Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(9):1207-1214. doi:10.7150/ijms.36131 This issue Cite
Research Paper
Parvalbumin Promoter Methylation Altered in Major Depressive Disorder
Benjamard Thaweethee-Sukjai1,2, Sirijit Suttajit3, Samur Thanoi1,2, Caroline F. Dalton4, Gavin P. Reynolds2,4, Sutisa Nudmamud-Thanoi1,2 ![]()
1. Department of Anatomy, Faculty of Medical Science, Naresuan University, Phitsanulok, 65000, Thailand
2. Centre of Excellence in Medical Biotechnology, Faculty of Medical Science, Naresuan University, Phitsanulok, 65000, Thailand
3. Department of Psychiatry, Faculty of Medicine, Chiang Mai University, Chiang Mai, 50200, Thailand
4. Biomolecular Sciences Research Centre, Sheffield Hallam University, Sheffield, S1 1WB, UK
Received 2019-4-28; Accepted 2019-7-10; Published 2019-8-14
Abstract
Aims: To determine the extent of DNA methylation of parvalbumin gene (PVALB) promoter in major depressive disorder (MDD) patients with and without suicide attempt in comparison with healthy controls.
Methods: The extracted DNA from dried blood spots of MDD patients (n = 92) including non-suicidal MDD and suicidal-MDD subgroups (n = 45 and n = 47, respectively) and age-matched control subjects (n = 95) was used for DNA methylation analysis at four CpG sites in the promoter sequence of PVALB by pyrosequencing.
Results: The PVALB methylation was significantly increased at CpG2 and decreased at CpG4 in the MDD group compared to the control group, while there was no difference between non-suicidal MDD and suicidal-MDD subgroups. A significant inverse correlation of severity of MDD was indicated only for CpG4.
Conclusion: This study provides the first evidence of abnormalities of PVALB promoter methylation in MDD and its correlation with MDD severity indicating a role for epigenetics in this psychiatric disorder.
Keywords: DNA methylation, major depressive disorder, parvalbumin, pyrosequencing, suicide attempt
Introduction
Major depressive disorder (MDD) is a severe neuropsychiatric disorder which is a common public health issue for people worldwide. The World Health Organization (WHO) has reported that 300 million people worldwide suffer from MDD [1]. Around 800,000 people die from suicide every year [1]. Several studies have reported that psychiatric disorders, especially MDD, have been strongly linked to suicide [2-6]. Moreover, patients with MDD have an almost 20-fold greater prevalence of suicide attempt than other patients hospitalized with no history of MDD [7].
Recently, the etiology of MDD and suicide remains unclear. Nevertheless, there is strong evidence for neurobiological deficits in the pathophysiology in MDD and suicide. Abnormalities of major neurotransmitter systems (serotonin, catecholamine, γ-aminobutyric acid (GABA) and glutamate) have been widely reported in both MDD patients [8,9] and suicidal subjects [10,11]. Epigenetic mechanisms, which can be modified by environmental factors, may play an important role in MDD pathology by genetic alterations in response to susceptibility or development to diseases [12-14]. DNA methylation is the most widely studied epigenetic factor, for which the main mechanism is an addition of a methyl group at the cytosine 5' position in cytosine-phosphate-guanine dinucleotide (CpG) sites [14,15]. DNA methylation occurring in or near promoter regions influences, and generally represses, gene transcription. Therefore, DNA methylation is a key mechanism that may contribute to the changes in gene expression seen in psychiatric disorders. Chen et al. have recently reviewed the evidence for the involvement of DNA methylation in depression, demonstrating particularly how brain-derived neurotrophic factor (BDNF) and solute carrier family 6 member 4 (SLC6A4) genes are implicated and that changes in methylation in several other genes emerging from whole-genome methylation studies have been reported [13].
