Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials & Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(14):2063-2076. doi:10.7150/ijms.48244 This issue Cite
Research Paper
Identification of Key Genes and Pathways in Myeloma side population cells by Bioinformatics Analysis
Qin Yang1, Kaihu Li2, Xin Li1 ![]() , Jing Liu1
, Jing Liu1 ![]()
1. Department of Hematology, the Third Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China.
2. Department of Orthopaedics, Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China.
*These authors have contributed equally to this work.
Received 2020-5-15; Accepted 2020-7-12; Published 2020-7-25
Abstract
Background: Multiple myeloma (MM) is the second most common hematological malignancy, which is still incurable and relapses inevitably, highlighting further understanding of the possible mechanisms. Side population (SP) cells are a group of enriched progenitor cells showing stem-like phenotypes with a distinct low-staining pattern with Hoechst 33342. Compared to main population (MP) cells, the underlying molecular characteristics of SP cells remain largely unclear. This bioinformatics analysis aimed to identify key genes and pathways in myeloma SP cells to provide novel biomarkers, predict MM prognosis and advance potential therapeutic targets.
Methods: The gene expression profile GSE109651 was obtained from Gene Expression Omnibus database, and then differentially expressed genes (DEGs) with P-value <0.05 and |log2 fold-change (FC)| > 2 were selected by the comparison of myeloma light-chain (LC) restricted SP (LC/SP) cells and MP CD138+ cells. Subsequently, gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis, protein-protein interaction (PPI) network analysis were performed to identify the functional enrichment analysis of the DEGs and screen hub genes. Cox proportional hazards regression was used to select the potential prognostic DEGs in training dataset (GSE2658). The prognostic value of the potential prognostic genes was evaluated by Kaplan-Meier curve and validated in another external dataset (MMRF-CoMMpass cohort from TCGA).
Results: Altogether, 403 up-regulated and 393 down-regulated DEGs were identified. GO analysis showed that the up-regulated DEGs were significantly enriched in innate immune response, inflammatory response, plasma membrane and integral component of membrane, while the down-regulated DEGs were mainly involved in protoporphyrinogen IX and heme biosynthetic process, hemoglobin complex and erythrocyte differentiation. KEGG pathway analysis suggested that the DEGs were significantly enriched in osteoclast differentiation, porphyrin and chlorophyll metabolism and cytokine-cytokine receptor interaction. The top 10 hub genes, identified by the plug-in cytoHubba of the Cytoscape software using maximal clique centrality (MCC) algorithm, were ITGAM, MMP9, ITGB2, FPR2, C3AR1, CXCL1, CYBB, LILRB2, HP and FCER1G. Modules and corresponding GO enrichment analysis indicated that myeloma LC/SP cells were significantly associated with immune system, immune response and cell cycle. The predictive value of the prognostic model including TFF3, EPDR1, MACROD1, ARHGEF12, AMMECR1, NFATC2, HES6, PLEK2 and SNCA was identified, and validated in another external dataset (MMRF-CoMMpass cohort from TCGA).
Conclusions: In conclusion, this study provides reliable molecular biomarkers for screening, prognosis, as well as novel therapeutic targets for myeloma LC/SP cells.
Keywords: Multiple myeloma, Cancer stem cell, Side population cells, Bioinformatics analysis, differentially expressed gene
Introduction
Multiple myeloma (MM) is a B-cell malignancy characterized by the aberrant expansion of clonal plasma cells within bone marrow, which is the second most common hematological malignancy [1]. Despite remarkable progress of biology and recent development of novel therapy [2, 3], MM continues to remain incurable due to the emergence of drug resistance and frequent relapses, highlighting the further understanding of the possible mechanisms.
Cancer stem cells (CSCs) are thought to have the distinctive properties of constituting a small fraction of tumor cells with self-renewal capacity and be able to propagate the disease [4, 5]. Besides, CSCs are considered to be more resistant to chemo- and radio-therapy and have better DNA repair mechanisms and increased anti-apoptotic activity, just like hematopoietic stem cells [6]. Previously, CSCs have been identified in MM [7] by the evidence that the CD138-/CD19+ fraction of MM has a greater clonogenic potential and the phenotype of a memory B-cell (CD19+, CD27+), resulting in the development of refractory clones and disease relapse[8]. Then, it has been defined that possible stem cell populations include light-chain restricted cells with a CD138-/CD19+/CD27+ phenotype [7, 9, 10], CD138+/CD34+/B7-H1+ subpopulations [11] and CD38++/CD45- plasma cells [12, 13]. Despite these phenotypes have been described, the distinct CSCs marker in MM is still controversial.
Side population (SP) cells, first described by Goodell et al. [14], are a group of enriched progenitor cells showing stem-like phenotypes and a distinct low-staining pattern with Hoechst 33342, and have been widely used as a unique source for studying CSCs in the absence of specific markers [4, 15-21]. Although lots of previous studies have explored the stem-like properties and tumorigenicity of myeloma SP cells, a better understanding of SP cells still remains largely unclear [10, 22-25]. Thus, it is vital to elucidate the key molecular characteristics expressed within myeloma SP cells.
It is generally known that gene expression profiling analysis based on microarray technology enables the possibilities for identifying certain disease-related biomarkers. Recently, many studies have been carried out on the base of microarray data profiles to identify the pathogenesis of MM [26-28]. Nevertheless, the key molecular characteristics of myeloma SP cells in comparison to MP cells have not yet been explored. This bioinformatics analysis was performed to elucidate key candidate genes and pathways in myeloma SP cells, provide novel biomarkers, predict MM prognosis and advance potential therapeutic targets.
In this study, we downloaded microarray dataset GSE109651 (Zhan et al., 2018) from Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/), which is a public functional genomics data repository with array- and sequence-based data. By comparing myeloma light-chain (LC) restricted SP (LC/SP) cells with myeloma MP cells based on R software and Bioconductor, differentially expressed genes (DEGs) were identified. Gene Ontology (GO) analysis, Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis and protein-protein interaction (PPI) network analysis were performed to identify the functional enrichment analysis of the DEGs and screen hub genes. Subsequently, we constructed a prognostic model to predict survivals of MM patients. This study provides reliable molecular biomarkers for screening, prognosis, as well as novel therapeutic targets for LC/SP cells of MM.
Materials & Methods
Microarray data profile
The gene expression dataset GSE109651 was obtained from GEO database. The microarray data of GSE109651, based on the GPL570 platform ([HG-U133_Plus_2] Affymetrix Human Genome U133A Plus 2.0 Array) and normalized using the MAS5 algorithm of the Affymetrix expression console version1.1 software (Affymetrix), includes 7-paired LC/SP cells and MP CD138+ cells of myeloma bone marrow from 7 diagnosed MM patients isolated by fluorescence-activated cell sorting (FACS) using Hoechst 33342 and CD138 antibody. To perform survival analysis, GSE2658 dataset of 559 MM patients and TCGA MM RNA sequencing dataset (MMRF-CoMMpass) of 787 cases with MM including clinicopathological information were downloaded from GEO database and TCGA (https://tcga-data.nci.nih.gov/) databases, respectively. For the retrospective cohort, the patients' characteristics were estimated by Pearson test χ2 or Fisher's exact test, indicating no significant statistical difference.
Data processing and identification of DEGs of GSE109651
Firstly, we detected the quality of raw data by R statistical software (version 3.6.3, https://www.r-project.org/), including a quality control overview diagram based on the “simpleaffy” package, weights and residuals plot, relative log expression (RLE) boxplot and normalized unscaled standard errors (NUSE) box plot based on the "affyPLM" and "RColorBrewer" packages, RNA degradation curve based on the "affy" package and clustering analysis diagram based on the "gcrma", "graph" and "affycoretools" packages.
Then, DEGs between LC/SP cells and MP CD138+ cells of MM were identified by an empirical Bayes method based on the “limma” package in R. The process included six main steps: construction of a gene expression matrix, construction of an experimental design matrix, construction of a contrast matrix, fitting of a linear model, Bayes test, and generation of results. In this study, genes with P-value < 0.05 and |log2 fold-change (FC)| > 2 were defined as DEGs.
GO and KEGG pathway enrichment analysis of DEGs
To explore the functional roles of the above DEGs, DAVID database (https://david.ncifcrf.gov/) was used to perform GO term enrichment analysis of molecular function (MF), biological process (BP), and cellular component (CC) and KEGG pathway enrichment analysis. P-value < 0.05 was considered as the cut-off criterion.
PPI network construction and modular analysis
Search Tool for the Retrieval of Interacting Genes (STRING) database (http://www.string-db.org/) was used to construct the PPI network. The visualization and analysis of the PPI network were based on Cytoscape software version 3.7.2. Then, the plug-ins Molecular Complex Detection (MCODE) and Biological Network Gene Ontology tool (BiNGO) in Cytoscape software were used to screen significant modules of the PPI network (the parameters were set to default) and perform GO analysis that the module genes were significantly enriched in.
