Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(1):60-67. doi:10.7150/ijms.27442 This issue Cite
Research Paper
A Novel Model for Studying Voltage-Gated Ion Channel Gene Expression during Reversible Ischemic Stroke
Yun-Shen Tai1, Shih-Chieh Yang2, Yi-Chun Hsieh3, Yaw-Bin Huang4,5, Pao-Chu Wu4, Ming-Jun Tsai6,7,8, Yi-Hung Tsai4*, Ming-Wei Lin5,9 ![]() *
*
1. Department of Surgery, E-Da Hospital, Kaohsiung, Taiwan
2. Department of Orthopedic Surgery, E-Da Hospital, Kaohsiung, Taiwan
3. Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD, USA.
4. School of Pharmacy, Kaohsiung Medical University, Kaohsiung, Taiwan
5. Center for Stem Cell Research, Kaohsiung Medical University, Kaohsiung, Taiwan
6. Department of Neurology, China Medical University Hospital, Taichung, Taiwan
7. School of Medicine, China Medical University, Taichung, Taiwan
8. Department of Neurology, China Medical University, An-Nan Hospital, Tainan, Taiwan
9. Department of Medical Research, E-Da Hospital/ E-Da Cancer Hospital, Kaohsiung, Taiwan
*Equal contributors
Received 2018-5-23; Accepted 2018-10-31; Published 2019-1-1
Abstract
The dysfunction of voltage-gated ion channels contributes to the pathology of ischemic stroke. In this study, we developed rat models of transient ischemic attack (TIA) and reversible ischemic neurological deficit (RIND) that was induced via the injection of artificial embolic particles during full consciousness, that allow us to monitor the neurologic deficit and positron emission tomography (PET) scans in real-time. We then evaluated the infarction volume of brain tissue was confirmed by 2,3,5-triphenyl tetrazolium chloride (TTC) staining, and gene expressions were evaluated by quantitative real-time PCR (qPCR). We found that rats with TIA or RIND exhibited neurological deficits as determined by negative TTC and PET findings. However, the expression of voltage-gated sodium channels in the hippocampus was significantly up-regulated in the qPCR array study. Furthermore, an altered expression of sodium channel β-subunits and potassium channels, were observed in RIND compared to TIA groups. In conclusion, to our knowledge, this is the first report of the successful evaluation of voltage-gated ion channel gene expression in TIA and RIND animal models. This model will aid future studies in investigating pathophysiological mechanisms, and in developing new therapeutic compounds for the treatment of TIA and RIND.
Keywords: animal model, embolic stroke, transient ischemic attack, reversible ischemic neurological deficit, voltage-gated ion channels
Introduction
Stroke is the leading cause of disabilities in adults and the primary cause of death worldwide. Additionally, stroke-induced brain damage can often lead to seizures and epilepsy [1-3].
A TIA is a brief episode of neurological dysfunction caused by the loss of blood flow to the brain, spinal cord, or retinal. This usually arises from a blood clot or a dislodged atherosclerotic plaque blocking a blood vessel in the brain. A TIA exhibits the same underlying characteristics as a stroke, i.e., a disruption of cerebral blood flow, and is frequently referred to as a mini-stroke. Additionally, TIAs and strokes share similar symptoms, such as contralateral paralysis and epilepsy [4, 5]. However, unlike a stroke, the symptoms of a TIA resolve quickly ranging from a few minutes to 24 h [6]. Stroke symptoms that last longer than 24 h but fewer than 72 h are called RIND [4]. Importantly, the very early risk of stroke after a first TIA is highlight in previous study [7]. Therefore, medical attention is sought more urgently. Currently, there is no suitable animal model for studying TIA and RIND, so the molecular mechanisms underlying these disorders remain unclear.
The hippocampus is a key brain structure for memory, learning, and cognition. It has been implicated in many aspects of the neuroendocrine responses during stress [8]. Lesion on the hippocampus could lead to the impaired persistent memory which was considered as the usual consequence of posterior cerebral artery ischemia. Moreover, stroke lesions in the hippocampus may become even more relevant in the presence of a second pathology like Alzheimer dementia [9]. Therefore, the hippocampus function may involve in the ischemia-induced neurologic deficit.
