Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(4):441-450. doi:10.7150/ijms.5765 This issue Cite
Research Paper
The Anti-Fibrotic Effect of Bone Morphogenic Protein-7(BMP-7) on Liver Fibrosis
Lan Zhong2, Xiaolin Wang3, Shenglan Wang1, Li Yang1, Hengjun Gao1, Changqing Yang1 ![]()
1. Division of Gastroenterology and Institute of Digestive Disease, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China;
2. Division of Gastroenterology, East Hospital of Tongji University School of Medicine, Shanghai 200120, China;
3. Special Outpatient Clinic, The Affiliated Hospital of Medical College Qingdao University, Qingdao 266003, China.
Received 2012-12-25; Accepted 2013-2-26; Published 2013-3-2
Abstract
Background/Aims: Transforming growth factor-β1 (TGF-β1) plays an important role in the pathogenesis of liver fibrosis and cirrhosis. Recombinant human bone morphogenic protein-7 (rhBMP-7) alleviates renal fibrosis and improves kidney function. However, the beneficial effect of BMP-7 on hepatic fibrosis and cirrhosis remains unknown. The purpose of this study was to investigate the prophylactic and therapeutic effects of rhBMP-7 on liver fibrosis and the underlying mechanisms.
Methods: Liver fibrosis in the rat model was induced by peritoneal injection of porcine-serum (0.5ml/kg body weight) twice a week over 8 weeks. The effect of rhBMP-7 on hepatic fibrosis was monitored in rhBMP-7 pre-treated and non-treated rats. Pathologic changes were determined by immunohistolocial staining. TGF-β1 expression was investigated by immunohistolocial staining, western blotting, and real-time PCR. Collagen secretion was measured by enzyme-linked immunosorbent assay.
Results: Liver fibrosis was significantly reduced by rhBMP-7. The secretion of collagen type-I and -III was decreased by rhBMP-7 in hepatic stellate cells (HSCs) but not in hepatocytes. The anti-fibrotic effect of rhBMP-7 on liver fibrosis was resulted by blocking the nuclear accumulation of Smad2/3 or by inhibiting TGF-β1 expression in HSCs or hepatocytes.
Conclusions: The anti-fibrogenic mechanism of rhBMP-7 in the rat liver fibrosis was depended on the reduction of TGF-β1 overexpression and the inhibition of TGF-β1 triggered intracellular signalling in hepatic cells.
Keywords: liver fibrosis, hepatic stellate cell, hepatocytes, bone morphogenic protein-7, transforming growth factor beta 1, epithelial-to-mesenchymal transition, extracellular matrix.
Introduction
Chronic liver diseases are characterized by the gradual destruction of the parenchyma structure and the excessive accumulation of extracellular matrix (ECM). It is assumed that the increased synthesis and decreased degradation of ECM distorts the hepatic architecture and results in liver fibrosis and cirrhosis (1-3).
Several studies provided evidence of hepatic fibrogenesis and increased expression of transforming growth factor-β1 (TGF-β1), which resulted from inflammation and injury. The complex role of TGF-β1 in the pathology of liver fibrosis includes the deregulation of cell proliferation, differentiation, apoptosis, increased cytokine expression, and the deregulated synthesis and degradation of ECM (4-15). Overexpression of TGF-β1 in transgenic mice induced hepatic fibrosis and apoptosis of hepatocytes (16), which could be inhibited by neutralizing TGF-β1 (17). TGF-β1 increased the accumulation of collagen type-I, a major pro-inflammatory ECM component and plasminogen activator inhibitor-1 (PAI-1), the inhibitor of ECM degradation. The TGF-β/Smad intercellular signalling pathway is regarded to be the major mediator in TGF-β1 induced hepatic fibrosis (18-21). Smad2/3 associates with Smad4 is required for TGF-β1 induced collagen type-I synthesis and epithelial mesenchymal transition (EMT) activation (22). In contrast, Smad7 inhibited collagen and alpha smooth muscle actin (α-SMA) expression and reduced the ECM content by inhibiting Smad2/3 phosphorylation (23). In addition, hepatic stellate cell (HSC) generated TGF-β1 promoted the upregulation of ECM expression and the increase of chemokine and cytokine expression during liver fibrosis (1). Moreover, the inhibition of TGF-β1 by siRNA transfection or shRNA significantly decreased the expression of tissue inhibitors of metalloproteinases-I(TIMP-1), α-SMA and collagen type-I and of inflammatory cytokines (7).