GABA is the most important and abundant inhibitory neurotransmitter in the central nervous system. Recently, psychiatric disorders have been linked with abnormalities of GABAergic neurotransmission in schizophrenia, bipolar disorder and MDD. In MDD, there have been multiple reports of altered GABA content in plasma, cerebrospinal fluid (CSF) and brains of depressed patients as well as changes in GABA-related gene and protein expression [9,16-18]. Moreover, altered mRNA GABAA receptor subunit expression was found in postmortem frontal cortex in depressed suicide victims [19]. Postmortem microarray studies in a cohort of suicides reported that GABA-related gene expression was altered in suicides both with and without depression in prefrontal cortex [20], limbic system [21] and also global brain [22]. Moreover, an association of depression with GABA-related genes indicated by differences in gene expression between suicides with and without depression has been reported [20-22]. These findings indicate that GABAergic neurotransmission is likely to be dysregulated in depression and suicide.
GABAergic interneurons can be subdivided into three types based on the expression of the calcium-binding proteins calbindin, calretinin and parvalbumin (PV) [23]. Specific deficits of PV-immunoreactive cells are found in the brain in both schizophrenia and its animal models, and recently an increase of PV gene (PVALB) promoter methylation has been recently reported in hippocampal tissue from both schizophrenia [24] and its sub-chronic phencyclidine rat model [25]. Further evidence implicating PVALB promoter hypermethylation in psychosis comes from the study of methamphetamine-induced psychosis subjects [26]. While frontal cortical PV protein or PVALB expression are not reportedly changed in MDD [27-29], deficits of PVALB expression in anterior cingulate cortex have been observed [30]. Thus, as there are reported changes in genes and proteins related to GABAergic neurotransmission in MDD, the present study hypothesized that DNA methylation of PVALB may be abnormal in this disorder. This study examined DNA methylation within the promoter sequence of PVALB in MDD patients both with and without a history of attempted suicide. Exploration of the relationship between DNA methylation in the PVALB promoter with symptom severity of MDD was a secondary objective of this study.
Materials and methods
Subjects
The subject groups collected for this study comprised 100 unmedicated MDD patients and 100 age- and sex-matched control subjects (18-65 years old). MDD patients were diagnosed by a researcher psychiatrist in Clinical Psychiatry, Maharaj Nakorn Chiang Mai Hospital, Thailand based on criteria in the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5). Exclusion criteria for patients included a previous history of schizophrenia, bipolar disorder, or drug abuse. The subjects in the MDD group were divided into two subgroups of non-suicidal MDD and suicidal-MDD groups based on their suicide attempt history determined by the psychiatrist according to the Mini International Neuropsychiatric Interview (M.I.N.I.) (Thai version 5.0.0 - Revised 2007). The 17 Item Hamilton Depression Rating Scale Thai version (HAMD-7) was used for assessing severity of depression. The control subjects completed the Thai Mental Health Indicator (TMHI-66) for evaluation of mental health. Volunteers with previous history of schizophrenia, mood disorder, anxiety, or drug addiction and abnormal mental health were excluded from this study.
The experimental procedures of this study were approved by the Naresuan University Institutional Review Board, Thailand and the Research Ethics Committee, Faculty of Medicine, Chiang Mai University, Thailand. Informed consent forms were obtained from the participants involved.
Blood sample collection and DNA extraction
Fingertip blood samples were collected on Whatman FTA™ cards (GE Healthcare) using Chelex® 100 resin (Bio-Rad) following a modified protocol from application note 28-9822-22 AA by GE Healthcare (GE Healthcare). Then FTA discs were punched from dried blood spots using a Harris Micro Punch 2.0 mm (GE Healthcare, USA) and used for DNA extraction. Briefly, FTA discs were washed three times in 1 ml high-purity water by incubation at room temperature for 10 min with occasional inverting, centrifugation at 20,000 x g for 3 min and discarding the water in each time. Subsequently, 5% Chelex® 100 resin (Bio-Rad) was added and incubated at 56°C for 20 min. The samples were mixed by vortexing, further incubated at 100°C for 8 min and then centrifuged at 20,000 x g for 3 min. The supernatant containing genomic DNA was carefully collected and the concentration determined using NanoDrop® ND-1000 (Thermo Fisher Scientific).