Identification of hub genes
The plug-in cytoHubba in Cytoscape was used to identify key (hub) genes among the above DEGs by maximal clique centrality (MCC) computing method. The hub genes were selected to discuss their function and effect on myeloma LC/SP cells.
Survival analysis
DEGs significantly associated with myeloma-specific survival in the training dataset (GSE2658) were identified using univariate Cox proportional hazards analysis with P-value < 0.01 by “survival” package [29]. Then, the final genes significantly correlated with survival at a P-value of less than 0.05 were identified by multivariate Cox proportional hazards analysis. Subsequently, the risk score on the base of the aforementioned candidate genes and survival information was calculated as follows: Risk score = ∑β i × ExpGene i (β i was the coefficient value and ExpGene i was the gene expression level). According to the median risk score, the cohort was dichotomized into low-risk and high-risk group, then survival time was compared by the Kaplan-Meier analysis and the log-rank test with a P-value of less than 0.01. Another external dataset (MMRF-CoMMpass cohort) was used to assess the prognostic value through a process similar to the training dataset.
Results
Identification of DEGs
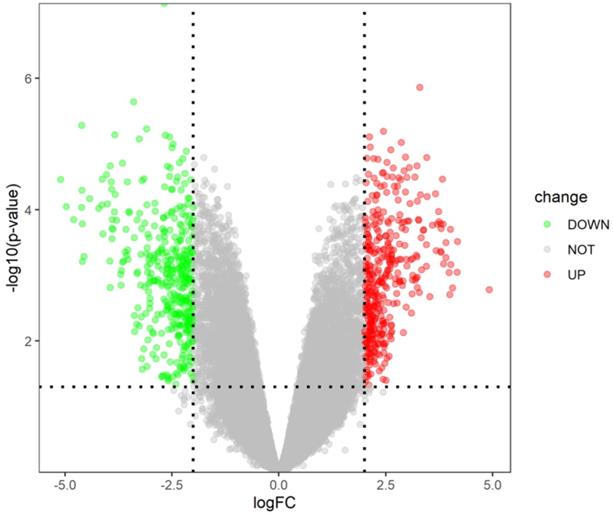
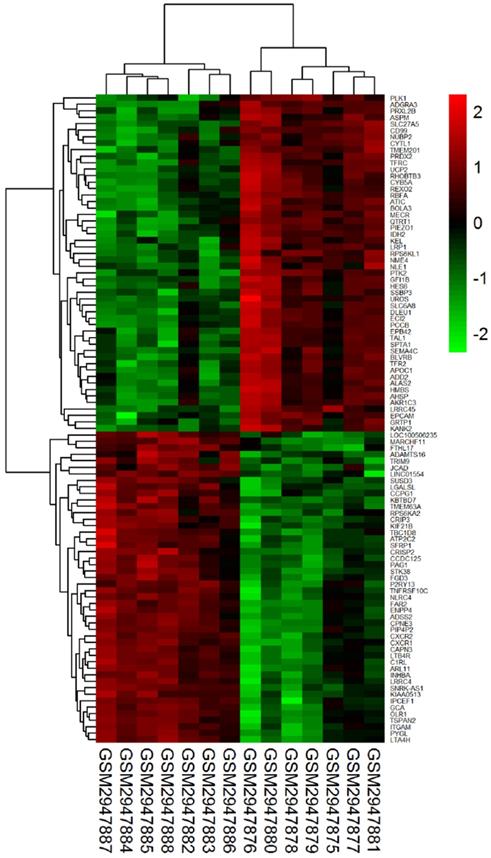
The gene expression dataset GSE109651 included 7-paired LC/SP cells samples and MP CD138+ cells samples of myeloma bone marrow. On the basis of cut-off criterion of DEGs described previously, there were 796 DEGs in LC/SP cells compared with MP CD138+ cells of myeloma bone marrow, among which 393 DEGs were significantly down-regulated and 403 DGEs were significantly up-regulated. The volcano plot of DEGs was shown in Figure 1. The expression heat map of the top 100 DEGs (including 52 significantly down-regulated genes and 48 significantly up-regulated genes) was depicted in Figure 2, which could effectively distinguish LC/SP cells from MP CD138+ cells and might function as biomarker and target of MM. The detailed information of the top 10 DEGs was shown in Table 1.
Volcano plot of DEGs (393 down-regulated genes and 403 up-regulated genes).
Heat map of the top 100 DEGs (52 down-regulated genes and 48 up-regulated genes).
Detailed information on the top 10 DEGs in the analysis
| Symbol | Name | AveExpr | t | P-Value | adj.P-Val | B | logFC |
|---|---|---|---|---|---|---|---|
| SLC27A5 | solute carrier family 27 member 5 | 6.641 | -9.704 | 7.23E-08 | 1.46E-03 | 7.969 | -2.685 |
| MARCHF11 | membrane associated ring-CH-type finger 11 | 4.541 | 7.686 | 1.38E-06 | 1.16E-02 | 5.451 | 3.293 |
| RBFA | ribosome binding factor A | 6.812 | -7.379 | 2.26E-06 | 1.16E-02 | 5.015 | -3.401 |
| GFI1B | growth factor independent 1B transcriptional repressor | 6.468 | -6.874 | 5.21E-06 | 1.16E-02 | 4.267 | -4.616 |
| ECI2 | enoyl-CoA delta isomerase 2 | 9.317 | -6.799 | 5.91E-06 | 1.16E-02 | 4.152 | -3.099 |
| SNRK-AS1 | SNRK antisense RNA 1 | 7.699 | 6.755 | 6.38E-06 | 1.16E-02 | 4.084 | 2.448 |
| PTK2 | protein tyrosine kinase 2 | 6.761 | -6.680 | 7.25E-06 | 1.16E-02 | 3.968 | -3.842 |
| RPS6KL1 | ribosomal protein S6 kinase like 1 | 6.489 | -6.673 | 7.34E-06 | 1.16E-02 | 3.957 | -2.658 |
| LRRC45 | leucine rich repeat containing 45 | 6.612 | -6.639 | 7.78E-06 | 1.16E-02 | 3.903 | -2.562 |
| KBTBD7 | kelch repeat and BTB domain containing 7 | 10.589 | 6.635 | 7.83E-06 | 1.16E-02 | 3.898 | 2.122 |
GO term enrichment analysis of DEGs
To explore the functional roles of the DEGs, we performed GO enrichment analysis of up-regulated and down-regulated DEGs by using DAVID gene annotation tool. It turned out that obvious differences were enriched in BPs, MFs and CCs among the 796 DEGs. For BPs, the up-regulated DEGs were primarily enriched in immune response, including innate, adaptive immune response and T cell differentiation involved in immune response, suggesting that these DEGs could significantly associate with the immune system of myeloma LC/SP cells. Besides, these genes were also significantly enriched in inflammatory response, leukotriene metabolic process and neutrophil chemotaxis. The down-regulated DEGs were significantly enriched in protoporphyrinogen IX biosynthetic process, heme biosynthetic process and erythrocyte differentiation, indicating that the down-regulated DEGs may be relevant to the development and differentiation of erythrocytes. In the CCs group, the up-regulated DEGs were significantly involved in plasma membrane, integral component of membrane and extracellular space. In addition, down-regulated genes were largely enriched in the extracellular exosome and hemoglobin complex. Regarding MFs category, the up-regulated genes were mainly enriched in the binding of carbohydrate, calcium ion and arachidonic acid. Moreover, the most significantly enriched GO terms for down-regulated genes were immunoglobulin receptor binding, oxygen transporter activity and NAD activity. GO enrichment analysis results were displayed in Figure 3 and Table 2.
Bubble plots of GO enrichment analysis of DEGs. (A) Bubble plot of GO enrichment analysis of up-regulated DEGs. (B) Bubble plot of GO enrichment analysis of down-regulated DEGs.