Bone morphogenetic protein-7 (BMP-7), a 35KDa homodimeric protein, was initially described for its ability to induce bone formation (24). Renal injury rat models suggested that BMP-7 prohibited tubuloinsterstitial fibrogenesis (25, 26) and reversed renal fibrosis by counteracting TGF-β1-induced EMT (26-30). In addition, the exogenous administration of BMP-7 antagonized the expression of collagen type-IV, fibronectin, thrombospondin, and connective tissue growth factor (CTGF) in murine mesangial cells (28). BMP-7 prevented the TGF-β-driven ECM overexpression associated with fibrogenesis in mesangial cells (29). Zeisberg et al demonstrated that in renal chronic fibrosis the mechanism underlying the inhibitory effect of BMP-7 on TGF-β dependent EMT involved Smad-1/-3 and -5, but not mitogen activated protein kinases (MAPK) Erk1/2, p38, and JNK (25-26, 31, 32). However the detailed mechanism of TGF-β1 signalling in the pathophysiology of hepatic fibrosis and cirrhosis remains unclear.
Material and methods
Animal model
All experimental procedures have been approved by the Institutes Animal Care and Use committee and the local experimental ethics committee. Female Sprague-Dawley rats (body weight 150±10 g) were purchased from Dept. of Experimental Animals, Fudan University (Shanghai, China). Rats were randomly divided into five groups: negative control (n=10), positive control (n=8), prophylactic group (n=9), two treatment groups: early-stage treatment group (n=10) and late-stage treatment group (n=8). Rats treated with intraperitoneal injection of 0.5 ml/kg weight of saline twice a week served as negative control. Rats injected with porcine serum (0.5 ml/kg weight) represented the fibrotic positive control. In the prophylactic group, rats were pretreated with rhBMP-7 (100 μg/kg weight) 1 week before porcine serum injection. In the two treatment groups, rats were intraperitoneal injected with rhBMP-7 (100 μg/kg weight) every 48 h starting at week 2 and week 4 after porcine serum injection respectively. In the period of 8 weeks, rats were sacrificed and livers were collected.
Cell isolation and cell culture
Rat HSCs were isolated as described previously (30). Briefly, livers were perfused with D-Hanks buffer via the portal vein followed by perfusion for 10 min with DMEM medium (Shanghai Qibio Science and Technologies Co., Ltd., China) containing type-IV collagenase (0.05% for negative control and 0.1% for fibrotic rats respectively) and 0.1% protease E (Shanghai Joy Biopharm. Co., Ltd., China). Livers were cut into 2 mm3 pieces which were incubated in digesting solution (0.05% type-IV collagenase, 0.02% protease E and 0.01% DNase in DMEM) for 10 min at 37oC with frequent vortex. The supernatant was centrifuged (25,000*g, 20 min) over a 9.2% Nycodenz density gradient and the top pale cell ring was gently collected. After two washes with D-Hanks buffer, HSCs were collected by centrifuge (800*g, 5 min) and then resuspended in DMEM supplemented with 20% of fetal calf serum (Shanghai Qibio Science and Technologies Co., Ltd., China) for culture.
Rat hepatocytes were isolated and purified with the modified two-step procedures method as described by Seglen (32, 33). Rat livers were perfused with D-Hanks buffer (contains 10 units/ml heparin) via the portal vein and were dissected. Isolated livers were incubated 30 min at 25 °C after a perfusion with D-hanks buffer containing 0.0025% collagenase type-I and 0.075% CaCl2.
The tissue digestion was dispersed through an 80 micron sieve and the digestion was neutralized with an equal volume of DMEM with 20% FBS. Cells were pelleted (800*g, 5 min) and washed with D-Hanks buffer. Cells were purified by a centrifugation through Nycodenz D-hanks solution (18% w/v) for 30 min at 1200*g to avoid the contamination of stellate cells. Obtained hepatocytes were washed with 10 ml DMEM containing 20% FBS three times.
HSCs and hepatocytes (1 x 104 cells/cm2) were seeded in DMEM supplemented with 20% FBS and were grown under regular cell culture condition until 60-70% confluence. Cells were then starved in serum free DMEM medium for 24 h before treatment.
Histological staining
Rat liver tissue was fixed in 10% formaline and then embedded in paraffin following standard procedures. Tissue sections were stained with Hematoxylin-Eosin and Masson's trichrome. The extent of liver fibrosis was estimated by Knodell scores (34).
Real-time (RT)-PCR
Total RNA was obtained by the phenolic-acid guandinum method (35). RNA (1 µg) was reverse transcribed using AMV First Strand cDNA Synthesis kit and random oligodeoxynucleotide primers (Roche Diagnostics Corporation, Indianapolis, IN). Quantitative real-time PCR amplifications were performed for TGF-β1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was employed as an internal control. Forward and reverse primer sequences for TGF-β1 were 5'-ACTACACGCGAAGGCACAGC-3' and 5'-CTGCTCCTTCTTGTCTCCCGA-3' (TOYOBO Biotech Co., Ltd., JPN) respectively. The TGF-β1 probe was fam-5'- CTGGAGCCCGCGGTGATTGTAGAGC-3'-tamara 73. RT-PCR (Icycler, Bio-Rad laboratories, USA) was performed using 60 cycles as follows: 20s at 95℃ thermal cycling and 20s at 60℃ detection.
