nucleotide binding and adenyl ribonucleotide binding were in the first three enriched places. Pathway enrichment analysis indicated that the top three significantly enriched pathways were DNA replication, cell cycle and mismatch repair (Figure 5).
Functional and pathway enrichment analysis using the DAVID database. The GO items and pathways were differentiated by colors. The position of a circle corresponds to the enrichment significance of each term and the number in the circle indicates the enriched gene count. GO, Gene Ontology; CC, cellular component; BP, biological process; MF, molecular function.
Weighted co-expression network construction and module selection
Using the expression data of the 2000 significant correlated genes in 307 samples in CESC, the WGCNA algorithm was used to generate the co-expression module. The hclust function was used to perform sample clustering analysis, and no outlier samples were found (Figure S2A). As topological analysis was required to construct the network, the soft threshold power value was set to 5 according to the scale-free network standard (Figure S2B). According to the determined power value, six effective modules with gene number more than 30 were detected with one gray module containing genes that cannot be clustered as a module (Figure S3).
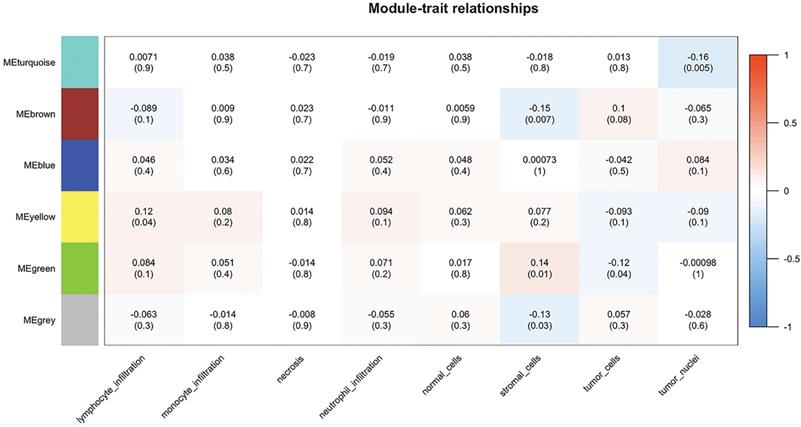
By importing the clinical characteristics including lymphocyte infiltration, monocyte infiltration, necrosis, neutrophil infiltration, the number of normal/stromal/tumor cells and the number of tumor nuclei, module trait association analysis was performed to identify modules that were significantly related to clinical traits. The yellow module was found to positively correlate with lymphocyte infiltration (R = 0.12, P = 0.04), whereas the brown module was negatively correlated with the number of stromal cells (R = -0.15, P = 0.007). In addition, the green module was negatively correlated with the number of stromal cells (R = 0.14, P = 0.01) and the number of tumor cells (R = -0.12, P = 0.04), and the turquoise obtained similar results for the number of tumor nuclei (R = -0.16, P = 0.005) (Figure 6).
Module-patient trait associations. The relationship between modules and patient traits was analyzed using WGCNA. Each row corresponds to a module eigengene, each column to a trait. Each cell contains the corresponding correlation and p-value. The table is color-coded by correlation according to the color legend. WGCNA, Weighted Gene Co-expression Network Analysis; ME, module eigengene.