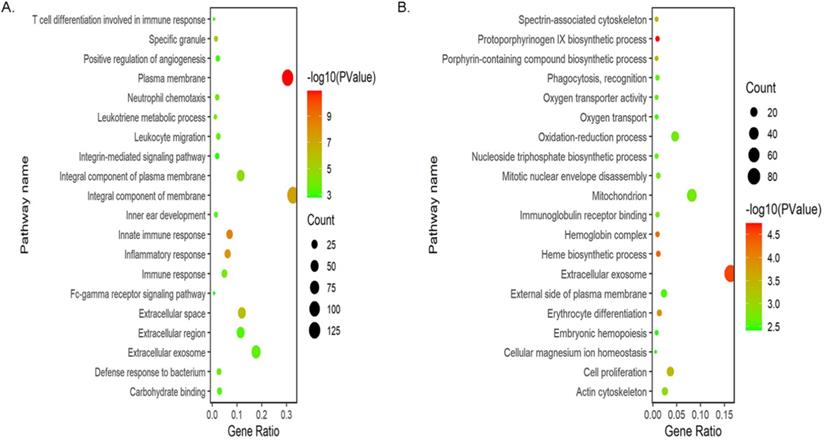
Gene Ontology term enrichment analysis of DEGs in LC/SP cells of MM.
| Expression | Category | ID | Term | Count | PValue |
|---|---|---|---|---|---|
| BP | GO:0045087 | innate immune response | 28 | 3.13E-09 | |
| BP | GO:0006954 | inflammatory response | 25 | 2.07E-08 | |
| BP | GO:0006955 | immune response | 20 | 8.02E-05 | |
| BP | GO:0006691 | leukotriene metabolic process | 5 | 8.69E-05 | |
| BP | GO:0030593 | neutrophil chemotaxis | 8 | 1.07E-04 | |
| CC | GO:0005886 | plasma membrane | 122 | 1.23E-11 | |
| CC | GO:0016021 | integral component of membrane | 131 | 5.17E-08 | |
| UP-DEGs | CC | GO:0005615 | extracellular space | 48 | 1.22E-06 |
| CC | GO:0042581 | specific granule | 6 | 1.47E-06 | |
| CC | GO:0005887 | integral component of plasma membrane | 46 | 2.27E-05 | |
| MF | GO:0030246 | carbohydrate binding | 12 | 4.29E-04 | |
| MF | GO:0005509 | calcium ion binding | 24 | 1.91E-03 | |
| MF | GO:0050544 | arachidonic acid binding | 3 | 2.63E-03 | |
| MF | GO:0004198 | calcium-dependent cysteine-type endopeptidase activity | 4 | 4.76E-03 | |
| MF | GO:0005536 | glucose binding | 3 | 1.36E-02 | |
| BP | GO:0006782 | protoporphyrinogen IX biosynthetic process | 5 | 1.81E-05 | |
| BP | GO:0006783 | heme biosynthetic process | 6 | 4.80E-05 | |
| BP | GO:0030218 | erythrocyte differentiation | 7 | 1.31E-04 | |
| BP | GO:0006779 | porphyrin-containing compound biosynthetic process | 4 | 4.09E-04 | |
| BP | GO:0008283 | cell proliferation | 19 | 4.20E-04 | |
| CC | GO:0070062 | extracellular exosome | 84 | 2.77E-05 | |
| CC | GO:0005833 | hemoglobin complex | 5 | 5.86E-05 | |
| DOWN-DEGs | CC | GO:0014731 | spectrin-associated cytoskeleton | 4 | 3.66E-04 |
| CC | GO:0015629 | actin cytoskeleton | 13 | 1.08E-03 | |
| CC | GO:0005739 | mitochondrion | 42 | 1.84E-03 | |
| MF | GO:0034987 | immunoglobulin receptor binding | 5 | 1.51E-03 | |
| MF | GO:0005344 | oxygen transporter activity | 4 | 2.28E-03 | |
| MF | GO:0004029 | aldehyde dehydrogenase (NAD) activity | 4 | 4.07E-03 | |
| MF | GO:0051015 | actin filament binding | 9 | 4.36E-03 | |
| MF | GO:0004064 | arylesterase activity | 3 | 5.39E-03 |
KEGG pathway enrichment analysis of DEGs
According to the KEGG pathway enrichment analysis of up- and down-regulated DEGs, the up-regulated DEGs were mainly enriched in osteoclast differentiation, cytokine-cytokine receptor interaction, Staphylococcus aureus infection, leukocyte transendothelial migration and cell adhesion molecules. Furthermore, enrichment of down-regulated DEGs was mostly in the porphyrin and chlorophyll metabolism, hematopoietic cell lineage and metabolic pathways. KEGG analysis results were displayed in Figure 4 and Figure 5, and the detailed analysis results of the top 5 pathways were shown in Table 3.
KEGG pathway analysis of DEGs.
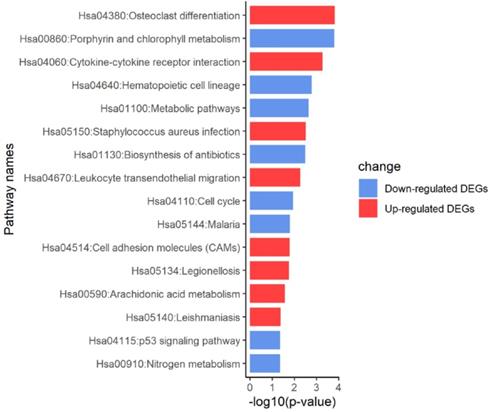
Distribution of DEGs in myeloma LC/SP cells for the top 5 KEGG enriched pathways.
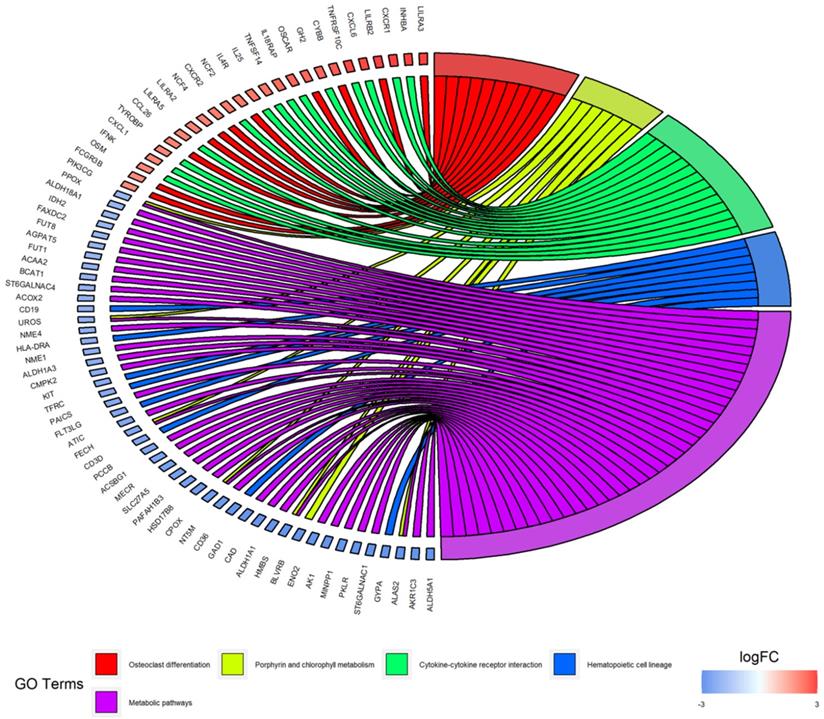
KEGG pathway enrichment analysis of DEGs in LC/SP cells of MM
| Expression | Category | Term | Count | P-Value |
|---|---|---|---|---|
| hsa04380 | Osteoclast differentiation | 11 | 1.50E-04 | |
| hsa04060 | Cytokine-cytokine receptor interaction | 14 | 5.51E-04 | |
| UP-DEGs | hsa05150 | Staphylococcus aureus infection | 6 | 3.15E-03 |
| hsa04670 | Leukocyte transendothelial migration | 8 | 5.44E-03 | |
| hsa04514 | Cell adhesion molecules (CAMs) | 8 | 1.64E-02 | |
| hsa00860 | Porphyrin and chlorophyll metabolism | 7 | 1.63E-04 | |
| hsa04640 | Hematopoietic cell lineage | 8 | 1.65E-03 | |
| DOWN-DEGs | hsa01100 | Metabolic pathways | 39 | 2.24E-03 |
| hsa01130 | Biosynthesis of antibiotics | 12 | 3.24E-03 | |
| hsa04110 | Cell cycle | 8 | 1.16E-02 |
PPI network construction and modular analysis
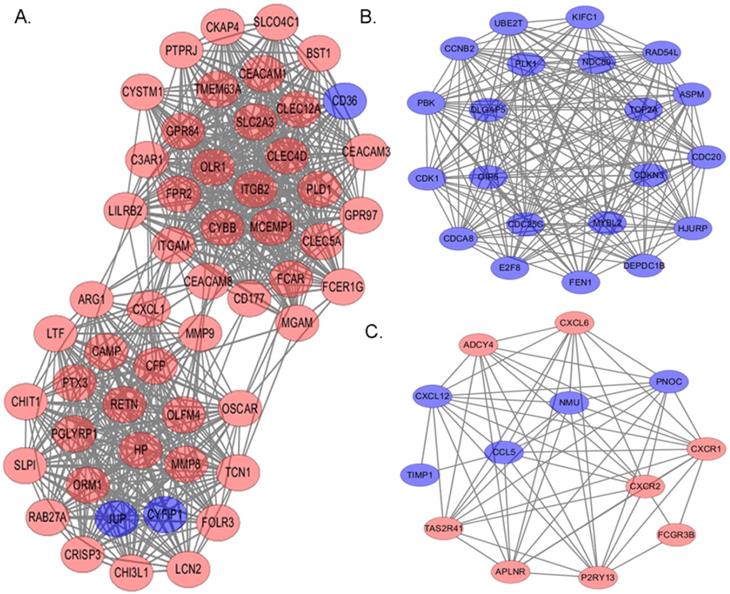
On the base of STRING online database and Cytoscape software, we established a PPI network of these DEGs in myeloma LC/SP cells, with 610 nodes and 2922 edges identified, including 288 up-regulated and 322 down-regulated genes. Then, PPI module analysis was implemented by plug-ins MCODE in Cytoscape, and three significant modules were identified from the whole network. The top 3 modules with high scores were selected for display: module 1 contained 53 nodes and 684 edges (Figure 6A), module 2 contained 21 nodes and 197 edges (Figure 6B) and module 3 contained 13 nodes and 60 edges (Figure 6C). Subsequently, corresponding GO term enrichment analysis was performed by plug-ins BiNGO in Cytoscape. Genes in Module 1 were significantly enriched in the defense response, immune system process and immune response. Moreover, genes in Module 2 were mainly enriched in cell cycle phase, cell cycle and M phase of mitotic cell cycle. Additionally, genes in module 3 were primarily enriched in G-protein coupled receptor protein, signaling pathway and chemotaxis. The detailed information of the top 3 modules was shown in Table 4.