Western blots
The expression of TGF-β1 by HSCs was evaluated by western blots (36). Total protein was extracted and an equal amount of protein was size fractionated in a 12% poly-acrylamide SDS-gel. Standard transfer to PVDF membranes (Millipore Corp., Billerica, MA, USA) was processed and monitored by Ponceau's staining. Membranes were blocked in blocking buffer (0.2% casein in 0.1% TBS-Tween, pH 7.5) for 1 h and then were incubated with the primary anti-TGF-β1 antibody (1:200, SC-146, Santa Cruz Biotechnology, CA, USA) for 2 h at room temperature. After 3 washes (10 min each) with 0.1% TBS-Tween, membranes were incubated with a species specific horseradish-peroxidase linked secondary antibody (1:5000, SC-2004, Santa Cruz Biotechnology, CA, USA) for 2 h at room temperature. Following 3 washes with 0.1% TBS-Tween, membranes were soaked in enzyme-linked chemiluminescence detection reagent (GE Healthcare, NJ, USA) and protein bands were visualized on x-ray films.
Enzyme-Linked Immunosorbent Assay (ELISA)
Serum starved cells were treated with rhBMP-7 (10-100ng/ml) for 24 h or 96 h. The culture medium was collected and cell debris was discarded after centrifugation (2,000*g, 5min). The quantity of collagen type-I, -III and -IV in supernatant was measured with ELISA kit (BP-E30413, BP-E30458 and BP-E30178, Shanghai Lengton Biological Technology, China) following the manufacturer's instructions.
Immunohistostaining
HSCs and hepatocytes were grown to 80% confluence in cell culture chambers. Quiescent cells were incubated with rhBMP-7 for 24 h or 96 h respectively. Cells were fixed and permeabilized with cold methanol/acetone (1:1) for 10 min followed by 2 washes with phosphate buffered saline (PBS)-Triton x-100 (0.1%) for 10 min. Slides were blocked with 10% FBS in PBS for 1 h at room temperature and then incubated with the primary anti-Smad 2/3 antibody (1:200, SC-6033 Santa Cruz Biotechnology, CA, USA) over night at 4°C. After 3 washes with PBS-Triton x-100, slides were incubated with a species specific secondary antibody (1:1000, SC-2361 Santa Cruz Biotechnology, CA, USA) for 2 h at room temperature and nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI).
Statistical analysis
To assess intergroup differences: variance analysis and Chi2-square test were performed with SPSS 10.0 for windows. P<0.05 was considered as statistical significance. The null hypothesis was: no difference between groups.
Results
Establishment of animal model
As shown in Fig. 1, the liver fibrosis was established by intraperitoneal injection of porcine serum. The severity of fibrosis in the animal model was time dependent or increased over time. Livers of the negative control group (panel A) showed normal hepatic lobule structure with the terminal hepatic venule at the centre and the portal tracts around the lobule. In the positive fibrosis control group, the pathological changes induced by porcine serum injection were observed. After 2 weeks, a significant increase of hepatic interstitial proliferation and fibrotic structures in form of enriched deposition of collagen (panel B-1: 2 weeks) was observed. After 6 weeks the typical septal fibrosis structure was observed, in which the hepatic lobules were clipped by complete and incomplete fibrotic septaes (panel B-2: 6 weeks). Late stages of liver fibrosis were found 8 weeks after porcine serum injection as massive fibrous proliferation, pseudo-lobules, and the start of nodular cirrhosis (panel B-3: 8 weeks). HSCs and hepatocytes were isolated.
Masson Trichrome and Hematoxylin-Eosin staining observed in rat liver tissue. (original magnification 40 X). (A)Negative control: Normal liver; (B) Positive control : liver fibrosis was induced by intraperitoneal injection with 0.5ml/kg weight porcine serum twice weekly for 8 weeks. Pictures were taken under at different time points. Week 2: Hepatic interstitial proliferation and fibrosis; Week 6: Septal fibrosis; Week 8: Massive fibrous proliferation in liver, pseudo-lobules and early nodular cirrhosis; (C) early-stage rhBMP-7 treatment; (D) late-stage treatment; (E) prophylactic.