The Voltage-gated ion channels are responsible for maintaining neuronal homeostasis and function [10-12]. Consequently, while the influx of Na+ and/or Ca2+ ions modulate neuronal excitability, the sudden influx of these ions may also trigger apoptosis or give rise to cytotoxic edema [13]. Voltage-gated ion channels underlie the cell membrane conductance of Ca2+, K+, and Na+ ions and directly prorogate, inhibit, or modify electrical signals. Since these channels are responsible for ion influx or efflux, it is evident that these are the channels that are key to neuronal action potential generation [10]. Ischemic brain damage, like TIA or RIND, may activate signal transduction cascades that result in changes in gene expression, including the genes responsible for encoding ion channels. The alteration of ion channel gene expression may affect neuronal function and could also result in stroke-like behavior. Recently, voltage-gated ion channel modulators proved as promising candidates for the treatment of stroke [14]. Studies such as these demonstrate the importance of obtaining molecular mechanistic information about these disorders in order to apply it to translational channelopathy with the goal of identifying new therapeutic strategies for neuronal protection and repair of damage caused by TIA and RIND.
In this study, we used a novel animal model by using temperature-sensitive melted solid lipid microparticles to induce reversible stroke in order to mimic clinical TIA and RIND conditions, as we published previously [15]. Moreover, we utilized qPCR array analysis to understand how voltage-gated ion channel-encoded gene regulation plays a critical role in TIA and RIND. Understanding the role of these channels in stroke-associated pathologies is important for further new therapeutic strategy development.
Materials and Methods
Characterization of the melted solid lipid microparticles
The solid lipid microparticles were produced by emulsifying the oil phase, prepared from a mixture of different melting point of Gelucire, and the water phase, containing hydrophilic surfactant. We mixed Gelucire 33/01 and Gelucire 43/01 at a ratio of 9:1 to form solid lipid microparticles with a melting point of 34 and hydrophilic balances of 1 (34/01).
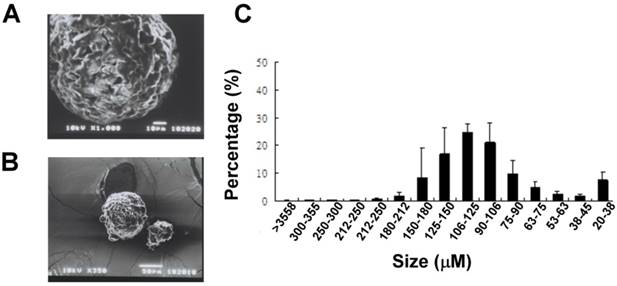
Gradation sieving of the melted solid lipid microparticles yielded 15 size fractions ranging from 20 μm to >355 μm (Fig. 1A). The distribution of the amount of each size fraction was showed; moreover half of the particles produced by our method were of sizes between 90 and 180 μm. The blended Gelucire (34/01) solid lipid microparticles had uniform round shapes with a porous surface represented by SEM analysis (Fig 1B).
Physical characteristics of the lipid microparticles. (A) Representative SEM analysis showed the surface morphology of the particles. (Upper panel, bar = 10 μm; lower panel, bar = 50 μm) (C) Distribution of the sizes of lipid microparticles. Particles size was classified using different sizes of mesh. The distribution tends to have a skewed type (n = 3).
Lipid particle preparation
The detailed procedure to prepare lipid microparticles has been described previously [16]. The oil phase was prepared by mixing Gelucire 43/01 and Gelucire 33/01. A ratio of 1:9 of Gelucire 43/01 and Gelucire 33/01 was prepared for the blending of solid lipid microparticles (a melting point of about 34°C was estimated by using a melting point apparatus Mel-Temp II, Laboratory Devices, INC, USA). To obtain the blends, the oil phase was homogenously mixed by heating to a temperature that was over the melting point of the Gelucire lipid.