The top 3 modules with relatively high scores from the protein-protein interaction network. Red: up-regulation; Blue: down-regulation. (A) Module 1 with 53 nodes and 684 edges was significantly enriched in defense response, immune system process and immune response. (B) Module 2 with 21 nodes and 197 edges was significantly enriched in cell cycle phase, cell cycle and M phase of mitotic cell cycle. (C) Module 3 with 13 nodes and 60 edges was significantly enriched in G-protein coupled receptor protein, signaling pathway and chemotaxis.
The top 5 significantly enriched GO terms and corresponding gene information in module analysis
| Modules | GO-ID | P-value | corr P-value | x | Description | Genes in test set |
|---|---|---|---|---|---|---|
| 6952 | 2.31E-16 | 2.51E-13 | 20 | defense response | ORM1|CRISP3|ITGB2|HP|CYBB|RAB27A|CXCL1|FPR2|LILRB2|PLD1|CFP|CLEC4D|CLEC5A|C3AR1|LCN2|OLR1|PTX3|PGLYRP1|CAMP|LTF | |
| 2376 | 6.99E-13 | 3.80E-10 | 20 | immune system process | ITGAM|ARG1|CRISP3|ITGB2|CYBB|RAB27A|CXCL1|LILRB2|CFP|MMP9|FCAR|CHIT1|BST1|CLEC4D|C3AR1|LCN2|PTX3|CEACAM8|PGLYRP1|LTF | |
| module 1 | 6955 | 1.24E-11 | 4.49E-09 | 16 | immune response | ARG1|CRISP3|CYBB|RAB27A|CXCL1|LILRB2|CFP|FCAR|CHIT1|BST1|CLEC4D|LCN2|PTX3|CEACAM8|PGLYRP1|LTF |
| 5576 | 3.52E-10 | 9.56E-08 | 24 | extracellular region | ORM1|ARG1|CRISP3|HP|CXCL1|RETN|OLFM4|MMP8|CFP|MMP9|FCAR|CHIT1|TCN1|CEACAM1|SLPI|LCN2|OLR1|CHI3L1|PTX3|CEACAM8|FOLR3|PGLYRP1|CAMP|LTF | |
| 30246 | 6.48E-09 | 1.38E-06 | 11 | carbohydrate binding | CHIT1|ITGAM|CLEC4D|CLEC12A|ARG1|CLEC5A|OLR1|CHI3L1|PTX3|PGLYRP1|LTF | |
| 22403 | 2.21E-18 | 1.13E-15 | 14 | cell cycle phase | PLK1|CDCA8|CDC25C|NDC80|CDC20|CCNB2|ASPM|KIFC1|CDK1|PBK|RAD54L|OIP5|DLGAP5|CDKN3 | |
| 7049 | 3.54E-18 | 1.13E-15 | 16 | cell cycle | PLK1|HJURP|CDCA8|CDC25C|NDC80|CDC20|CCNB2|ASPM|KIFC1|CDK1|PBK|RAD54L|OIP5|DLGAP5|CDKN3|E2F8 | |
| module 2 | 87 | 6.92E-18 | 1.48E-15 | 12 | M phase of mitotic cell cycle | CDC20|CCNB2|ASPM|KIFC1|PLK1|CDK1|PBK|CDCA8|OIP5|CDC25C|NDC80|DLGAP5 |
| 279 | 9.72E-18 | 1.56E-15 | 13 | M phase | PLK1|CDCA8|CDC25C|NDC80|CDC20|CCNB2|ASPM|KIFC1|CDK1|PBK|RAD54L|OIP5|DLGAP5 | |
| 278 | 2.74E-17 | 3.51E-15 | 13 | mitotic cell cycle | PLK1|CDCA8|CDC25C|NDC80|CDC20|CCNB2|ASPM|KIFC1|CDK1|PBK|OIP5|DLGAP5|CDKN3 | |
| 7186 | 1.94E-08 | 8.23E-06 | 7 | G-protein coupled receptor protein signaling pathway | CXCL12|CXCR1|CXCR2|PNOC|NMU|ADCY4|APLNR | |
| 42330 | 8.82E-08 | 1.25E-05 | 5 | taxis | CXCL6|CXCL12|CXCR1|CCL5|CXCR2 | |
| module 3 | 6935 | 8.82E-08 | 1.25E-05 | 5 | chemotaxis | CXCL6|CXCL12|CXCR1|CCL5|CXCR2 |
| 7610 | 4.59E-07 | 4.18E-05 | 6 | behavior | CXCL6|CXCL12|CXCR1|CCL5|CXCR2|NMU | |
| 4918 | 4.93E-07 | 4.18E-05 | 2 | interleukin-8 receptor activity | CXCR1|CXCR2 |
Selection of hub genes from the PPI network
Among the previously described DEGs, significant hub genes were identified by plug-in cytoHubba of Cytoscape using MCC algorithm. The top 10 hub genes were ITGAM, MMP9, ITGB2, FPR2, C3AR1, CXCL1, CYBB, LILRB2, HP and FCER1G. The top 10 hub genes and their most relevant functions were displayed in Table 5.
The top 10 hub genes and their most relevant functions
| Symbol | Gene name | Degree | Relevant function | Reference |
|---|---|---|---|---|
| ITGAM | integrin subunit alpha M | 78 | A poor prognostic factor in MM and AML patients; | [57-61] |
| MMP9 | Matrix metallopeptidase 9 | 64 | Participates in the breakdown of extracellular matrix; Promotes invasion of MM; | [73, 79-84] |
| ITGB2 | Integrin subunit beta 2 | 62 | Involved in cell adhesion and cell-surface mediated signaling; Associated with drug resistance to chemotherapy in MM cell line; | [63, 66] |
| FPR2 | Formyl-peptide receptor-2 | 52 | Associated with invasion and metastasis of some cancers; | [96-98] |
| C3AR1 | complement C3a receptor 1 | 51 | Involved in drug resistance to chemotherapy in AML cell; Predicts overall survival of AML; | [99] |
| CXCL1 | C-X-C motif chemokine ligand 1 | 51 | Associated with the growth and progression of some cancers; | [100-102] |
| CYBB | cytochrome b-245 beta chain | 48 | Involved in the progression of some cancers by promotion of angiogenesis; | [103, 104] |
| LILRB2 | leukocyte immunoglobulin like receptor B2 | 46 | Inhibits stimulation of an immune response; Promotes tumor progression; | [105-107] |
| HP | haptoglobin | 44 | NA | - |
| FCER1G | Fc fragment of IgE receptor Ig | 43 | Associated with disease progression in lymphoma and some solid cancers; Deficient expression represents T-cell immunodeficiency in CLL. | [108-110] |
Survival analysis of DEGs
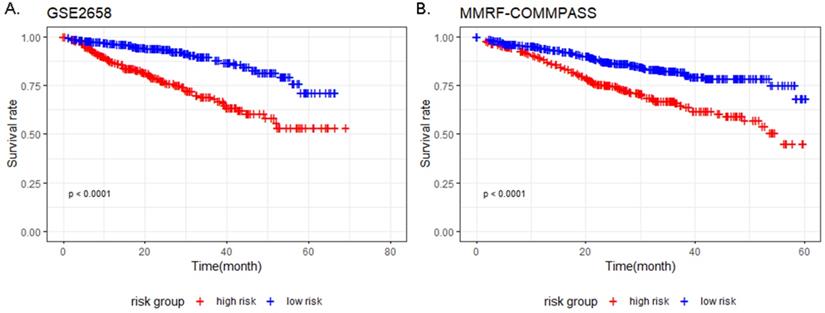
The result of univariate Cox analysis showed 76 survival related genes (Table S1; 19 up-regulated and 57 down-regulated) (P-value < 0.01). Afterwards, the 76 genes were fitted into the multivariate Cox proportional hazards analysis, and 9 genes including TFF3, EPDR1, MACROD1, ARHGEF12, AMMECR1, NFATC2, HES6, PLEK2 and SNCA were identified with P-value < 0.05 (Table 6). The prognostic models of training and validation dataset containing 9 genes were constructed by discriminating the low-risk group from the high-risk group based on the respective median risk score. Kaplan-Meier curve showed that high-risk group had worse survival compared to the low-risk group in both training and validation dataset (Figure 7).