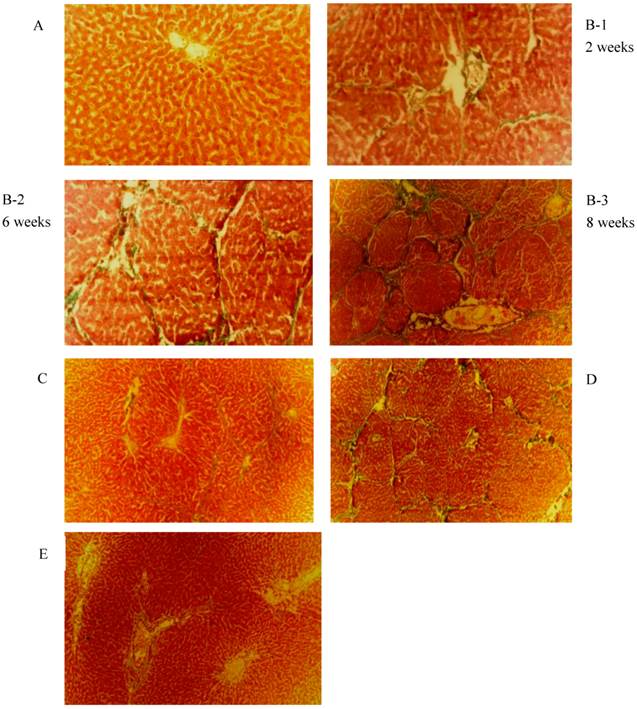
The effect of rhBMP-7 on liver fibrosis development in rats
Liver tissue sections sampled from different groups were stained with Masson-trichrome to measure collagen accumulation and to score fibrosis (Fig.1). The degree of liver fibrosis was decreased in animals treated with rhBMP-7 in a time dependent manner. The highest density of collagen in the liver was observed in the porcine serum treated group and the lowest expression of collagen was found in animals treated with rhBMP-7 for 6 weeks. Semi-quantitative analysis of the collagen contents showed a significant inhibitory effect of rhBMP-7 on porcine serum induced liver fibrosis scored by Masson's Trichrome-stain (Table 1). Notably, the level of liver fibrosis was significantly alleviated in rats that were pre-treated with rhBMP-7 comparing to those injected with rhBMP-7 after the initiation of fibrosis (Table 1).
The Knodell scores in liver tissue sections from different groups. The density of collagen was obtained by Masson-trichrome satin. Data were means ± SEM of independent experiments. Animal numbers with different Knodell classification was presented. *P<0.05, compared with group B (Positive control); + P<0.05, compared with group D (late-stage treatment).
| Group | Knodell classification | n | ||||
|---|---|---|---|---|---|---|
| 0 | I | II | III | IV | ||
| A: Negative control | 10 | 10 | ||||
| B: Positive control | 2 | 6 | 8 | |||
| C: early-stage treatment* | 2 | 6 | 2 | 10 | ||
| D: late-stage treatment* | 1 | 5 | 2 | 8 | ||
| E: prophylactic * ╅ | 2 | 3 | 3 | 1 | 9 | |
* P<0.05, compared with group B (Positive control); + P<0.05, compared with group D (late-stage treatment).
The effect of rhBMP-7 on the collagen secretion by primary HSCs and hepatocytes
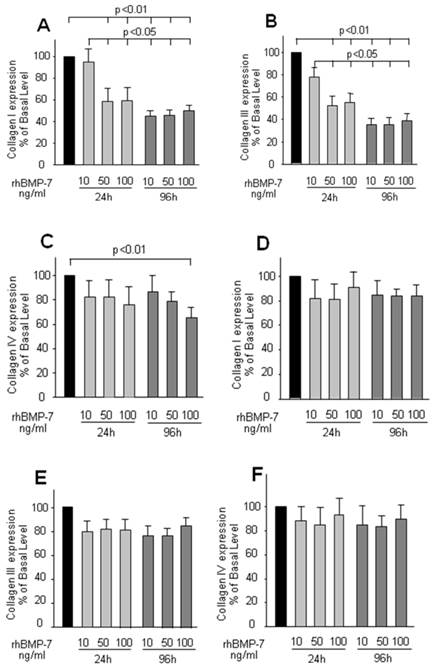
The regulatory effect of rhBMP-7 on collagen secretion was examined by dose and time course experiments. In HSCs, the expression of collagen type-I and -III was significantly and dose dependently decreased by rhBMP-7 (Fig 2A, 2B, 2C), in which this effect also presented as a time dependent manner. Interestingly, the expression of collagen type-IV was not affected by rhBMP-7 at any experimental concentration in the first 24 h, but a higher concentration of rhBMP-7 (100 ng/ml) with a longer treatment (96 h) reduced the collagen type-IV expression. In contrast to HSC cells, the expression of three kinds of collagen in hepatocytes was not modified by rhBMP-7 (Fig 2D, 2E, 2F).