The water phase was prepared by adding sodium dodecyl sulfate in purified water to get a final concentration of 1%. Both the oil and water phase were heated up to 70 °C. The oil phase (3 ml) was kept at 70°C and was mixed with the water phase (300 ml) by slowly dropping the oil into the water phase while stirring at a speed of 300 rpm for 5 min. The water phase was then slowly brought down to room temperature. Gelled lipid microparticles were then allowed to harden in the cool water phase while stirring at a speed of 300 rpm for 24 h. After hardening, the microparticles were filtered, rinsed with deionized water and air dried overnight before sieving the particles using standard U.S. size meshes (from 40 to 400 mesh). Finished microparticles were kept at 4°C and final particles sizes ranging from 75 to 125 μm were used as emboli for inducing transient brain ischemia.
Scanning electron microscopy (SEM)
Lipid microparticles were attached onto double-sided adhesive tape, fixed to a stage, and coated with gold so that the size and morphology of the microparticles could be observed and photographed using JEOL JSM-5300 scanning electron microscopy (Tokyo, Japan).
Animal model and surgical preparation
All animals of experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University. Three-month-old male Wistar rats weighing 300 -350 g were used for this study. Male rats were housed in a well-controlled animal care facility under diurnal lighting cycles, and given free access to food and water.
All animals were food-restricted overnight and were anesthetized the next day by an intra-peritoneal injection of 300 mg/kg choral hydrate. Body temperature was monitored by a rectal thermometer and kept at 37°C by an automated temperature regulator system. The rat was then fixed in the supine position on an operation plate and a midline excision of the ventral neck was used to explore the bifurcation of the right carotid artery. A PE-10 catheter (Becton Dickinson, NJ, USA) was inserted through the excised external carotid artery at the bifurcation of the common carotid artery concomitantly with ligation of the ipsilateral four collateral cerebral arteries. The PE-10 catheter was preserved by being carefully fixed to the external carotid artery and exposed on the neck as an indwelling injection tube after the closure of the surgical wound; heparin was used for preventing clot on the PE-10 tubing. In order to prevent infections, surgical wounds were carefully cleared, and all rats received postoperative neurologic evaluations. If any focal neurologic deficits were found, rats were excluded from further studies.
The stroke model induced by melted solid lipid in conscious animals was established in our previous study [15]. Briefly, we injected solid lipid microparticles within the range of 75 -125 μm via the indwelling catheter tube, injections were made into the ipsilateral bifurcation of the common carotid artery without anesthesia. Neurologic deficit was evaluated immediately after injecting solid lipid microparticles, and the total duration of neurologic deficit induced by transient ischemia was recorded.
In order to induce RIND, we injected solid lipid microparticles 12 h after recovery from the transiently induced ischemic episode using the same process described above. Neurological deficit was again evaluated immediately after injecting the solid lipid microparticles until the symptoms of the stroke disappeared.
Following the transient ischemic episode, TTC staining and quantitative real-time PCR (qPCR) were used to measure the infarct volume in the brain.
Neurologic deficit evaluation
All rats received neurological testing every 2 min in the first hour, then at 1.5, 2, 2.5, 3, 3.5, 4, and 24 h after operation. The neurologic deficits were scored as 0 (no neurologic defects), 1 (one paw clumsiness), 2 (tilt), 3 (rounding in only a unilateral circle), 4 (akinesia), 5 (seizure), 6 (absence of any spontaneous movement), and 7 (death). All neurologic behaviors were evaluated by the same investigator.
Calculation of infarction volume
After neurological evaluations, rats were deeply anesthetized, and brains were removed and then positioned in a rat brain matrix (Activational Systems, Ann Arbor, MI). In cases where rats died prior to the end of evaluations, brains were removed within 30 min of death. Each brain was cut into twelve 2-mm-thick coronal sections. We then used TTC staining to measure infarct size by incubating sections in a 0.05% TTC solution for 30 min at 30°C before being fixed in a 4% buffered formaldehyde solution for 24 h.