Bisulfite conversion and methylation analysis
Bisulfite conversion of 500 ng genomic DNA template for each reaction was performed using EpiTect® Fast DNA Bisulfite Conversion kit (QIAGEN) according to the manufacturer's protocol.
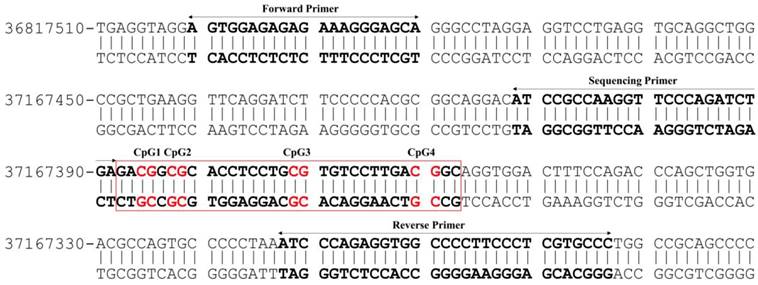
The extent of DNA methylation within a promoter sequence of PVALB (Homo sapiens chromosome 22, GRCh38.p12 Primary Assembly, NCBI Reference Sequence: NC_000022.11 (36800701..36819473, complement)) (Figure 1) and long interspersed nucleotide element-1 (LINE-1) genes was evaluated using the pyrosequencing technique according to the protocol recommended by the manufacturer (QIAGEN). The present study used a set of primers: forward primer: 5'-AGTGGAGAGAGAAAGGGAGTA-3', biotinylated reverse primer: 5'-AACACCAAAAAAAAAACCACCTCTAAAAT-3' and sequencing primer: 5'-ATTAGTTAAGGTTTTTAGATTTGA-3' (Eurofins MWG Operon) [24,26] to quantify methylation of four CpG sites of PVALB. In addition, predesigned PyroMark® CpG LINE-1 Assay (QIAGEN) containing proprietary forward, biotinylated reverse and sequencing primers to detect three CpG sites located at positions 331 to 318 of LINE-1 (GenBank accession number X58075) was used.
Illustration of the CpG sites and primers used for DNA methylation analysis of PVALB promotor region on chromosome 22. The target region consists of 217 bp containing four CpG sites.
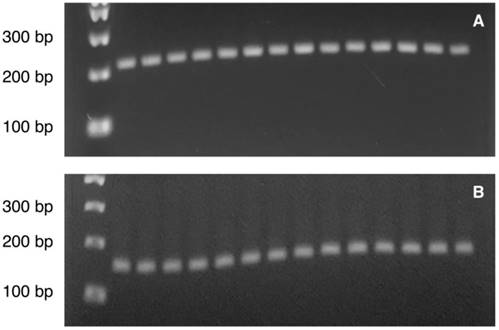
The PCR reaction included 1x PyroMark PCR Master Mix, 1x CoralLoad Concentrate (only for the reaction of PVALB), 0.2 µM of forward and reverse primers, RNase-free water and 1 ng bisulfite-converted DNA template in 25 µl PCR final volume. The PCR amplification was conducted with initial PCR activation step at 95°C for 15 min, 45 cycles of denaturation at 94°C for 30 s, annealing at 56°C (for PVALB) / 50°C (for LINE-1) for 30 s and extension at 72°C for 30 s, and final extension step at 72°C for 10 min. A quality of PCR product was checked by agarose gel analysis with ethidium staining prior to Pyrosequencing analysis (Figure 2A-2B). Then, ten microliter of PCR product was immobilized with Streptavidin SepharoseTM High Performance (GE Healthcare) and purified using the PyroMark Q24 Vacuum workstation following the manufacturer's instructions (QIAGEN). The samples then underwent pyrosequencing to determine methylation using the PyroMark Q24 system according to the instructions from the manufacturer (QIAGEN). The percentage methylation at each CpG site in a pyrogram was calculated by Pyromark® Q24 2.0.6.20 software (QIAGEN). All results which were labelled as “check” or “failed” were repeated from the PCR amplification step. All samples that failed to provide reliable results after replication were rejected.