ELISA results of collagen I, II, IV expression in cell culture supernatants in HSCs and hepatocytes. HSCs and hepatocytes were treated with rhBMP-7 in different concentration (10ng/ml, 50ng/ml, and 100ng/ml) for 24 h and 96 h respectively. The collagen expressed in untreated cells was serviced as control. Black bars, light grey bars, and grey bars indicated no treatment, rhBMP-7 for 24 h and rhBMP-7 for 96 h respectively. Data were means ± SEM of 10 independent experiments (n=10). Chart A, B, C were results from HSCs, and D, E, F were driven in hepatocytes respectively.
Effects of rhBMP-7 on the expression of TGF-β1 in rat liver
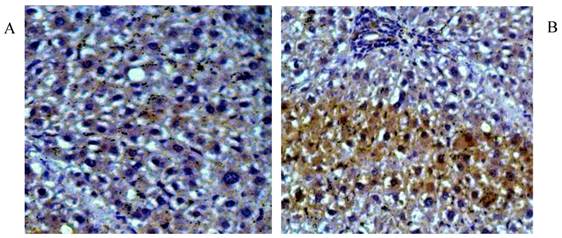
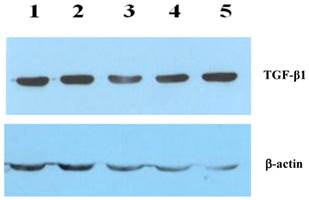
As a potential stimulus of collagen secretion, the expression and location of TGF-β1 was studied in liver tissue sections by immunohistochemical staining and in liver protein extraction by western-blot. Higher level of cytosolic TGF-β1 was observed in liver fibrosis (Fig 3A), which was suppressed by rhBMP-7 treatment (Fig 3B). In line with the immunohistochemical pattern, western blots showed a time dependent reduction of TGF-β1 expression by rhBMP-7 (Fig 4). Compared to the negative control (lane 1) and the positive fibrosis group (lane 5), the expression of TGF-β1 in HSCs was reduced by rhBMP-7 (lane 2-4) and this inhibitory effect correlated with the length of rhBMP-7 treatment.
Immunohistochemical staining for TGF-β1 in rat liver tissues. Pictures were obtained under 200 X magnification. (A) Liver tissues from positive fibrosis control; (B) Liver section from early-stage treatment. Brown positive staining were the localizations of TGF-β1.
The expression of TGF-β1 using the protein extraction in rat liver tissues isolated from different studied group. Lane 1, negative control; lane 2, prophylactic group; lane 3, early-stage treatment; lane 4, late-stage treatment; and lane 5, positive fibrosis control.
The effect of rhBMP-7 on TGF-β1 receptor expression by HSCs
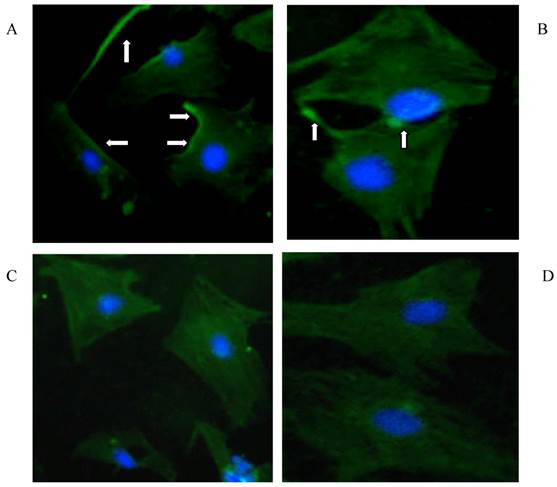
The expression of TGF-β1 receptors (TβR-I, TβR-II) was measured by immunofluorescence staining (Fig 5) in isolated primary HSCs. Both TβR-I (panel A and B) and TβR-II (panel C and D) were mainly located on the membrane of HSCs; however their expression was not affected by the presence of rhBMP-7 within 96 h.
Immunofluorescence staining for TGF-β1 binding receptors (TβR-I, TβR-II) in HSCs cultured for 96 h with (B and D) and without (A and C) 50 ng/ml rhBMP-7 treatment. The arrows indicated the expression of TβR-I, TβR-II on the cell membrane of HSCs.
The effect of rhBMP-7 on TGF-β1 mRNA expression by primary HSCs
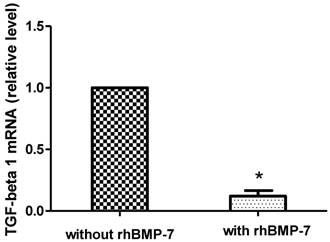
RT-PCR analysis of TGF-β1 mRNA expression in growing HSCs was performed after 96 h rhBMP-7 (50ng/ml) treatment and showed that rhBMP-7 significantly inhibited the expression of TGF-β1 mRNA by 8.33 fold compared to without rhBMP-7 treatment (2-∆∆CT=8.33, P<0.05). (Fig. 6)
rhBMP-7 affects the relative level of mRNA TGF-β1. TGF-β1 mRNA expression was reduced treatment with rhBMP-7 compared without rhBMP-7 as detected by real-time RT-PCR. * P<0.05.