Multivariate Cox regression analysis of 9 genes used for constructing the prognostic model
| Symbol | Coefficient | HR | Lower 95%CI | Upper 95%CI | P-value |
|---|---|---|---|---|---|
| TFF3 | 0.566 | 1.762 | 1.240 | 2.503 | 0.002 |
| EPDR1 | 0.492 | 1.636 | 1.132 | 2.364 | 0.009 |
| MACROD1 | 0.745 | 2.107 | 1.189 | 3.734 | 0.011 |
| ARHGEF12 | -0.540 | 0.583 | 0.389 | 0.874 | 0.009 |
| AMMECR1 | 0.862 | 2.369 | 1.142 | 4.913 | 0.020 |
| NFATC2 | 0.679 | 1.972 | 1.258 | 3.090 | 0.003 |
| HES6 | 0.513 | 1.671 | 1.091 | 2.560 | 0.018 |
| PLEK2 | 0.527 | 1.694 | 1.041 | 2.758 | 0.034 |
| SNCA | 0.759 | 2.135 | 1.216 | 3.749 | 0.008 |
Kaplan-Meier survival analysis of 9 prognostic genes in MM patients in the training and validation datasets. (P-value < 0.0001 in both GSE2658 and MMRF-COMMPASS). (A) Kaplan-Meier survival analysis of 9 prognostic genes in MM patients in GSE2658. (B) Kaplan-Meier survival analysis of 9 prognostic genes in MM patients in MMRF-COMMPASS.
Discussion
In the present study, we performed a bioinformatics analysis to identify DEGs between myeloma LC/SP cells and MP CD138+ cells to explore the molecular characteristics of LC/SP cells. Based on the gene expression profile, we screened a total of 796 DEGs, including 403 up-regulated and 393 down-regulated genes. Subsequently, deeper exploration of these DEGs were performed by bioinformatics methods, including GO and KEGG pathway enrichment analysis, PPI network construction and modules analysis, selection of hub genes and survival analysis.
GO enrichment analysis demonstrated that the up-regulated DGEs were significantly enriched in innate immune response, inflammatory response, plasma membrane and integral component of membrane. Firstly, according to our enrichment analysis, up-regulated DEGs were most enriched in innate immune response. As reported by Grivennikov SI et al., components of innate immunity such as macrophages, and DCs can either induce anti-tumor immune responses or promote tumor growth and progression depending on their morphological and phenotypic subtypes [30]. In multiple solid tumor models, the presence of tumor infiltrating macrophages (TAM) in tumor lesions can promote “stemness” property of cancer cells [31]. However, for MM, the association between SP cells and innate immune response has not been explained clearly yet. With regard to up-regulated DEGs enriched in immune response, it is now well-established that FGR, CXCL1, NLRC4 and S100A9 influence the pathogenesis of cancer by modulating immune responses and promoting progression, aggressiveness and cell survival [32-35]. Besides, up-regulated DGEs were also significantly enriched in inflammatory response. Then, the up-regulated DEGs were significantly involved in plasma membrane and integral component of membrane. It has been reported that the unique and specific makeup and arrangement of cell membranes of cancer cells are critical for cells to survive, grow and proliferate[36]. The enrichment analysis indicated that myeloma LC/SP cells may have unique plasma membrane and integral component of membrane compared to MP cells, and targeting the uniqueness may lead to the reduction of SP cells. Additionally, the down-regulated DEGs were significantly enriched in protoporphyrinogen IX and heme biosynthetic process, hemoglobin complex and erythrocyte differentiation, indicating that the down-regulated DEGs may be relevant to the development and differentiation of erythrocytes. Moreover, extracellular exosome was also enriched significantly. Exosomes are membranous structures that carry signaling molecules and regarded as important mediators of inter-cellular communication in health and disease [37]. Studies have revealed a strong cross-talk between the MM cells and their microenvironment in the bone marrow, which leads to the final phenotype of a typical MM patient [38, 39]. This result demonstrated that extracellular exosome may function significantly in this small fraction of MM cells.
KEGG pathway enrichment analysis showed some DEGs were significantly enriched in osteoclast differentiation, porphyrin and chlorophyll metabolism and cytokine-cytokine receptor interaction. Osteolytic bone disease is the hallmark of MM, which deteriorates the quality of life of myeloma patients. It has been demonstrated that increased osteoclast activity is one of the important mechanisms [40]. Among the DEGs, some studies found that PIK3CG, LILRB2 and CYBB could regulate the differentiation of osteoclast, which highlighted the possible biological significance of LC/SP cells in osteoclast differentiation [41-43]. ALAS2, significantly enriched in porphyrin and chlorophyll metabolism pathway, plays a key role in erythropoiesis by regulation of erythroid heme synthesis [44]. As already described in GO analysis, parts of the down-regulated DEGs may associate with the erythropoiesis, indicating that myeloma LC/SP cells may impair the erythropoiesis. CXCL1, significantly enriched in cytokine-cytokine receptor interaction pathway, could result in the enhancement of MM cell viability and migration [45]. Additionally, Staphylococcus aureus infection was enriched in KEGG pathway analysis. It's well known that infectious complications are a frequent cause of morbidity and mortality of MM [46]. A prospective study observed the rate of infections varied in different phases of MM, and the most infections were clinically diagnosed as pneumonia and bronchopneumonia caused by Haemophilus influenzae or Streptococcus pneumonia in early-stage MM [47]. As to Staphylococcus aureus infection, a recent study showed that Staphylococcus aureus bacteremia (SAB) may be an early prognostic indicator of cancer because of the phenomenon that patients with SAB were more likely to die from cancer than the general population [48]. Furthermore, an association between SAB and risk of multiple myeloma was described [49]. According to our results, it is presumable that myeloma SP cells may be relevant to infectious complications, especially the SAB infection, which opens a fundamental direction to understand infections for patients suffering from MM. However, the results need to be confirmed in further basic and clinical research. In brief, the enriched GO and KEGG pathways partly clarified the specific molecular characteristics of myeloma SP cells.
Then a PPI network of these DEGs in myeloma LC/SP cells was established, containing 610 nodes and 2922 edges. In the network, we selected three significant modules through the degree of importance and corresponding GO term enrichment analysis was performed, which indicated that myeloma LC/SP cells was significantly associated with immune system process, immune response and cell cycle, basically consistent with what we mentioned above. Subsequently, ten significant hub genes have been identified, containing ITGAM, MMP9, ITGB2, FPR2, C3AR1, CXCL1, CYBB, LILRB2, HP and FCER1G.
ITGAM was identified as the top 1 hub gene and had the highest degree of connectivity. ITGAM encodes CD11b, a component of the macrophage-1 antigen complex (Mac1, also known as complement receptor 3 [CR3]), which together with CD18, form Mac-1 or CR3, a protein that mediates leukocyte adhesion, migration, and phagocytosis in different cells [50-53]. CD11b contributes to the phagocytosis of opsonized particles, including apoptotic cells and immune complex [53]. What's more, CD11b is defined as a marker for myeloid-derived suppressor cells, which is reported to be harnessed by malignant cells to restrain antitumor immunity and promote malignant expansion or refractoriness to treatment [54-56]. It has been considered as a poor prognostic factor in MM [57] and AML patients [58-61]. But association between SP cells and CD11b remains unclear, and it is presumable that CD11b may participate in the regulation of biology of LC/SP cells and its up-regulation may promote expansion of MM. ITGB2 produces a protein, known as CD18, which is a cell surface marker expressed on lymphocytes [62] and is involved in cell adhesion and cell-surface mediated signaling [63]. It has been demonstrated that mutation in the ITGB2 gene could lead to leukocyte adhesion deficiency [64]. And its expression in CLL cells predicts disease progression [65]. In MM cell line, ITGB2 is overexpressed in vincristine resistant cell line [66]. Nonetheless, the correlation of drug resistance and ITGB2 requires further analysis.