Illustration of the PCR product bands of (A) PVALB (217 bp) and (B) LINE-1 (146 bp) on agarose gel visualized under ultraviolet (UV) illumination. Lane 1: 100 bp DNA ladder. Lane 2-8: Control sample 1-7. Lane 9-15: MDD sample 1-7.
Statistical analysis
The Statistical Package for the Social Sciences (SPSS) software version 17.0 for Windows (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. A difference in sex distribution was tested using Chi-square tests. Comparisons of differences in age, duration of illness, HAMD-17 score and percentage of DNA methylation were analyzed using non-parametric Mann-Whitney test or Kruskal-Wallis test. Correlation was evaluated using Spearman's rank correlation. The significance level was set at p < 0.05. GraphPad Prism® version 8.0 for Windows (La Jolla, CA, USA) was used to create scatter dot plots.
Results
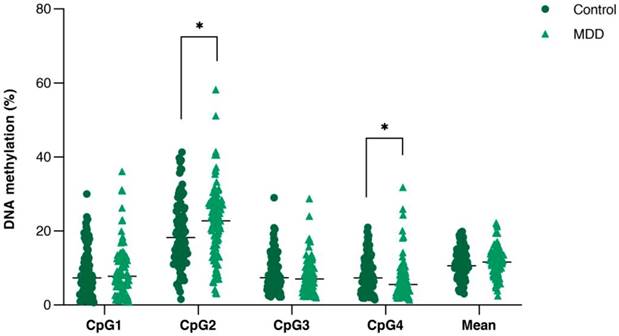
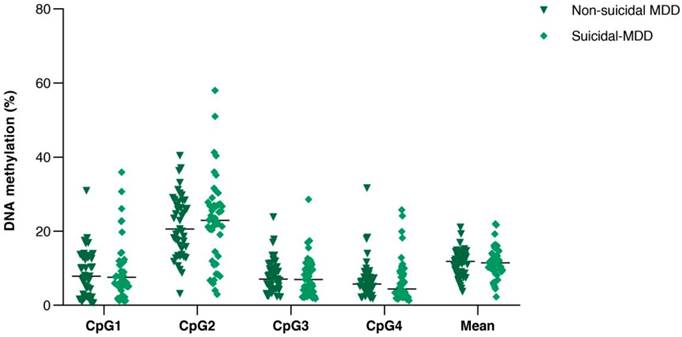
All samples showed single PCR bands without evidence of DNA degradation before determination of PVALB and LINE-1 methylations (Figure 2). Eight MDD and five control samples were rejected on the basis of unreliable pyrosequencing results for either PVALB or LINE-1, leaving an MDD group of 68 females and 24 males (43.13 ± 14.72 years old) and a control group of 71 females and 24 males (43.06 ± 15.03 years old) with complete data. DNA methylation status of the four CpG sites in the PVALB promoter sequence of MDD and control groups is shown in Figure 3. While the mean percentage methylation at the four CpG sites showed no significant effect in MDD group (Z = -0.762; p = 0.446), a significant increase in percentage methylation at CpG2 (Z = -2.021; p = 0.043) and a significant decrease in percentage methylation at CpG4 (Z = -2.161; p = 0.031) were seen. The methylation at each of these two CpG sites was not significantly different when comparing non-suicidal MDD and suicidal-MDD groups (Z = -0.144; p = 0.885 and Z = -1.172; p = 0.241, respectively) (Figure 4), nor between these MDD subgroups and control group (ꭓ2 = 4.090; df = 2; p = 0.129 and ꭓ2 = 5.571; df = 2; p = 0.062, respectively).
Comparison of DNA methylation (%) of the four consecutive CpG sites in PVALB promoter between control and MDD groups. Scatter dot plots of each groups are horizontally lined with the median value. * p < 0.05. MDD: Major depressive disorder.