The role of rhBMP-7 in the activation of Smad 2 and 3 (Smad2/3) in HSCs and hepatocytes
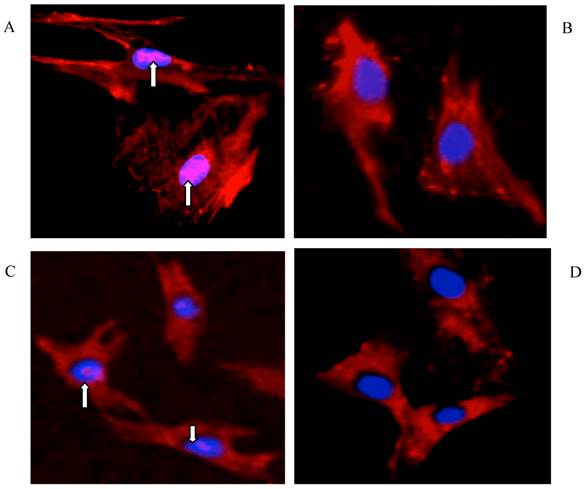
Immunofluorescence staining in Fig. 7 clearly showed the nuclear accumulation of Smad2/3 in growing HSCs (A) and hepatocytes (C), whereas this signal was eliminated by rhBMP-7 (50ng/ml) in HSCs (B) and in hepatocytes (D) within 96 h.
Immunofluorescence staining for Smad2/3 in HSCs and hepatocytes. Primary rat HSCs and hepatocytes were cultured for 96 h with (B and D) and without (A and C) 50ng/ml of rhBMP-7 for 96 h. Smad2/3 expression was represented as pink staining and was indicated by arrows. The nuclear was shown as blue staining.
Discussion
In this study, a stable animal model of liver fibrosis has been established by repeated intraperitoneal injection of porcine serum. Compared to adenoviral vectors or other viral transduction systems induced liver fibrosis (37), our method avoided problems due to the immune responses against to viruses. Additionally the combination of in vivo and in vitro models has been for the first time used to investigate mechanism of liver fibrosis. Evidence claimed HSC as the major extracellular matrix producer in liver and its dysfunction resulted in a deregulated deposition of ECM (38-40). Activity of HSC by cytokines, such as TGF-β1, was demonstrated playing an important role in the progression of liver fibrosis. Hence the investigation in the function of HSC and the underlying mechanism are reasonable in liver fibrosis (1).
Liver fibrosis in rat was triggered by porcine serum in a time and dose dependent manner, in which the earliest lesion was observed after two weeks. Porcine serum induced liver fibrosis in rat was significantly inhibited by rhBMP-7, the effect of which was dependent on the duration of porcine serum treatment and the length of rhBMP-7 treatment. Correlated with in vivo results, rhBMP-7 significantly reduced the expression of collagen type-I and -III by primary HSCs, which is considered as the major source of fibrillar collagens and of other ECM proteins (7, 38-40).
A strong TGF-β1 signal was obtained either in rat liver tissue or in primary HSCs derived from fibrotic liver and this was decreased by rhBMP-7. The variation of the TGF-β1 signal correlated with the level of collagen deposition and the gradient of liver fibrosis in vivo and in vitro and might indicate that TGF-β1 underlied the porcine serum induced liver fibrosis.
TGF-β1 plays an important role in initiating and maintaining fibrogenesis in many organs including the liver (41-43). In hepatic fibrogenesis, TGF-β1 is believed to be involved in the synthesis and deposition of extracellular matrix components like fibronectin, collagens type I, III, and IV via an autocrine or a paracrine mechanism; however the detailed mechanism still remains unclear (44, 45). As a member of the TGF-β superfamily, BMP-7 was assumed to reverse fibrosis in the kidney by decreasing pro-inflammatory cytokines, inhibiting EMT and inducing matrix metalloproteinase activity (25-26, 31). In addition, the BMP-7 dependent resistance on the TGF-β1 induced liver fibrosis in rats was explained by the inhibition of EMT in a cell culture model (32) and by the block of collagen expression in an animal model (27). Moreover, the upregulatory effect of TGF-β1 on ECM expression together with the transdifferentiation to myofibroblasts in HSCs was considered as important fibrogenic contributions to fibrotic liver diseases (7, 11-12, 46-48). Furthermore, the pathological change of hepatocytes during the fibrogenic process was related to TGF-β1 driven apoptosis and mesenchymal transition of hepatocytes into fibroblasts or fibroblast like cells (32).