Protein-protein interaction network analysis
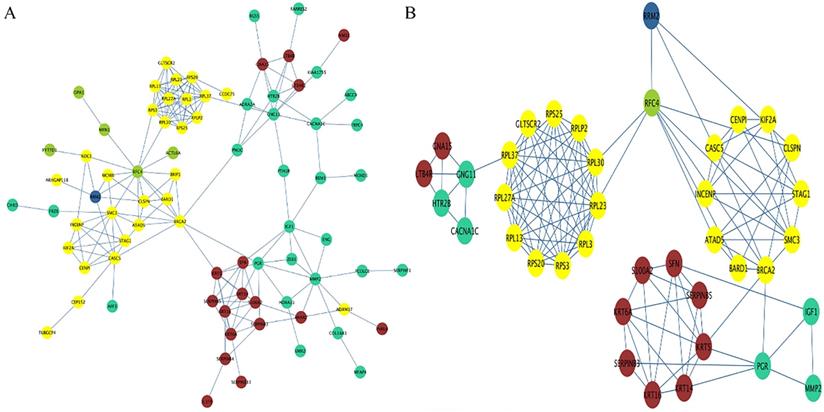
By extracting the genes in each identified module, a preliminary PPI with 79 nodes and 190 edges was constructed, with the color of each node representing their corresponding module (Figure 7A). Using degree, closeness and betweenness scores of nodes higher than the mean value, a subnetwork containing 39 nodes and 125 edges was extracted (Figure 7B). Combining the subnetwork with the result of module trait association analysis, the possible mechanisms for RRM2 to promote the progression of cervical cancer formed as follows. RRM2 may co-express with RFC44 (correlated with stromal cells and tumor cells) to increase the number of tumor cells and reduce the number of stromal cells, and subsequently regulate the expression of the RPL family (RPL30, RPL23 and RPL3 et al.) or CENPI, KIF2A and CASCS (correlated with lymphocyte infiltration) to promote lymphocyte infiltration. After the lymphocyte infiltration, the expression of CNG11 or PGR (correlated with tumor nuclei) would be regulated to enhance the malignant behavior by increasing the number of tumor nuclei, and further regulate the expression of GNA15/LTB4R or SFN/KRT14 (correlated with stromal cells) to reduce the number of stromal cells. In another route, RRM2 may also directly interact with the genes in the yellow module to exert a promoted effect on it. These processes may be structured as a vicious circle.
PPI network. (A) The preliminary PPI network of these genes in each identified module. The color of each node represents its corresponding module. (B) The subnetwork containing 39 nodes and 125 edges was extracted based on the criteria of degree, closeness and betweenness scores of nodes being higher than the mean value. RRM2 may co-express with RFC44 to increase the number of tumor cells and reduce the number of stromal cells, and subsequently regulate the expression of the RPL family (RPL30, RPL23 and RPL3 et al.) or CENPI, KIF2A and CASCS to promote lymphocyte infiltration. After the lymphocyte infiltration, CNG11 or PGR would be regulated to enhance the malignant behavior by increasing the number of tumor nuclei, and further regulate the expression of GNA15/LTB4R or SFN/KRT14 to reduce the number of stromal cells. In another route, RRM2 may also directly interact with the genes in the yellow module to exert a promoted effect on it. Circles represent genes; lines represent interactions between these selected genes. PPI, protein-protein interaction.
Discussion
Cervical cancer is the third most commonly diagnosed gynecological malignant tumor and the fourth major cause of cancer‐related death, with especially high morbidity in developing countries [34]. Although the etiological role of HPV in the development of cervical cancer has been confirmed [35], the specific molecular mechanism remains unclear. Previous study had revealed that RRM2 was a downstream target for the HPV E7 gene, and that HPV E7 can induce upregulation of RRM2 and then promotes cervical carcinogenesis via ROS-ERK1/2-HIF-1α-VEGF-induced angiogenesis [6]. To date, RRM2 had been reported as having an important impact on cervical cancer [18, 36, 37]. Furthermore, several non-coding RNA, such as long non-coding RNA (LncRNA) or micro RNA (miRNA), regulate the occurrence and development of cervical cancer through RRM2 [38, 39]. However, the mechanisms showing how RRM2 promote cervical cancer are still unclear. In the present study, we investigated the promotion role of RRM2 for cervical cancer using WGCNA.