Comparison of DNA methylation (%) of the four consecutive CpG sites in PVALB promoter between non-suicidal MDD and suicidal-MDD groups. Scatter dot plots of each groups are horizontally lined with the median value. MDD: Major depressive disorder.
Mean LINE-1 methylation showed no significant difference between MDD and control groups (70.85 ± 1.63 vs 70.95 ± 1.68; Z = -0.389; p = 0.697), nor between non-suicidal MDD and suicidal-MDD groups (70.77 ± 1.33 vs 70.93 ± 1.89; Z = -0.894; p = 0.371). Methylation of CpG2, but no other PVALB sites, was significantly negatively correlated with LINE-1 methylation in the control group (r = -0.230, p = 0.025), but not in the MDD group (r = -0.184, p = 0.079).
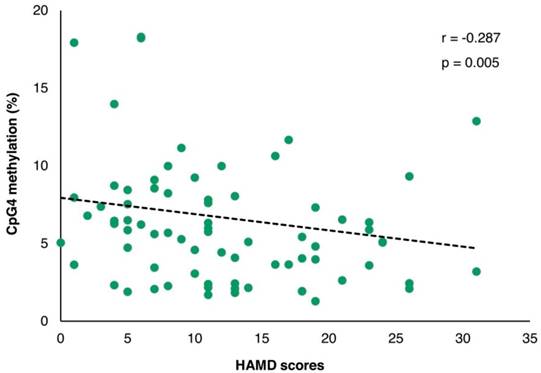
The demographic and clinical variables are shown in Table 1. A significant correlation between age and LINE-1 methylation was observed in both control and MDD groups (r = -0.331; p = 0.001 and r = 0.328; p = 0.001, respectively) with no difference in age neither between control and MDD groups nor between non-suicidal MDD and suicidal-MDD subgroups (data not shown). The duration of illness in the non-suicidal MDD group was significantly lower than the suicidal-MDD group (Z = -2.534; p = 0.011). Moreover, a significant correlation of methylation with duration of illness was found for LINE-1 (r = 0.266; p = 0.010), but not for any PVALB site (data not shown). Although no significant difference of HAMD-17 score was observed between non-suicidal MDD and suicidal-MDD groups (Z = -1.212; p = 0.225), a highly significant correlation with HAMD-17 score was apparent for CpG4 methylation (r = -0.287; p = 0.005), but not for CpG2 (r = -0.024; p = 0.820) (Figure 5). This effect remained after controlling for age and sex in a partial Pearson correlation analysis (p = 0.016).
Demographic data and clinical characteristics of control and MDD groups, and non-suicidal MDD and suicidal-MDD subgroups.
| Control (n = 95) (mean ± SD) | MDD (n = 92) (mean ± SD) | p-value | Non-suicidal MDD (n = 45) (mean ± SD) | Suicidal-MDD (n = 47) (mean ± SD) | p-value | |
|---|---|---|---|---|---|---|
| Age (years) | 43.06 ± 15.03 | 43.13 ± 14.72 | 0.928 | 42.62 ± 15.15 | 43.62 ± 14.45 | 0.860 |
| Sex (n, female/male) | 71/24 | 68/24 | 0.897 | 35/10 | 33/14 | 0.409 |
| Duration of illness (years) | 5.30 ± 5.75 | 10.15 ± 10.25 | 0.011* | |||
| HAMD-17 (scores) | 11.69 ± 7.43 | 13.47 ± 7.38 | 0.225 |
Data are represented as mean ± SD or number (n).
* p < 0.05.
HAMD: Hamilton depression rating scale; MDD: Major depressive disorder.
The correlation of PVALB methylation at CpG4 and HAMD-17 scores among MDD patients. HAMD: Hamilton depression rating scale.