MicroPET study
A high-resolution PET scanner (Concord Microsystems, Knoxville, TN) was used for brain imaging according to previous study [17]. Briefly, rats were anesthetized with isoflurane, administered 37 MBq 18F-FDG via the tail vein, and were allowed 30 min for uptake. Within 10 min of 18F-FDG administration, images were acquired in the PET scanner and reconstructed with a 3D ordered subset expectation-maximization (OSEM) iterative algorithm. In transverse brain sections, regions of interest (ROIs) in the lesion area were identified by changes in metabolism induced by artery occlusion. Following calibration of both ipsi- and contralateral insulated areas, the uptake of 18F-FDG in the ROIs was calculated as the average nCi/cc. Radioactivity in the contralateral area was used as a reference to normalize data obtained in the ipsilateral area of the same image.
RNA isolation and cDNA synthesis
Total RNA from the rat hippocampus was extracted using the TRIzol plus RNA purification kit (Invitrogen Life Technology, Carlsbad, CA), and was converted to cDNA using the SuperScript® III first-strand synthesis system (Invitrogen Life Technology, Carlsbad, CA).
Quantitative real-time PCR array
A customized qPCR Array (Invitrogen Life Technology, Carlsbad, CA) was performed according to the manufacturer's protocol to detect voltage-gated ion channels expressions. This array was developed to profile the expression of a panel of 96 genes encoding brain-related ion channels and transporters. The genes represented on the array were grouped according to their functional and structural features, and included Ca2+, K+, Na+, and Cl- channels. Expression of mRNA in the rat hippocampus was then quantified by real-time RT-PCR using Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems) to verify the mRNA targets.
Statistics
Statistical analysis was performed using a one-way ANOVA followed by Bonferroni's post-hoc comparison test and a student t-test. A p value of < 0.05 was considered statistically significant.
Results
Characterization of lipid microparticles
A SEM analysis was performed to reveal the morphology (Fig 1A) and size of the lipid microparticles. The size of the particles was determined to be around 100 μm (Fig 1B).
Neurological and histology studies
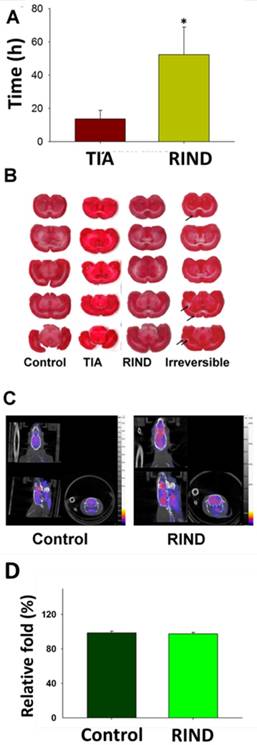
Rats that received the microparticle injections successfully displayed neurological deficits (Fig 2A). As shown in Fig 2A, TIA was considered to be induced when behavioral deficits (neurological score >1) lasted from a few minutes to 24 h. Importantly, neurological behavior deficits lasting from 24 to 72 h were categorized as RIND. As showed in Fig 2A, rats received the microparticles injection again after we inducing rats TIA successfully, the neurological behavior deficits of rats lasted more than 24h, and were categorized as RIND. The infarction area during RIND was then determined by TTC staining. However, no obvious region was demarcated suggesting that there was no significant neuronal death during RIND (Fig 2B). The irreversible stroke rats displayed as a positive control for the TTC staining.
(A) The duration of neurologic behaviors was repeatedly evaluated after injection of the particles. (B) The infarction area of rat brains after TIA, RIND, and irreversible ischemic damage was determined by TTC staining. Arrows indicate the infraction. (C) The metabolic changes in the brains after RIND were detected by PET scan. (D)The experiments were repeated three times and similar results were observed. All p-values are represented as (p < 0.05).
MicroPET studies
PET has been widely used in clinical diagnostics and treatment monitoring. Normal brain tissue has prominent uptake of 18F-FDG due to the constant need for glucose in the brain. Using the contralateral side as baseline, RIND-induced metabolic changes over the ipsilateral side of the rat brain were detected by PET scan after RIND (Fig. 2C). Radioactive quantification of the ROIs in transverse coronal brain sections indicated no significant changes in glucose uptake in the ipsi- and contralateral areas of the hippocampus (Fig. 2D).