To clarify the antifibrotic mechanism of rhBMP-7, we examined the activity of TGF-β1 intracellular signal pathway in primary HSCs. The expression of TGF-β1 binding receptor: TβR-I and TβR -II was not affected by rhBMP-7, while the expression of TGF-β1 either on protein level or the mRNA transcription was significantly down-regulated by rhBMP-7. It was logic that the nuclear translocation of Smad-2/-3 was also interrupted by rhBMP-7 both in HSCs and in hepatocytes. The transcription of collagen genes was trigged by Smad-2/-3 phosphorylation (49-52). Therefore the inhibitory effect of rhBMP-7 on liver fibrosis depended on the decrease of TGF-β1 and its signalling cascade, which was triggered by porcine serum.
In liver fibrosis, ECM accumulation was linked to activated fibroblasts which originated from hepatocytes by EMT that was induced by TGF-β1 and inhibited by BMP-7 (32). Our finding together with other studies suggested that the inhibitory effect of rhBMP-7 on liver fibrosis occurred by decreased collagen deposition and the resistance to EMT. Whether the regulation of rhBMP-7 on TGF-β1 signalling pathway occurred on the posttranscriptional level remained to be investigated.
In conclusion, we provided new evidence that rhBMP-7 could act as a prophylactic therapy for porcine-serum induced liver fibrosis through the decrease of TGF-β1 expression, and the inhibition of Smd2/3 in HSCs and hepatocytes, which led to reduce collagens deposition.
Abbreviations
BMP-7, bone morphogenic protein-7; HSC, hepatic stellate cells; TGF-β1, transforming growth factor beta1; ELISA, enzyme-linked immunosorbent assay; EMT, epithelial-to-mesenchymal transition; RT-PCR, reverse transcriptase-polymerase chain reaction; TβR I and II, TGF-β1 receptor type I and II; ECM, extracellular matrix.
Acknowledgements
This study was sponsored by a grant from Shanghai Pujiang Program (No. 05PJ14098) and National Natural Science Funds (No. 30570847).
Conflict of interest
There is no conflict of interest for any author with the content of this paper.
References
1. Brenner DA, Waterboer T, Choi SK. et al. New aspects of hepatic fibrosis. J Hepatol. 2000;32(1 Suppl):32-8
2. Marthinez-Hernandez A, Aamenta P.S. The extracellular matrix in hepatic regeneration. FASEB J. 1995;9:1401-10
3. Sato M, Suzuki S, Sonoo H. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct. 2003;28:105-12
4. Arias E, Anderson RN, Kung HC. et al. Deaths: final data for 2001. Natl Vital Stat Rep. 2003;52(3):1-115
5. Bauer M, Schuppan D. TGF-β1 in liver fibrosis: time to change paradigms? FEBS Letters. 2001;502:1-3
6. Breitkopf K, Godoy P, Ciuclan L. et al. TGF-beta/Smad signaling in the injured liver. J Gastroenterology. 2006;44(11):57-66
7. Chen K, Yang N, Mahato RI. TGF-β1 Gene Silencing for Treating Liver Fibrosis. Mol Pharm. 2009;6(3):772-79
8. de Gouville AC, Boullay V, Krysa G. et al. Inhibition of TGF-beta signaling by an ALK5 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br J Pharmacol. 2005;145(2):166-77
9. Friedman SL. Liver fibrosis—from bench to bedside. J Hepatol. 2003;38:S38-S53
10. George J, Roulot D, Koteliansky VE. et al. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA. 1999;96(22):12719-24
11. Gressner AM, Weiskirchen R, Breitkopf K. et al. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;1(7):d793-807
12. Kim KH, Kim HC, Hwang MY. et al. The antifibrotic effect of TGF-beta1 siRNAs in murine model of liver cirrhosis. Biochem Biophys Res Commun. 2006;343(4):1072-78
13. Okuno M, Akita K, Moriwaki H. et al. Prevention of rat hepatic fibrosis by the protease inhibitor, camostat mesilate, via reduced generation of active TGF-beta. Gastroenterology. 2001;120(7):1784-1800
14. Qi Z, Atsuchi N, Ooshima A. et al. Blockade of type beta transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc Natl Acad Sci USA. 1999;96(5):2345-49
15. Svegliati-Baroni G, Inagaki Y, Rincon-Sanchez AR. et al. Early response of α2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology. 2005;42(2):343-52
16. Sanderson N, Factor V, Nagy P. et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci USA. 1995;92(7):2572-76
17. George J, Roulot D, Koteliansky VE. et al. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA. 1999;96(22):12719-24
18. Zhao C, Chen W, Yang L. et al. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 2006;350(2):385-91
19. Latella G, Vetuschi A, Sferra R. et al. Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int. 2009;29(7):997-1009