As a key adhesion receptor, integrin CD11b/CD18 meditates leukocyte migration and immune functions [67]. Recently, several studies have investigated that the adhesion and angiogenesis system is vital to propagate MM progression with a vicious cycle by the endothelial-MM interaction. β integrin has been described to participate in the homing and adhesion of endothelial progenitor cells to sites of vascular remodeling [68, 69]. It has been uncovered that some integrins were detected in high levels in MM, while in non-detectable levels in non-active MM and MGUS patients, suggesting the adhesion molecules support the interactions between MM and the microvasculature and facilitate disease progression [70]. Furthermore, junctional adhesion molecule A has been identified as a key mediator of MM progression by promoting MM-associated angiogenesis and an independent prognostic factor for both newly diagnosed MM and relapsed/refractory MM [71, 72]. Similarly, our enrichment analysis of up-regulated DEGs had identified positive regulation of angiogenesis and cell adhesion, demonstrating that myeloma SP cells may be relevant to angiogenesis and cell adhesion to propagate MM progression.
The significantly up-regulated MMP9 gene (matrix metallopeptidase 9), one of the most widely investigated matrix metalloproteinases, is a significant protease which plays vital roles in many biological processes and cancer cell invasion, metastasis and angiogenesis [73]. Recently, MMP9 has been identified as a potential biomarker for several cancers [74-78]. As far as MM concerned, previous reports indicated that the expression of MMP9 in MM cells promote MM invasion [79-84], which may highlight the role of increased neovascularization in MM progression. In fact, it has been testified that angiogenesis, which is linked to aberrant expression of pro-angiogenic and down-regulation of anti-angiogenic genes [85], is a feature of MM progression through the transition from MGUS to MM, and plays a role in medullary and extramedullary dissemination [86, 87]. Recently, several angiogenic factors in active MM have been discovered, like VEGF (Vascular endothelial growth factor), FGF-2 (Fibroblast growth factor-2), HGF (Hepatocyte growth factor), MMP-2/9 and so on [88]. Additionally, data have shown that mTORC2 is involved in MM angiogenesis [89], and activation of the PI3K/AKT/mTOR pathway regulates pro-angiogenic factors of MMP-9 [90]. Besides, Notch signaling has been investigated in the cross talk between endothelial cells and MM cells to enable angiogenesis [91]. Consistently, in our GO enrichment analysis, the up-regulated DEGs were enriched in positive regulation of angiogenesis, suggesting the role of angiogenesis of myeloma SP cells. In a conclusion, neovascularization and positive regulation of angiogenesis may be regarded as potential factors in modulating MM progression and deserving prognostic roles. Nevertheless, its biological mechanisms have not clearly revealed yet. In the future, additional studies are needed to further confirm the mechanisms of angiogenesis in myeloma SP cells.
Extramedullary disease of MM remains a key area of therapeutic challenge, and the expression of adhesion molecules and changes in angiogenesis concerning mostly VEGF, MMP-9 and others are involved in extramedullary spread of MM cells [92, 93]. Plasma cells from extramedullary plasmacytomas showed angiogenesis related expression [94]. What's more, neovascularization can promote the growth of plasmacytomas [95]. These discoveries supported the idea that increased angiogenesis could facilitate malignant plasma cells growth outside the BM microenvironment. However, there are still unsolved questions on extramedullary myeloma involvement, especially on the relevant association with myeloma SP cells, which require further study.
In our survival analysis, to evaluate the association between the DEGs and clinical survival of MM patients and predict the prognosis of MM patients, we revealed 9 DEGs including TFF3, EPDR1, MACROD1, ARHGEF12, AMMECR1, NFATC2, HES6, PLEK2 and SNCA to be significantly associated with survival and established a survival prediction model based on the 9 genes. Stratified by risk score, a significantly different clinical outcome of MM patients were showed by the Kaplan-Meier curve in both training and validation datasets. However, further investigation of these genes in clinical research is warranted.
There are some limitations in our study. Firstly, the identification of DEGs profile was performed without external validation of other databases because of the absence of available data about SP cells compared to MP cells in MM. Second, we didn't evaluate the correlation of the prognostic model with clinicopathological characteristics. Thirdly, our study was only analyzed based on bioinformatics analysis. Hence, further investigations are warranted to validate the results and enhance our understanding of the biological role of these genes in MM.
Conclusions
To sum up, we performed a comprehensive bioinformatics analysis on microarray data of myeloma LC/SP cells. DEGs were identified to be significantly enriched in various pathways, especially positive regulation of angiogenesis and cell adhesion. The results of this study increase our understanding of novel biomarkers of myeloma LC/SP cells, prediction of MM prognosis and potential therapeutic targets. Nevertheless, further relevant studies are needed to confirm the identified DEGs and pathways in LC/SP cells of MM.
Supplementary Material
Supplementary table.
Supplementary raw data and code.
Acknowledgements
This work was supported by the National Natural Science Funds of China (No. 81870166).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
2. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7:585-98
3. Tan D, Chng WJ, Chou T, Nawarawong W, Hwang SY, Chim CS. et al. Management of multiple myeloma in Asia: resource-stratified guidelines. Lancet Oncol. 2013;14:e571-81
4. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
5. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253-61
6. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-60
7. Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y. et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103:2332-6
8. Delude C. Tumorigenesis: Testing ground for cancer stem cells. Nature. 2011;480:S43-5
9. Boucher K, Parquet N, Widen R, Shain K, Baz R, Alsina M. et al. Stemness of B-cell progenitors in multiple myeloma bone marrow. Clin Cancer Res. 2012;18:6155-68
10. Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I. et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68:190-7
11. Kuranda K, Berthon C, Dupont C, Wolowiec D, Leleu X, Polakowska R. et al. A subpopulation of malignant CD34+CD138+B7-H1+ plasma cells is present in multiple myeloma patients. Exp Hematol. 2010;38:124-31
12. Kim D, Park CY, Medeiros BC, Weissman IL. CD19-CD45 low/- CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia. 2012;26:2530-7
13. Yaccoby S, Epstein J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood. 1999;94:3576-82
14. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797-806
15. Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ. et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028-34
16. Challen GA, Little MH. A side order of stem cells: the SP phenotype. Stem Cells. 2006;24:3-12
17. Haraguchi N, Utsunomiya T, Inoue H, Tanaka F, Mimori K, Barnard GF. et al. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells. 2006;24:506-13
18. Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R. et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006;103:11154-9
19. Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827-33
20. Tavaluc RT, Hart LS, Dicker DT, El-Deiry WS. Effects of low confluency, serum starvation and hypoxia on the side population of cancer cell lines. Cell Cycle. 2007;6:2554-62
21. Chiba T, Miyagi S, Saraya A, Aoki R, Seki A, Morita Y. et al. The polycomb gene product BMI1 contributes to the maintenance of tumor-initiating side population cells in hepatocellular carcinoma. Cancer Res. 2008;68:7742-9