Q-PCR array analysis
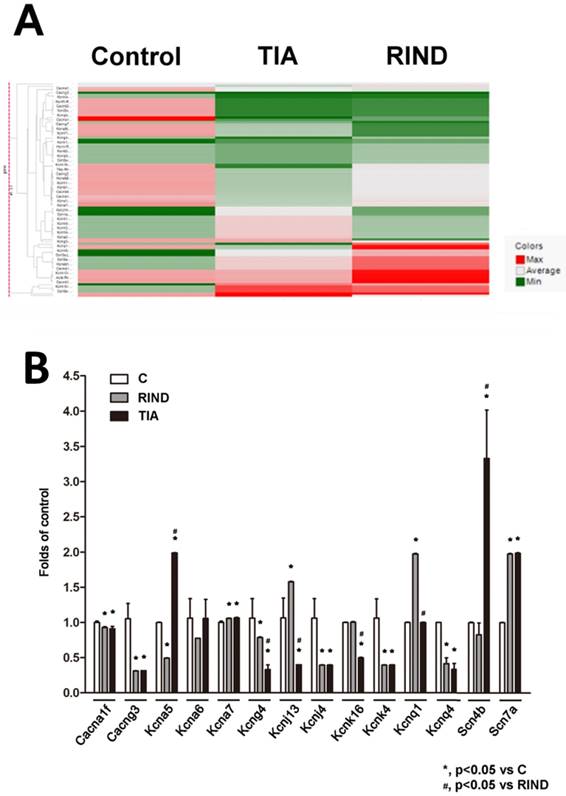
To examine the transcriptional response to TIA or RIND, we performed a qPCR array to compare the mRNA profiles between the hippocampus in control, TIA, and RIND rats. The genes that encode voltage-gated ion channels that were included in the custom array are listed in supplemental table 1. Expression of voltage-gated ion channel mRNA was considered significantly different when values for all samples from the hippocampus of experimental rats were more than double those for the controls (n=3 for each subgroup). The hierarchical cluster analysis of all significant mRNAs is shown in Fig 3A, which illustrates mRNAs differentially expressed in the hippocampus during TIA and RIND, compared to control conditions. Interestingly, the voltage-gated potassium channels, Kcnj4, were down-regulated in both TIA and RIND, as shown in Fig 3B. However, Cang3 and Kcnq1 gene expression decreased during RIND, but not in the TIA condition. The up-regulated mRNA targets that were more than those of the control, including significant changes in Scn7a and Kcna7, are shown in Fig 3B. Moreover, the genes exhibiting a greater changes during RIND compared to TIA were kcna5, kcna6, kcnj13, kcnk16, and the sodium channel modulatory β-subunit, scn4b.
(A) Hierarchical cluster analysis of significant expression of rat hippocampal voltage-gated ion channel mRNA in control, TIA and RIND conditions. (B) Genes exhibiting greater change following TIA or RIND. The experiments were repeated in triplicate on one occasion. All p-values are represented as (p < 0.05).
Statistical analysis
All results were presented as mean ± standard error of the mean, and the Student t test was used to evaluate inter-group differences. A p-value of <0.05 was considered statistically significant.
Discussion
TIA and RIND are transient episodes of neurological dysfunction caused by ischemia. These types of neurological abnormalities are reversible and recover in less than 72 h. However, suitable animal models of TIA and RIND are not currently available. Most of the currently available animal models have been created by inducing irreversible-ischemic damage and show obvious infarctions in the brain. In our previous study, we used artificial microparticles to successfully establish an animal model of embolic stroke, including a novel model that mainly expressed lacunar infraction [16]. Therefore, in our current study we used this technique with the aim of creating a more suitable animal model of TIA and RIND. Interestingly, while stroke-related neurological dysfunctions were observed in our TIA and RIND animal models, we obtained negative results in TTC-staining (Fig 2A). These results suggest that there was only minor cell death during our microparticle-induced TIA or RIND indicating that our model might be more suitable for studying reversible ischemic stroke.