Discussion
The main finding in this study was the site-specific change in PVALB promoter methylation which was increased at CpG2 and decreased at CpG4 in Thai MDD patients. This occurred along with no change of global DNA methylation represented by LINE-1 methylation. Moreover, we found a strong association of PVALB promoter methylation at CpG4 with the severity of depression, but no association with suicide attempt in MDD patients.
The present study has assessed DNA methylation at the same CpG sites of the PVALB promoter as previous studies in humans [24,26], and (for CpG2) the equivalent sequence in rats [25]. Interestingly, the finding of elevated DNA methylation at CpG2 was also previously observed associated with schizophrenia [24] and methamphetamine dependence, especially methamphetamine-induced psychosis [26], in humans, and at the equivalent site in brain tissue from rats receiving phencyclidine [25]. While the previous consistent findings indicate that hypermethylation of PVALB at CpG2 might be generally associated with psychotic illness and animal model of schizophrenia, these results suggest that this epigenetic change may extend to other psychiatric disorders, in this case MDD.
The decrease in methylation at CpG4 associated with MDD is in contrast to the increase at this site in post-mortem hippocampus in schizophrenia subjects [24], while no significant difference was identified in methamphetamine-induced psychosis subjects [26]. The validity of this finding is supported by the highly significant inverse correlation of CpG4 methylation with the severity of depression indicated by HAMD-17 scores, an independent effect of age or sex. Reanalysis of the data excluding outlying subjects gave essentially the same result (data not shown).
In the mammalian genome, most CpG sites are normally methylated while CpG residues enriched in CpG islands, particularly located in or near gene promoter regions, are relatively protected from methylation. An elevation of DNA methylation or hypermethylation at such CpG islands often results in blocking transcriptional initiation leading to repression of gene expression [31], while a reduction of DNA methylation or hypomethylation may lead to increased gene expression [32].
However, the direct relationship between DNA methylation and gene expression of PVALB is unknown, although the findings described above in schizophrenia and its animal model show both increased methylation and reduced expression of PVALB. In contrast, previous studies in postmortem brain in MDD have demonstrated no changes of PVALB gene expression or neuronal immunoreactivity in most brain regions studied [27-29,33], other than the anterior cingulate [30]. These findings imply that the PVALB methylation profiles might influence the pathology of MDD in specific brain regions. To support this hypothesis, hypermethylation of the PVALB promoter has been identified specifically in only hippocampus, but not prefrontal cortex, in schizophrenia [24].
LINE-1 methylation provides an indication of global DNA methylation levels. This study has found that the LINE-1 methylation was positively correlated with age, but not associated with MDD. A positive relationship between LINE-1 methylation and age is well established and has also been reported in psychiatric disorders [34]. However, unchanged LINE-1 methylation from this study is in agreement with a previous finding in blood samples in MDD [35]. The lack of any indication of a change in global DNA methylation indicates that the findings in PVALB are independent of such an effect and perhaps specific for that gene.
One important but inevitable limitation of this study is that the PVALB promoter methylation was determined in DNA derived from peripheral blood samples. While studies in post-mortem tissue from psychiatric patients have been undertaken [24], DNA methylation is most often investigated in blood samples, while there is evidence from previous studies of concordant changes in methylation between blood and brain [36]. We have also observed increased PVALB methylation in blood samples similar to reported changes in brain tissue in schizophrenia (Reynolds et al., unpublished data). There have been other findings of DNA methylation from blood samples of patients with MDD. Changes in methylation profiles of specific genes such as zinc finger and BTB domain containing 20 (ZBTBT20) [37], BDNF [38-42] and nuclear receptor subfamily 3, group C, member 1 (NR3C1) [43] have been reported. We do not know the direction of causality in these findings, i.e. whether the changes in PVALB methylation are a contributor to, or a consequence of, MDD and, for CpG4, depressive symptom severity. While some of these findings may also be confounded by drug treatment, we can exclude this influence this influence in the current study using unmedicated subjects.