Our results indicated that RRM2 is upregulated and significantly associated with poor prognosis in cervical cancer, and these results are in line with previous studies [18, 19, 36, 40]. With 192 samples (29 normal, 30 low-grade dysplasia, 30 high-grade dysplasia and 103 invasive cancer tissue specimens) and SiHa cervical cancer cells, Su et al. reported the expression of RRM2 in cancer tissues was increased when compared to high-grade dysplasia, low-grade dysplasia or normal cervical tissues [40]. Furthermore, higher expression of RRM2 was significantly correlated with worse survival. Wang et al. demonstrated that the RNA levels of RRM2 in cervical cancer tissues were significantly higher than paracarcinoma tissues by analyzing 38 pairs of cervical cancer and normal cervical tissues [19]. They also found that downregulated RRM2 promoted apoptosis and cell cycle arrest, as well as inhibiting tumor formation. In addition, bioinformatics analyses also indicated that OS was significantly better in the low-expression RRM2 group [18, 36].
The results of the present study found that RRM2 may exert its promoted effect in cervical cancer by reducing the number of stromal cells, increasing the number of tumor cells and promoting lymphocyte infiltration. At present, the study of mechanisms for RRM2 in cervical cancer is limited. Su et al. reported that RRM2 upregulation was significantly correlated with deep stromal invasion and parametrial invasion [40]. Furthermore, stromal invasion is considered as a significant predictive factor of OS for cervical cancer [41-43]. In other malignancies, Sun et al. reported that RRM2 was a positive regulator of glioma progression and it contributed to the migration and proliferation of glioma cells [44]. Zhang et al. found that overexpression of RRM2 promoted the growth of tumor cells, as well as positively affecting the overall angiogenic activity of tumor cells, in oropharyngeal carcinoma [45]. Li et al. presented similar results that increased expression of RRM2 is essential for cell proliferation in glioblastoma, while knockdown of RRM2 led to cancer cells arrested at the G1 phase and promoted apoptosis to inhibit cell growth [46]. Unfortunately, the study of the relationship between RRM2 and lymphocyte infiltration is also limited. Lu et al. reported that RRM2 may play an important role in the infiltration and metastasis of colorectal cancer by analyzing 56 pairs of cancerous and normal tissues [47]. Nevertheless, RRM2 still had potential association with the regulation of the stromal cells, tumor cells and lymphocyte infiltration.
In addition, there are some genes and proteins that have been associated with the effects and mechanisms of RRM2 in cervical cancer progression. As a tumor inhibitor, p53 plays a pivotal role in cervical cancer [48], He et al. indicate that both RRM1 and RRM2 were positively controlled by mTOR signaling while suppressed by p53 signaling [49]. The HPV E7 protein targets and inactivates the retinoblastoma protein (Rb), resulting in the release of active E2F and the activation of E2F target genes [50]. Research found that RRM2 was a novel downstream target activated by HPV E7, and promoted angiogenesis by producing ROS, activating the ERK1/2 signaling pathway and inducing expression of HIF-1a and VEGF, leading to cervical angiogenesis and carcinogenesis [6]. At present, evidence of the relationship between RRM2 and CDKN3 in cervical cancer is limited, but their relationship can be found in other cancers, such as hepatocellular carcinoma, breast cancer, renal carcinoma and lung cancer [51-54]. Qi et al. have found RRM2 and CDKN3 may become biomarkers for diagnosis and poor prognosis of breast cancer [51]. Zhou et al. have also found there is a correlation between RRM2 and CDKN3 in hepatocellular carcinoma [52]. Wei et al. found RRM2 and CDKN3 associated with the metastasis of Clear cell renal cell carcinoma [53]. MacDermed et al. have also found their relationship in lung adenocarcinoma [54]. Therefore, we speculate that there is an interaction between RRM2 and CDKN3 in cervical cancer, which needs to be confirmed by further study [55].
Our studies also had limitations. First, we selected the top 1000 positively/negatively correlated genes with RRM2 for WGCNA, and so the results may change if we enlarge the number of selected genes. Second, the hub nodes were identified based on the topological parameters such as degree, closeness and betweenness. Applying another algorithm may also lead to the alteration of the results. Third, as the present study is an in-silico study, the results need to be further validated.