During an ischemic episode, neurons exhibit a transient, early rise in intracellular Ca2+ and modulate calcium-dependent gene transcription.[18, 19] Calcium-response factor or sodium-calcium exchanger isoform 1 may responsible for this calcium-dependent gene transcription, and may be play a relevant role under pathophysiological conditions of stroke by regulating intraneuronal Ca2+ and Na+ homeostasis [20, 21]. In this study, we identified a novel group of genes that encode proteins that underlie the transmission of nerve impulses. We also expanded the analysis to profile the expression of all 90 genes listed as putative neuronal targets of stroke-related neurological dysfunction in the hippocampus. Consistent with earlier reports, we found that the expression of 14 of the 90 previously reported genes were statistically different, in the current study. These results may implicate the involvement of voltage-gated ion channel transcriptional activity in hippocampal pathologies, such as epilepsy. Indeed, previous studies have shown that epilepsy can arise because of stroke [2, 3]. Interestingly, a vast number of channelopathies, related to recurrent seizures have been associated with increased expression of genes responsible for encoding a component of voltage-gated ion channels, as well as the increased expression of many other genes that directly or indirectly control brain development, neuronal function, and neuronal activity [1]. Therefore, alterations in the expression of these genes may contribute to the pathophysiology of recurrent seizures.
Accumulating evidence suggests that voltage-gated ion channels are either up or down-regulated in ischemic stroke and that these alterations may play a direct role in the modulation of sodium or potassium-mediated neuronal function [11]. Our qPCR array analysis demonstrated that Scn7a, which encodes an atypical voltage-gated sodium channel, up-regulated during TIA and RIND. This is in line with previous findings of increased expression of Scn7a in the human and rat hippocampus during epileptogenic processes in temporal lobe epilepsy. The Scn7a gene, which encodes Nav2.x, has also been shown to be persistently up-regulated during a chronic epileptic phase in neurons of the hippocampus [22, 23]. The β-subunit of sodium channels functions as a modulator to control channel (α-subunit) gating. One previous study showed that functional deletion of the β-subunit in sodium channels resulted in Dravet Syndrome, a severe and intractable pediatric epileptic encephalopathy [24]. Therefore, the up-regulation of Scn7a may result in contralateral paralysis, sudden weakness or numbness, and/or epilepsy during TIA and RIND. And down-regulation of sodium channel modulatory β-subunit, Scn4b, may be an important transition point from TIA to RIND.
In our qPCR array study, the calcium and potassium channel genes were also found to have altered expression patterns during TIA and RIND. Calcium overload may cause neuronal cell death during ischemic damage [25, 26]. Therefore, the reduction expression of the voltage-gated calcium channels Cacna1f and Cacng3 in our study may also explain why we got negative TTC staining in TIA and RIND groups. Interestingly, a down-regulation of Cacna1f and Cacng3 channel genes has been reported to be associated with childhood absence epilepsy [27, 28].
Along with sodium and calcium channels, we also found an alteration in potassium channel expression in our animal models of TIA and RIND. The potassium channels, Kcna and Kcnj encodes the delayed rectifier K+ channels responsible for repolarization after an action potential [1, 29], and Kcnj and Kcnq encode inward rectifier K+ channel which play a role in controlling neuron cell membrane excitability [30]. Functional studies of the physiological role of these voltage-gated potassium channels in healthy cerebral vessels show that they contribute to vascular tone by regulating resting membrane potential, and limiting depolarization and Ca2+ influx by promoting K+ entry into the cells. Kv1.5 (encoded by Kcna5) has been reported to be down-regulated in the seizure-sensitive gerbil hippocampus [29]. Moreover, a recent study has suggested that the down-regulation of Kir2.3 channel expression, which is encoded by Kcnj4, may contribute to the pathogenesis of temporal lobe epilepsy [31]. Therefore, a disturbance of these voltage-gated channels could be a contributing factor to complex cases of neurological disease and disorders and may specifically result in seizures, a disruption in autonomic control, and can even lead to sudden death [1, 32].