22. Huff CA, Matsui W. Multiple myeloma cancer stem cells. J Clin Oncol. 2008;26:2895-900
23. Loh YS, Mo S, Brown RD, Yamagishi T, Yang S, Joshua DE. et al. Presence of Hoechst low side populations in multiple myeloma. Leuk Lymphoma. 2008;49:1813-6
24. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J. et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104:4048-53
25. Jakubikova J, Adamia S, Kost-Alimova M, Klippel S, Cervi D, Daley JF. et al. Lenalidomide targets clonogenic side population in multiple myeloma: pathophysiologic and clinical implications. Blood. 2011;117:4409-19
26. Sun J, Wen X, Jin F, Li Y, Hu J, Sun Y. Bioinformatics analyses of differentially expressed genes associated with bisphosphonate-related osteonecrosis of the jaw in patients with multiple myeloma. Onco Targets Ther. 2015;8:2681-8
27. Liu C, Gu X, Jiang Z. Identification of novel targets for multiple myeloma through integrative approach with Monte Carlo cross-validation analysis. J Bone Oncol. 2017;8:8-12
28. Yan H, Zheng G, Qu J, Liu Y, Huang X, Zhang E. et al. Identification of key candidate genes and pathways in multiple myeloma by integrated bioinformatics analysis. J Cell Physiol. 2019;234:23785-97
29. Wang P, Wang Y, Hang B, Zou X, Mao JH. A novel gene expression-based prognostic scoring system to predict survival in gastric cancer. Oncotarget. 2016;7:55343-51
30. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883-99
31. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H. et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108:12425-30
32. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG. et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165-78
33. Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Tumor-infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene. 2016;35:5735-45
34. Karki R, Man SM, Kanneganti TD. Inflammasomes and Cancer. Cancer Immunol Res. 2017;5:94-9
35. Roseweir AK, Powell A, Horstman SL, Inthagard J, Park JH, McMillan DC. et al. Src family kinases, HCK and FGR, associate with local inflammation and tumour progression in colorectal cancer. Cell Signal. 2019;56:15-22
36. Zalba S, Ten Hagen TL. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat Rev. 2017;52:48-57
37. Moloudizargari M, Abdollahi M, Asghari MH, Zimta AA, Neagoe IB, Nabavi SM. The emerging role of exosomes in multiple myeloma. Blood Rev. 2019;38:100595
38. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M. et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542-55
39. Wang X, Lu H, Li T, Yu L, Liu G, Peng X. et al. Kruppel-like factor 8 promotes tumorigenic mammary stem cell induction by targeting miR-146a. Am J Cancer Res. 2013;3:356-73
40. Terpos E, Ntanasis-Stathopoulos I, Gavriatopoulou M, Dimopoulos MA. Pathogenesis of bone disease in multiple myeloma: from bench to bedside. Blood Cancer J. 2018;8:7
41. Mori Y, Tsuji S, Inui M, Sakamoto Y, Endo S, Ito Y. et al. Inhibitory immunoglobulin-like receptors LILRB and PIR-B negatively regulate osteoclast development. J Immunol. 2008;181:4742-51
42. Sasaki H, Yamamoto H, Tominaga K, Masuda K, Kawai T, Teshima-Kondo S. et al. Receptor activator of nuclear factor-kappaB ligand-induced mouse osteoclast differentiation is associated with switching between NADPH oxidase homologues. Free Radic Biol Med. 2009;47:189-99
43. Kang H, Chang W, Hurley M, Vignery A, Wu D. Important roles of PI3Kgamma in osteoclastogenesis and bone homeostasis. Proc Natl Acad Sci U S A. 2010;107:12901-6
44. Sadlon TJ, Dell'Oso T, Surinya KH, May BK. Regulation of erythroid 5-aminolevulinate synthase expression during erythropoiesis. Int J Biochem Cell Biol. 1999;31:1153-67
45. De Veirman K, Wang J, Xu S, Leleu X, Himpe E, Maes K. et al. Induction of miR-146a by multiple myeloma cells in mesenchymal stromal cells stimulates their pro-tumoral activity. Cancer Lett. 2016;377:17-24
46. Blimark C, Holmberg E, Mellqvist UH, Landgren O, Bjorkholm M, Hultcrantz M. et al. Multiple myeloma and infections: a population-based study on 9253 multiple myeloma patients. Haematologica. 2015;100:107-13
47. Vacca A, Melaccio A, Sportelli A, Solimando AG, Dammacco F, Ria R. Subcutaneous immunoglobulins in patients with multiple myeloma and secondary hypogammaglobulinemia: a randomized trial. Clin Immunol. 2018;191:110-5
48. Gotland N, Uhre ML, Mejer N, Skov R, Petersen A, Larsen AR. et al. Long-term mortality and causes of death associated with Staphylococcus aureus bacteremia. A matched cohort study. J Infect. 2016;73:346-57
49. Gotland N, Uhre ML, Sandholdt H, Mejer N, Lundbo LF, Petersen A. et al. Increased risk of incident primary cancer after Staphylococcus aureus bacteremia: A matched cohort study. Medicine (Baltimore). 2020;99:e19984
50. Rhodes B, Furnrohr BG, Roberts AL, Tzircotis G, Schett G, Spector TD. et al. The rs1143679 (R77H) lupus associated variant of ITGAM (CD11b) impairs complement receptor 3 mediated functions in human monocytes. Ann Rheum Dis. 2012;71:2028-34
51. Maiti AK, Kim-Howard X, Motghare P, Pradhan V, Chua KH, Sun C. et al. Combined protein- and nucleic acid-level effects of rs1143679 (R77H), a lupus-predisposing variant within ITGAM. Hum Mol Genet. 2014;23:4161-76
52. Roberts AL, Thomas ER, Bhosle S, Game L, Obraztsova O, Aitman TJ. et al. Resequencing the susceptibility gene, ITGAM, identifies two functionally deleterious rare variants in systemic lupus erythematosus cases. Arthritis Res Ther. 2014;16:R114
53. Khan SQ, Khan I, Gupta V. CD11b Activity Modulates Pathogenesis of Lupus Nephritis. Front Med (Lausanne). 2018;5:52
54. Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S. et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911-20
55. De Veirman K, Van Valckenborgh E, Lahmar Q, Geeraerts X, De Bruyne E, Menu E. et al. Myeloid-derived suppressor cells as therapeutic target in hematological malignancies. Front Oncol. 2014;4:349
56. Younos IH, Abe F, Talmadge JE. Myeloid-derived suppressor cells: their role in the pathophysiology of hematologic malignancies and potential as therapeutic targets. Leuk Lymphoma. 2015;56:2251-63
57. Ruiz-Arguelles GJ, San Miguel JF. Cell surface markers in multiple myeloma. Mayo Clin Proc. 1994;69:684-90
58. Tucker J, Dorey E, Gregory WM, Simpson AP, Amess JA, Lister TA. et al. Immunophenotype of blast cells in acute myeloid leukemia may be a useful predictive factor for outcome. Hematol Oncol. 1990;8:47-58
59. Bradstock K, Matthews J, Benson E, Page F, Bishop J. Prognostic value of immunophenotyping in acute myeloid leukemia. Australian Leukaemia Study Group. Blood. 1994;84:1220-5
60. Junca J, Garcia-Caro M, Granada I, Rodriguez-Hernandez I, Torrent A, Morgades M. et al. Correlation of CD11b and CD56 expression in adult acute myeloid leukemia with cytogenetic risk groups and prognosis. Ann Hematol. 2014;93:1483-9
61. Xu S, Li X, Zhang J, Chen J. Prognostic Value of CD11b Expression Level for Acute Myeloid Leukemia Patients: A Meta-Analysis. PLoS One. 2015;10:e0135981
62. Goldin LR, McMaster ML, Rotunno M, Herman SE, Jones K, Zhu B. et al. Whole exome sequencing in families with CLL detects a variant in Integrin beta 2 associated with disease susceptibility. Blood. 2016;128:2261-3
63. Lucio PJ, Faria MT, Pinto AM, da Silva MR, Correia Junior ME, da Costa RJ. et al. Expression of adhesion molecules in chronic B-cell lymphoproliferative disorders. Haematologica. 1998;83:104-11
64. Yassaee VR, Hashemi-Gorji F, Boosaliki S, Parvaneh N. Mutation spectra of the ITGB2 gene in Iranian families with leukocyte adhesion deficiency type 1. Hum Immunol. 2016;77:191-5
65. Huang PY, Best OG, Almazi JG, Belov L, Davis ZA, Majid A. et al. Cell surface phenotype profiles distinguish stable and progressive chronic lymphocytic leukemia. Leuk Lymphoma. 2014;55:2085-92
66. Mutlu P, Ural AU, Gunduz U. Differential gene expression analysis related to extracellular matrix components in drug-resistant RPMI-8226 cell line. Biomed Pharmacother. 2012;66:228-31
67. Celik E, Faridi MH, Kumar V, Deep S, Moy VT, Gupta V. Agonist leukadherin-1 increases CD11b/CD18-dependent adhesion via membrane tethers. Biophys J. 2013;105:2517-27