20. Higashiyama H, Yoshimoto D, Kaise T. et al. Inhibition of activin receptor-like kinase 5 attenuates bleomycin-induced pulmonary fibrosis. Exp Mol Pathol. 2007;83(1):39-46
21. Liu X, Wang W, Hu H. et al. Smad3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-beta1 in cultured rat hepatic stellate cells. Pharm Res. 2006;23(1):82-9
22. Kaimori A, Potter J, Kaimori JY. et al. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282(30):22089-101
23. Dooley S, Hamzavi J, Breitkopf K. et al. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125(1):178-91
24. Heidin CH, Miyazono K, Dijke PT. TGFβ signaling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390(6659):465-71
25. Zeisberg M, Bottiglio C, Kumar N. et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse model. Am J Physiol Renal Physiol. 2003;285(6):F1060-7
26. Zeisberg M, Hanai J, Sugimoto H. et al. BMP-7 counteracts TGF-β1 -induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9(7):964-8
27. Kohji K, Iimuro Y, Otogawa K. et al. Adenovirus-mediated expression of BMP-7 suppresses the development of liver fibrosis in rats. Gut. 2007;56(5):706-14
28. Wang S, Hirschberg R. BMP7 antagonizes TGF-beta-dependent fibrogenesis in mesangial cells. Am J Physiol Renal Physiol. 2003;284(5):F1006-13
29. Wang S, Hirschberg R. Bone morphogenetic protein-7 signals opposing transforming growth factor β in mesangial cells. J Bio Chem. 2004;279(22):23200-6
30. Weiner FR, Blaner WS, Czaja MJ. et al. Ito cell expression of a nuclear retinoic acid receptor. Hepatology. 1992;15(2):336-42
31. Zeisberg M. Bone morphogenic protein-7 and the kidney: current concepts and open questions. Nephrol Dial Transplant. 2006;21(3):568-73
32. Zeisberg M, Yang C, Martino M. et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282(32):23337-47
33. Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29-83
34. Knodell RG, Ishak KG, Black WC. et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1(5):431-5
35. Chomezynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156-9
36. Sambrook J, Russell DW. Molecular cloning, a laboratory manual, 3rd ed. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press. 2001;3:15-35
37. Inagaki Y, Kushida M, Higashi K. et al. Cell type-specific intervention of transforming growth factor beta/Smad signaling suppress collagen gene expression and hepatic fibrosis in mice. Gastroenterology. 2005;129(1):259-68
38. Abdel Aziz MT, Atta HM, Mahfouz S. et al. Therapeutic potential of bone marrow-derived mesenchymal stem cells on experimental liver fibrosis. Clin Biochem. 2007;40(12):893-9
39. Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med. 1993;328(25):1828-35
40. Pinzani M. Novel insights into the biology and physiology of the Ito cell. Pharmcol Ther. 1995;66(2):387-412
41. Friedman SL. Mechanisms of disease: mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin PractGastroenterol Hepatol. 2004;1(2):98-105
42. Tomasek JJ, Gabbiani G, Hinz B. et al. Myofibroblasts and mechano-regulation of connective tissue remodeling. Nat Rev Mol Cell Biol. 2002;3(5):349-63
43. Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117(3):524-9
44. Liu X, Hu H, Yin JQ. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver int. 2006;26(1):8-22
45. Rachfal AW, Brigstock DR. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol Res. 2003;26(1):1-9
46. Gabele E, Brenner DA, Rippe RA. Liver fibrosis: signals leading to the amplification of of the fibrogenic hepatic stellate cell. Front Biosci. 2003;8:d69-77
47. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685-700
48. Wells RG. Fibrogenesis.V. TGF-beta signalling pathways. Am J Physiol Gastrointest Liver Physiol. 2000;279(5):G845-50
49. Clarke DC, Betterton MD, Liu X. Systems theory of Smad signalling. Syst Biol (Stevenage). 2006;153(6):412-24
50. Clarke DC, Liu X. Decoding the quantitative nature of TGF-b/Smad signaling. Trends in Cell Biology. 2008;18(9):430-43
51. Inman GJ, Nicolás FJ, Hill CS. Nucleocytoplasmic shuttling of Smads 2, 3 and 4 permits sensing of TGF-b receptor activity. Mol Cell. 2002;10(2):283-94
52. Lin X, Duan X, Liang YY. et al. PPM1A Functions as a Smad Phosphatase to Terminate TGFβ Signaling. Cell. 2006;125(5):915-28
Author contact
![]() Corresponding author: Changqing Yang, M.D., Ph.D., E-mail: cqyangedu.cn Tel: +86-21-6611-1076; Fax: +86-21-6611-1604.
Corresponding author: Changqing Yang, M.D., Ph.D., E-mail: cqyangedu.cn Tel: +86-21-6611-1076; Fax: +86-21-6611-1604.