Recently, some studies have suggested that the transcription factors, Pyk2 and REST, are activated following transient brain ischemia and reperfusion. These studies propose that these factors might play an important role in mediating post-ischemic signal transduction [33, 34]. Therefore, Pyk2 or REST-dependent remodeling may also modulate channel gene expression, such as those of Scn7a, Kcna or Kcnq in the hippocampus, leading to increased neuronal excitability and epileptiform activity during TIA or RIND. The mechanisms underlying the modulation of the expression of these genes still needs further investigation.
In conclusion, we performed a novel animal model of TIA and RIND, which resulted in a transient neurologic deficit without cell death in the brain. This animal model has indicated that specific ion channels may serve as potential biomarkers in neurological disorders such as stroke. Additionally, the data collected from qPCR screening, suggest that the voltage-gated sodium channels, SCN7A; the sodium channel regulatory subunit, SCN4B; the voltage-gated calcium channels, Cacna1f, Cacng3; the voltage-gated potassium channels, Kcna5, Kcna7, Kcng4, Kcnj13, Kcnj4, Kcnk4, Kcnq1 and Kcnq4, might be important molecular targets for brain repair after transient ischemic stroke. Profiling the array of genes that are dysregulated in response to neuronal disease may advance our understanding of the mechanisms underlying the pathophysiology of these disorders. Furthermore, it may lead to the identification of novel therapeutic strategies in TIA and RIND.
Supplementary Material
Supplementary information.
Acknowledgements
The authors thank the support provided by the MOST (106-2320-B-650-002-), KMUDK106, NCKUEDA10612, and NSYSU-KMU Joint Research Project (KMU-NSYSU 107-I001).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Misonou H, Mohapatra DP, Menegola M, Trimmer JS. Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci. 2005;25:11184-93
2. Brima T, Otahal J, Mares P. Increased susceptibility to pentetrazol-induced seizures in developing rats after cortical photothrombotic ischemic stroke at P7. Brain Res. 2013;1507:146-53
3. Arntz R, Rutten-Jacobs L, Maaijwee N, Schoonderwaldt H, Dorresteijn L, van Dijk E. et al. Post-stroke epilepsy in young adults: a long-term follow-up study. PLoS One. 2013;8:e55498
4. Calandre L, Gomara S, Bermejo F, Millan JM, del Pozo G. Clinical-CT correlations in TIA, RIND, and strokes with minimum residuum. Stroke. 1984;15:663-6
5. Johnston SC. Ischemic preconditioning from transient ischemic attacks? Data from the Northern California TIA Study. Stroke. 2004;35:2680-2
6. Sorensen AG, Ay H. Transient ischemic attack: definition, diagnosis, and risk stratification. Neuroimaging Clin N Am. 2011;21(x):303-13
7. Lovett JK, Dennis MS, Sandercock PA, Bamford J, Warlow CP, Rothwell PM. Very early risk of stroke after a first transient ischemic attack. Stroke. 2003;34:e138-40
8. Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65-75
9. Szabo K, Forster A, Jager T, Kern R, Griebe M, Hennerici MG. et al. Hippocampal lesion patterns in acute posterior cerebral artery stroke: clinical and MRI findings. Stroke. 2009;40:2042-5
10. Johnston J, Forsythe ID, Kopp-Scheinpflug C. Going native: voltage-gated potassium channels controlling neuronal excitability. J Physiol. 2010;588:3187-200
11. Fung ML. Role of voltage-gated Na+ channels in hypoxia-induced neuronal injuries. Clin Exp Pharmacol Physiol. 2000;27:569-74
12. Jarvis SE, Zamponi GW. Trafficking and regulation of neuronal voltage-gated calcium channels. Curr Opin Cell Biol. 2007;19:474-82