Conclusions
In conclusion, the expression of RRM2 is upregulated in cervical cancer and promotes cancerous progression. RRM2 may exert its promoted role in cervical cancer by reducing the number of stromal cells, increasing the number of tumor cells and promoting lymphocyte infiltration. It is likely to become a novel potential diagnostic and prognostic biomarker of cervical cancer.
Supplementary Material
Supplementary figures.
Acknowledgements
Availability of data and materials
The datasets used and/or analyzed during the current study are available in TCGA and GEO.
Author contributions
JW and YY established the concept of the present study and drafted the paper. YC and YX performed the literature research, data extraction and statistical analysis. WZ contributed to the literature research, data extraction and analysis, and reviewed and approved the final version of the paper. All authors have read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68:394-424
2. Parra S, Oden M, Schmeler K, Richards-Kortum R. Low-Cost Instructional Apparatus to Improve Training for Cervical Cancer Screening and Prevention. Obstetrics and gynecology. 2019;133:559-67
3. Pimple SA, Mishra GA. Global strategies for cervical cancer prevention and screening. Minerva ginecologica. 2019;71:313-20
4. Forouzanfar MH, Foreman KJ, Delossantos AM, Lozano R, Lopez AD, Murray CJ. et al. Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet (London, England). 2011;378:1461-84
5. Maguire R, Kotronoulas G, Simpson M, Paterson C. A systematic review of the supportive care needs of women living with and beyond cervical cancer. Gynecologic oncology. 2015;136:478-90
6. Wang N, Zhan T, Ke T, Huang X, Ke D, Wang Q. et al. Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. British journal of cancer. 2014;110:1034-44
7. Nordlund P, Reichard P. Ribonucleotide reductases. Annual review of biochemistry. 2006;75:681-706
8. Grolmusz VK, Karaszi K, Micsik T, Toth EA, Meszaros K, Karvaly G. et al. Cell cycle dependent RRM2 may serve as proliferation marker and pharmaceutical target in adrenocortical cancer. American journal of cancer research. 2016;6:2041-53
9. Grossi F, Dal Bello MG, Salvi S, Puzone R, Pfeffer U, Fontana V. et al. Expression of Ribonucleotide Reductase Subunit-2 and Thymidylate Synthase Correlates with Poor Prognosis in Patients with Resected Stages I-III Non-Small Cell Lung Cancer. Disease markers. 2015;2015:302649
10. Han P, Chen RH, Wang F, Zeng JY, Yu ST, Xu LH. et al. Novel chimeric transcript RRM2-c2orf48 promotes metastasis in nasopharyngeal carcinoma. Cell death & disease. 2017;8:e3047
11. Zhong Z, Cao Y, Yang S, Zhang S. Overexpression of RRM2 in gastric cancer cell promotes their invasiveness via AKT/NF-kappaB signaling pathway. Die Pharmazie. 2016;71:280-4
12. Li J, Pang J, Liu Y, Zhang J, Zhang C, Shen G. et al. Suppression of RRM2 inhibits cell proliferation, causes cell cycle arrest and promotes the apoptosis of human neuroblastoma cells and in human neuroblastoma RRM2 is suppressed following chemotherapy. Oncology reports. 2018;40:355-60
13. Dai L, Lin Z, Qiao J, Chen Y, Flemington EK, Qin Z. Ribonucleotide reductase represents a novel therapeutic target in primary effusion lymphoma. Oncogene. 2017;36:5068-74
14. D'Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y. et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023-34
15. Kang W, Tong JH, Chan AW, Zhao J, Wang S, Dong Y. et al. Targeting ribonucleotide reductase M2 subunit by small interfering RNA exerts anti-oncogenic effects in gastric adenocarcinoma. Oncology reports. 2014;31:2579-86
16. Morikawa T, Maeda D, Kume H, Homma Y, Fukayama M. Ribonucleotide reductase M2 subunit is a novel diagnostic marker and a potential therapeutic target in bladder cancer. Histopathology. 2010;57:885-92