In summary, this study provides the first evidence of changes in the PVALB promoter methylation in MDD. PVALB promoter methylation was increased at CpG2 and decreased at CpG4 in MDD patients, effects that were independent of a measure of global DNA methylation. Moreover, the decrease in PVALB methylation at CpG4 correlated strongly with severity of depression. Further studies need to determine how these findings might reflect changes in PVALB methylation within the brain and their impact on gene expression and, eventually, GABAergic neuronal function.
Acknowledgements
We are very grateful to all subjects for participation, the staff of Clinical Psychiatry, Maharaj Nakorn Chiang Mai Hospital, Thailand and the graduate students of Department of Anatomy, Faculty of Medical Science, Naresuan University, Thailand, for data and blood collections as well as Biomolecular Sciences Research Centre, Sheffield Hallam University, UK for facility support. This work was financially supported by the Naresuan University Research Fund and the funding from commission higher education by Naresuan University, and partially supported by Chiang Mai University. B Thaweethee-Sukjai was financially supported the Thailand Research Fund through the Royal Golden Jubilee Ph.D Program (Grant No. PHD/0297/2552) and the British Council through the Newton Fund: PhD Placement grant for scholars 2015/16.
Competing Interests
The authors have declared that no competing interest exists.
References
1. World Health Organization: Geneva, Switzerland. Depression. Revised 22 March 2018. https://www.who.int/en/news-room/fact-sheets/detail/depression
2. Rich CL, Young D, Fowler RC. San Diego suicide study. I. Young vs old subjects. Arch. Gen. Psychiatry. 1986;43:577-82
3. Henriksson MM, Aro HM, Marttunen MJ. et al. Mental disorders and comorbidity in suicide. Am. J. Psychiatry. 1993;150:935-40
4. Conwell Y, Duberstein PR, Cox C, Herrmann JH, Forbes NT, Caine ED. Relationships of age and axis I diagnoses in victims of completed suicide: a psychological autopsy study. Am. J. Psychiatry. 1996;153:1001-8
5. Harwood D, Hawton K, Hope T, Jacoby R. Psychiatric disorder and personality factors associated with suicide in older people: a descriptive and case-control study. Int. J. Geriatr. Psychiatry. 2001;16:155-65
6. Arsenault-Lapierre G, Kim C, Turecki G. Psychiatric diagnoses in 3275 suicides: a meta-analysis. BMC psychiatry. 2004;4:1-37
7. Chesney E, Goodwin GM, Fazel S. Risks of all-cause and suicide mortality in mental disorders: a meta-review. World Psychiatry. 2014;13:153-60
8. Gao SF, Bao AM. Corticotropin-releasing hormone, glutamate, and gamma-aminobutyric acid in depression. Neuroscientist. 2011;17:124-44
9. Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry. 2010;16:383-406
10. Sudol K, Mann JJ. Biomarkers of suicide attempt behavior: Towards a biological model of risk. Curr. Psychiatry Rep. 2017;19:31
11. Oquendo MA, Sullivan GM, Sudol K. et al. Toward a biosignature for sicide. Am. J. Psychiatry. 2014;171:1259-77
12. Saavedra K, Molina-Marquez AM, Saavedra N, Zambrano T, Salazar LA. Epigenetic modifications of major depressive disorder. Int. J. Mol. Sci. 2016;17:pii E1279
13. Chen D, Meng L, Pei F, Zheng Y, Leng J. A review of DNA methylation in depression. J. Clin. Neurosci. 2017;43(Supplement C):39-46
14. Portela A, Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010;28:1057-68
15. Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J. Cell. Physiol. 2007;213:384-90
16. Hasler G. Pathophysiology of depression: do we have any solid evidence. World Psychiatry. 2010;9:155-61
17. Croarkin PE, Levinson AJ, Daskalakis ZJ. Evidence for GABAergic inhibitory deficits in major depressive disorder. Neurosci. Biobehav. Rev. 2011;35:818-25