68. Tenreiro MM, Correia ML, Brito MA. Endothelial progenitor cells in multiple myeloma neovascularization: a brick to the wall. Angiogenesis. 2017;20:443-62
69. Caiado F, Dias S. Endothelial progenitor cells and integrins: adhesive needs. Fibrogenesis Tissue Repair. 2012;5:4
70. Vacca A, Di Loreto M, Ribatti D, Di Stefano R, Gadaleta-Caldarola G, Iodice G. et al. Bone marrow of patients with active multiple myeloma: angiogenesis and plasma cell adhesion molecules LFA-1, VLA-4, LAM-1, and CD44. Am J Hematol. 1995;50:9-14
71. Solimando AG, Da Via MC, Leone P, Borrelli P, Croci GA, Tabares P. et al. Halting the vicious cycle within the multiple myeloma ecosystem: blocking JAM-A on bone marrow endothelial cells restores the angiogenic homeostasis and suppresses tumor progression. Haematologica. 2020
72. Solimando AG, Brandl A, Mattenheimer K, Graf C, Ritz M, Ruckdeschel A. et al. JAM-A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia. 2018;32:736-43
73. Huang H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors (Basel). 2018 18
74. Roy R, Yang J, Moses MA. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol. 2009;27:5287-97
75. Shao W, Wang W, Xiong XG, Cao C, Yan TD, Chen G. et al. Prognostic impact of MMP-2 and MMP-9 expression in pathologic stage IA non-small cell lung cancer. J Surg Oncol. 2011;104:841-6
76. Hu X, Li D, Zhang W, Zhou J, Tang B, Li L. Matrix metalloproteinase-9 expression correlates with prognosis and involved in ovarian cancer cell invasion. Arch Gynecol Obstet. 2012;286:1537-43
77. Li LN, Zhou X, Gu Y, Yan J. Prognostic value of MMP-9 in ovarian cancer: a meta-analysis. Asian Pac J Cancer Prev. 2013;14:4107-13
78. Liang S, Chang L. Serum matrix metalloproteinase-9 level as a biomarker for colorectal cancer: a diagnostic meta-analysis. Biomark Med. 2018;12:393-402
79. Barille S, Akhoundi C, Collette M, Mellerin MP, Rapp MJ, Harousseau JL. et al. Metalloproteinases in multiple myeloma: production of matrix metalloproteinase-9 (MMP-9), activation of proMMP-2, and induction of MMP-1 by myeloma cells. Blood. 1997;90:1649-55
80. Kaushal GP, Xiong X, Athota AB, Rozypal TL, Sanderson RD, Kelly T. Syndecan-1 expression suppresses the level of myeloma matrix metalloproteinase-9. Br J Haematol. 1999;104:365-73
81. Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R. et al. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood. 1999;93:3064-73
82. Van Valckenborgh E, Bakkus M, Munaut C, Noel A, St Pierre Y, Asosingh K. et al. Upregulation of matrix metalloproteinase-9 in murine 5T33 multiple myeloma cells by interaction with bone marrow endothelial cells. Int J Cancer. 2002;101:512-8
83. Vande Broek I, Asosingh K, Allegaert V, Leleu X, Facon T, Vanderkerken K. et al. Bone marrow endothelial cells increase the invasiveness of human multiple myeloma cells through upregulation of MMP-9: evidence for a role of hepatocyte growth factor. Leukemia. 2004;18:976-82
84. Parmo-Cabanas M, Molina-Ortiz I, Matias-Roman S, Garcia-Bernal D, Carvajal-Vergara X, Valle I. et al. Role of metalloproteinases MMP-9 and MT1-MMP in CXCL12-promoted myeloma cell invasion across basement membranes. J Pathol. 2006;208:108-18
85. Hose D, Moreaux J, Meissner T, Seckinger A, Goldschmidt H, Benner A. et al. Induction of angiogenesis by normal and malignant plasma cells. Blood. 2009;114:128-43
86. Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F. et al. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994;87:503-8
87. Vacca A, Ribatti D. Bone marrow angiogenesis in multiple myeloma. Leukemia. 2006;20:193-9
88. Ribatti D, Vacca A. New Insights in Anti-Angiogenesis in Multiple Myeloma. Int J Mol Sci. 2018 19
89. Lamanuzzi A, Saltarella I, Desantis V, Frassanito MA, Leone P, Racanelli V. et al. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget. 2018;9:20563-77
90. Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ. et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res. 2009;39:177-86
91. Saltarella I, Frassanito MA, Lamanuzzi A, Brevi A, Leone P, Desantis V. et al. Homotypic and Heterotypic Activation of the Notch Pathway in Multiple Myeloma-Enhanced Angiogenesis: A Novel Therapeutic Target? Neoplasia. 2019;21:93-105
92. Blade J, Fernandez de Larrea C, Rosinol L, Cibeira MT, Jimenez R, Powles R. Soft-tissue plasmacytomas in multiple myeloma: incidence, mechanisms of extramedullary spread, and treatment approach. J Clin Oncol. 2011;29:3805-12
93. Weinstock M, Aljawai Y, Morgan EA, Laubach J, Gannon M, Roccaro AM. et al. Incidence and clinical features of extramedullary multiple myeloma in patients who underwent stem cell transplantation. Br J Haematol. 2015;169:851-8
94. Hedvat CV, Comenzo RL, Teruya-Feldstein J, Olshen AB, Ely SA, Osman K. et al. Insights into extramedullary tumour cell growth revealed by expression profiling of human plasmacytomas and multiple myeloma. Br J Haematol. 2003;122:728-44
95. Nakayama T, Yao L, Tosato G. Mast cell-derived angiopoietin-1 plays a critical role in the growth of plasma cell tumors. J Clin Invest. 2004;114:1317-25
96. Chakravarti N, Peddareddigari VG, Warneke CL, Johnson MM, Overwijk WW, Hwu P. et al. Differential expression of the G-protein-coupled formyl Peptide receptor in melanoma associates with aggressive phenotype. Am J Dermatopathol. 2013;35:184-90
97. Hou XL, Ji CD, Tang J, Wang YX, Xiang DF, Li HQ. et al. FPR2 promotes invasion and metastasis of gastric cancer cells and predicts the prognosis of patients. Sci Rep. 2017;7:3153
98. Xiang Y, Yao X, Chen K, Wang X, Zhou J, Gong W. et al. The G-protein coupled chemoattractant receptor FPR2 promotes malignant phenotype of human colon cancer cells. Am J Cancer Res. 2016;6:2599-610
99. Wu SY, Fan J, Hong D, Zhou Q, Zheng D, Wu D. et al. C3aR1 gene overexpressed at initial stage of acute myeloid leukemia-M2 predicting short-term survival. Leuk Lymphoma. 2015;56:2200-2
100. Yang C, Yu H, Chen R, Tao K, Jian L, Peng M. et al. CXCL1 stimulates migration and invasion in ERnegative breast cancer cells via activation of the ERK/MMP2/9 signaling axis. Int J Oncol. 2019;55:684-96
101. Hsu YL, Chen YJ, Chang WA, Jian SF, Fan HL, Wang JY. et al. Interaction between Tumor-Associated Dendritic Cells and Colon Cancer Cells Contributes to Tumor Progression via CXCL1. Int J Mol Sci. 2018 19
102. Yuan M, Zhu H, Xu J, Zheng Y, Cao X, Liu Q. Tumor-Derived CXCL1 Promotes Lung Cancer Growth via Recruitment of Tumor-Associated Neutrophils. J Immunol Res. 2016;2016:6530410
103. Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731-9
104. Wang P, Shi Q, Deng WH, Yu J, Zuo T, Mei FC. et al. Relationship between expression of NADPH oxidase 2 and invasion and prognosis of human gastric cancer. World J Gastroenterol. 2015;21:6271-9
105. Shao H, Ma L, Jin F, Zhou Y, Tao M, Teng Y. Immune inhibitory receptor LILRB2 is critical for the endometrial cancer progression. Biochem Biophys Res Commun. 2018;506:243-50
106. Cai Z, Wang L, Han Y, Gao W, Wei X, Gong R. et al. Immunoglobulinlike transcript 4 and human leukocyte antigenG interaction promotes the progression of human colorectal cancer. Int J Oncol. 2019;54:1943-54
107. Zhang Y, Zhao J, Qiu L, Zhang P, Li J, Yang D. et al. Co-expression of ILT4/HLA-G in human non-small cell lung cancer correlates with poor prognosis and ILT4-HLA-G interaction activates ERK signaling. Tumour Biol. 2016;37:11187-98
108. Chen L, Yuan L, Wang Y, Wang G, Zhu Y, Cao R. et al. Co-expression network analysis identified FCER1G in association with progression and prognosis in human clear cell renal cell carcinoma. Int J Biol Sci. 2017;13:1361-72
109. Liao Z, Zhou L, Wang C, He Z, Wang X, Luo X. et al. Characteristics of TCRzeta, ZAP-70, and FcvarepsilonRIgamma gene expression in patients with T- and NK/T-cell lymphoma. DNA Cell Biol. 2015;34:201-7
110. Huang L, Chen S, Zha X, Yang L, Li B, Yu Z. et al. Expression feature of CD3, FcepsilonRIgamma, and Zap-70 in patients with chronic lymphocytic leukemia. Hematology. 2012;17:71-5
Author contact
![]() Corresponding authors: Jing Liu, E-mail: jingliu0318com and Xin Li, E-mail: lixiner1975com.
Corresponding authors: Jing Liu, E-mail: jingliu0318com and Xin Li, E-mail: lixiner1975com.