17. Shah KN, Wilson EA, Malla R, Elford HL, Faridi JS. Targeting Ribonucleotide Reductase M2 and NF-kappaB Activation with Didox to Circumvent Tamoxifen Resistance in Breast Cancer. Molecular cancer therapeutics. 2015;14:2411-21
18. Yi Y, Liu Y, Wu W, Wu K, Zhang W. Reconstruction and analysis of circRNAmiRNAmRNA network in the pathology of cervical cancer. Oncology reports. 2019;41:2209-25
19. Wang N, Li Y, Zhou J. Downregulation of ribonucleotide reductase subunits M2 induces apoptosis and G1 arrest of cervical cancer cells. Oncology letters. 2018;15:3719-25
20. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M. et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic acids research. 2013;41:D991-5
21. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1
22. Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemporary oncology (Poznan, Poland). 2015;19:A68-77
23. Xia L, Su X, Shen J, Meng Q, Yan J, Zhang C. et al. ANLN functions as a key candidate gene in cervical cancer as determined by integrated bioinformatic analysis. Cancer management and research. 2018;10:663-70
24. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B. et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia (New York, NY). 2017;19:649-58
25. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC bioinformatics. 2011;12:77
26. Moolgavkar SH, Chang ET, Watson HN, Lau EC. An Assessment of the Cox Proportional Hazards Regression Model for Epidemiologic Studies. Risk analysis: an official publication of the Society for Risk Analysis. 2018;38:777-94
27. George B, Seals S, Aban I. Survival analysis and regression models. Journal of nuclear cardiology: official publication of the American Society of Nuclear Cardiology. 2014;21:686-94
28. Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic acids research. 2018;46:D956-d63
29. Jiao X, Sherman BT, Huang da W, Stephens R, Baseler MW, Lane HC. et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics (Oxford, England). 2012;28:1805-6
30. Whitehead MJ, McCanney GA, Willison HJ, Barnett SC. MyelinJ: an ImageJ macro for high throughput analysis of myelinating cultures. Bioinformatics (Oxford, England). 2019;35:4528-30
31. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics. 2008;9:559
32. Scardoni G, Tosadori G, Faizan M, Spoto F, Fabbri F, Laudanna C. Biological network analysis with CentiScaPe: centralities and experimental dataset integration. F1000Research. 2014;3:139
33. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498-504
34. Arbyn M, Weiderpass E, Bruni L, de Sanjose S, Saraiya M, Ferlay J. et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. The Lancet Global health. 2020;8:e191-e203
35. Rong D, Sun H, Li Z, Liu S, Dong C, Fu K. et al. An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget. 2017;8:73271-81
36. Tang X, Xu Y, Lu L, Jiao Y, Liu J, Wang L. et al. Identification of key candidate genes and small molecule drugs in cervical cancer by bioinformatics strategy. Cancer management and research. 2018;10:3533-49
37. Xue JM, Liu Y, Wan LH, Zhu YX. Comprehensive Analysis of Differential Gene Expression to Identify Common Gene Signatures in Multiple Cancers. Medical science monitor: international medical journal of experimental and clinical research. 2020;26:e919953
38. Ma J, Zhang F, Sun P. miR-140-3p impedes the proliferation of human cervical cancer cells by targeting RRM2 to induce cell-cycle arrest and early apoptosis. Bioorganic & medicinal chemistry. 2020;28:115283
39. Zhao H, Zheng GH, Li GC, Xin L, Wang YS, Chen Y. et al. Long noncoding RNA LINC00958 regulates cell sensitivity to radiotherapy through RRM2 by binding to microRNA-5095 in cervical cancer. Journal of cellular physiology. 2019;234:23349-59