18. Pehrson AL, Sanchez C. Altered γ-aminobutyric acid neurotransmission in major depressive disorder: a critical review of the supporting evidence and the influence of serotonergic antidepressants. Drug Des. Devel. Ther. 2015;9:603-24
19. Merali Z, Du L, Hrdina P. et al. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J. Neurosci. 2004;24:1478-85
20. Klempan TA, Sequeira A, Canetti L. et al. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol. Psychiatry. 2009;14:175-89
21. Sequeira A, Klempan T, Canetti L. et al. Patterns of gene expression in the limbic system of suicides with and without major depression. Mol. Psychiatry. 2007;12:640-55
22. Sequeira A, Mamdani F, Ernst C. et al. Global brain gene expression analysis Links glutamatergic and GABAergic alterations to suicide and major depression. PloS one. 2009;4:e6585
23. Lund JS, Lewis DA. Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. J. Comp. Neurol. 1993;328:282-312
24. Fachim HA, Srisawat U, Dalton CF, Reynolds GP. Parvalbumin promoter hypermethylation in postmortem brain in schizophrenia. Epigenomics. 2018;10:519-24
25. Fachim HA, Srisawat U, Dalton CF. et al. Subchronic administration of phencyclidine produces hypermethylation in the parvalbumin gene promoter in rat brain. Epigenomics. 2016;8:1179-83
26. Veerasakul S, Watiktinkorn P, Thanoi S. et al. Increased DNA methylation in the parvalbumin gene promoter is associated with methamphetamine dependence. Pharmacogenomics. 2017;18:1317-22
27. Sibille E, Morris HM, Kota RS, Lewis DA. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int. J. Neuropsychopharmacol. 2011;14:721-34
28. Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol. Psychiatry. 2002;52:708-15
29. Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32:471-82
30. Tripp A, Oh H, Guilloux JP, Martinowich K, Lewis DA, Sibille E. Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am. J. Psychiatry. 2012;169:1194-202
31. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23-38
32. Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15:R37
33. Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr. Res. 2002;55:1-10
34. Li S, Yang Q, Hou Y. et al. Hypomethylation of LINE-1 elements in schizophrenia and bipolar disorder. J. Psychiatr. Res. 2018;107:68-72
35. Nantharat M, Wanitchanon T, Amesbutr M, Tammachote R, Praphanphoj V. Glucocorticoid receptor gene (NR3C1) promoter is hypermethylated in Thai females with major depressive disorder. Genet. Mol. Res. 2015;14:19071-79
36. Masliah E, Dumaop W, Galasko D, Desplats P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics. 2013;8:1030-38
37. Davies MN, Krause L, Bell JT. et al. Hypermethylation in the ZBTB20 gene is associated with major depressive disorder. Genome Biol. 2014;15:R56
38. Januar V, Ancelin ML, Ritchie K, Saffery R, Ryan J. BDNF promoter methylation and genetic variation in late-life depression. Transl. Psychiatry. 2015;5:e619
39. Fuchikami M, Morinobu S, Segawa M. et al. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PloS one. 2011;6:e23881
40. D'Addario C, Dell'Osso B, Galimberti D. et al. Epigenetic modulation of BDNF gene in patients with major depressive disorder. Biol. Psychiatry. 2013;73:e6-7
41. Dell'Osso B, D'Addario C, Carlotta Palazzo M. et al. Epigenetic modulation of BDNF gene: differences in DNA methylation between unipolar and bipolar patients. J. Affect. Disord. 2014;166:330-33
42. Kang HJ, Kim JM, Lee JY. et al. BDNF promoter methylation and suicidal behavior in depressive patients. J. Affect. Disord. 2013;151:679-85
43. Na K-S, Chang HS, Won E. et al. Association between glucocorticoid receptor methylation and hippocampal subfields in major depressive disorder. PloS one. 2014;9:e85425
Author contact
![]() Corresponding author: Tel: +66 55 964 672 Fax: +66 55 964 770 E-mail: sutisatac.th
Corresponding author: Tel: +66 55 964 672 Fax: +66 55 964 770 E-mail: sutisatac.th