13. Simard JM, Tarasov KV, Gerzanich V. Non-selective cation channels, transient receptor potential channels and ischemic stroke. Biochim Biophys Acta. 2007;1772:947-57
14. Gribkoff VK, Winquist RJ. Voltage-gated cation channel modulators for the treatment of stroke. Expert Opin Investig Drugs. 2005;14:579-92
15. Tsai MJ, Kuo YM, Tsai YH. Transient ischemic attack induced by melted solid lipid microparticles protects rat brains from permanent focal ischemia. Neuroscience. 2014;275:136-45
16. Tsai MJ, Tsai YH, Kuo YM. Characterization of the pattern of ischemic stroke induced by artificial particle embolization in the rat brain. Biomaterials. 2011;32:6381-8
17. Fu YK, Chang CJ, Chen KY, Hwang LC, Wu KH, Chang KW. et al. Imaging of regional metabolic activity by (18)F-FDG/PET in rats with transient cerebral ischemia. Appl Radiat Isot. 2009;67:1743-7
18. Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846-60
19. Colbran RJ. Regulation and role of brain calcium/calmodulin-dependent protein kinase II. Neurochem Int. 1992;21:469-97
20. West AE. Biological functions and transcriptional targets of CaRF in neurons. Cell Calcium. 2011;49:290-5
21. Valsecchi V, Pignataro G, Sirabella R, Matrone C, Boscia F, Scorziello A. et al. Transcriptional regulation of ncx1 gene in the brain. Adv Exp Med Biol. 2013;961:137-45
22. Gorter JA, Zurolo E, Iyer A, Fluiter K, van Vliet EA, Baayen JC. et al. Induction of sodium channel Na(x) (SCN7A) expression in rat and human hippocampus in temporal lobe epilepsy. Epilepsia. 2010;51:1791-800
23. Okumura A, Yamamoto T, Shimojima K, Honda Y, Abe S, Ikeno M. et al. Refractory neonatal epilepsy with a de novo duplication of chromosome 2q24.2q24.3. Epilepsia. 2011;52:e66-9
24. Brackenbury WJ, Isom LL. Na Channel beta Subunits: Overachievers of the Ion Channel Family. Front Pharmacol. 2011;2:53
25. Luo CX, Zhu XJ, Zhang AX, Wang W, Yang XM, Liu SH. et al. Blockade of L-type voltage-gated Ca channel inhibits ischemia-induced neurogenesis by down-regulating iNOS expression in adult mouse. J Neurochem. 2005;94:1077-86
26. Bano D, Nicotera P. Ca2+ signals and neuronal death in brain ischemia. Stroke. 2007;38:674-6
27. Everett KV, Chioza B, Aicardi J, Aschauer H, Brouwer O, Callenbach P. et al. Linkage and association analysis of CACNG3 in childhood absence epilepsy. Eur J Hum Genet. 2007;15:463-72
28. Yalcin O. Genes and molecular mechanisms involved in the epileptogenesis of idiopathic absence epilepsies. Seizure. 2012;21:79-86
29. Lee SM, Kim JE, Sohn JH, Choi HC, Lee JS, Kim SH. et al. Down-regulation of delayed rectifier K+ channels in the hippocampus of seizure sensitive gerbils. Brain Res Bull. 2009;80:433-42
30. Sun HS, Feng ZP. Neuroprotective role of ATP-sensitive potassium channels in cerebral ischemia. Acta Pharmacol Sin. 2013;34:24-32
31. Xu L, Hao Y, Wu X, Yu P, Zhu G, Hong Z. Tenidap, an agonist of the inwardly rectifying K(+) channel Kir2.3, delays the onset of cortical epileptiform activity in a model of chronic temporal lobe epilepsy. Neurol Res. 2013;35:561-7
32. Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6
33. Cheong A, Bingham AJ, Li J, Kumar B, Sukumar P, Munsch C. et al. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol Cell. 2005;20:45-52
34. Hwang JY, Aromolaran KA, Zukin RS. Epigenetic mechanisms in stroke and epilepsy. Neuropsychopharmacology. 2013;38:167-82
Author contact
![]() Corresponding author: Ming-Wei Lin, Department of Medical Research, E-Da Hospital/ E-Da Cancer Hospital, Kaohsiung 824, Taiwan. TEL: 886-7-6151100 ext.5413. E-mail: ta990074com
Corresponding author: Ming-Wei Lin, Department of Medical Research, E-Da Hospital/ E-Da Cancer Hospital, Kaohsiung 824, Taiwan. TEL: 886-7-6151100 ext.5413. E-mail: ta990074com