40. Su YF, Wu TF, Ko JL, Tsai HT, Tee YT, Chien MH. et al. The expression of ribonucleotide reductase M2 in the carcinogenesis of uterine cervix and its relationship with clinicopathological characteristics and prognosis of cancer patients. PloS one. 2014;9:e91644
41. Grisaru DA, Covens A, Franssen E, Chapman W, Shaw P, Colgan T. et al. Histopathologic score predicts recurrence free survival after radical surgery in patients with stage IA2-IB1-2 cervical carcinoma. Cancer. 2003;97:1904-8
42. Kristensen GB, Abeler VM, Risberg B, Trop C, Bryne M. Tumor size, depth of invasion, and grading of the invasive tumor front are the main prognostic factors in early squamous cell cervical carcinoma. Gynecologic oncology. 1999;74:245-51
43. Sapp H, Goetzl L, Creasman W, Kohler M, Underwood P, Esnaola N. Combined adverse effect of African American race and deep stromal invasion on survival following radical hysterectomy for cervical cancer. American journal of obstetrics and gynecology. 2008;199:196.e1-7
44. Sun H, Yang B, Zhang H, Song J, Zhang Y, Xing J. et al. RRM2 is a potential prognostic biomarker with functional significance in glioma. International journal of biological sciences. 2019;15:533-43
45. Zhang K, Hu S, Wu J, Chen L, Lu J, Wang X. et al. Overexpression of RRM2 decreases thrombspondin-1 and increases VEGF production in human cancer cells in vitro and in vivo: implication of RRM2 in angiogenesis. Molecular cancer. 2009;8:11
46. Li YF, Wang ZY, Tang LY, Hu RH, Huang L, Ding JW. RRM2 overexpression in glioblastoma enhances the proliferation and invasion of cancer cells. International Journal of Clinical and Experimental Pathology. 2016;9:11623-30
47. Lu AG, Feng H, Wang PX, Han DP, Chen XH, Zheng MH. Emerging roles of the ribonucleotide reductase M2 in colorectal cancer and ultraviolet-induced DNA damage repair. World journal of gastroenterology. 2012;18:4704-13
48. Liu Y, Li L, Liu Y, Geng P, Li G, Yang Y. et al. RECK inhibits cervical cancer cell migration and invasion by promoting p53 signaling pathway. Journal of cellular biochemistry. 2018;119:3058-66
49. He Z, Hu X, Liu W, Dorrance A, Garzon R, Houghton PJ. et al. P53 suppresses ribonucleotide reductase via inhibiting mTORC1. Oncotarget. 2017;8:41422-31
50. Huh K, Zhou X, Hayakawa H, Cho JY, Libermann TA, Jin J. et al. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. Journal of virology. 2007;81:9737-47
51. Qi L, Zhou B, Chen J, Hu W, Bai R, Ye C. et al. Significant prognostic values of differentially expressed-aberrantly methylated hub genes in breast cancer. Journal of Cancer. 2019;10:6618-34
52. Zhou Z, Li Y, Hao H, Wang Y, Zhou Z, Wang Z. et al. Screening Hub Genes as Prognostic Biomarkers of Hepatocellular Carcinoma by Bioinformatics Analysis. Cell transplantation. 2019;28:76s-86s
53. Wei W, Lv Y, Gan Z, Zhang Y, Han X, Xu Z. Identification of key genes involved in the metastasis of clear cell renal cell carcinoma. Oncology letters. 2019;17:4321-8
54. MacDermed DM, Khodarev NN, Pitroda SP, Edwards DC, Pelizzari CA, Huang L. et al. MUC1-associated proliferation signature predicts outcomes in lung adenocarcinoma patients. BMC medical genomics. 2010;3:16
55. Chen J, Rajasekaran M, Xia H, Zhang X, Kong SN, Sekar K. et al. The microtubule-associated protein PRC1 promotes early recurrence of hepatocellular carcinoma in association with the Wnt/β-catenin signalling pathway. Gut. 2016;65:1522-34
Author contact
![]() Corresponding author: Wei Zhang, E-mail: zw6676com; Tel.: (+86) 13035106467.
Corresponding author: Wei Zhang, E-mail: zw6676com; Tel.: (+86) 13035106467.