International Journal of Medical Sciences 3.2
Impact Factor
ISSN: 1449-1907
Impact Factor
ISSN: 1449-1907
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Issue 1; 2026
- Issue 16; 2025
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Funding
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Funding
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(5):757-765. doi:10.7150/ijms.33532 This issue Cite
Research Paper
Inhibition of N822K T>A mutation-induced constitutive c-KIT activation in AML cells triggers apoptotic and autophagic pathways leading to death
Jianping Xu1, Jinyuan Zheng1, Xiaomeng Fu1, Wei Wu1, Linfen Tao1, Dan Li2, Donghong Lin1 ![]()
1. Department of Laboratory Medicine, School of Medical Technology and Engineering, Fujian Medical University, Fuzhou, Fujian, 350001 China
2. Department of Clinical Laboratory, Fujian Cancer Hospital, Fujian Medical University Cancer Hospital, Fuzhou, Fujian, 350014 China
Received 2019-1-26; Accepted 2019-4-11; Published 2019-5-21
Citation:
Xu J, Zheng J, Fu X, Wu W, Tao L, Li D, Lin D. Inhibition of N822K T>A mutation-induced constitutive c-KIT activation in AML cells triggers apoptotic and autophagic pathways leading to death. Int J Med Sci 2019; 16(5):757-765. doi:10.7150/ijms.33532. https://www.medsci.org/v16p0757.htm
Other stylesAbstract
Background: The D816V mutation of c-KIT can constitutively activate tyrosine kinase, thereby promote core binding factor acute myeloid leukemia (CBF-AML) cell proliferation and inhibit apoptosis. Previous studies have indicated similar proliferation and apoptosis between N822K and D816V mutations.The current study aims to determine the occurrence and potential functions of N822K mutation-induced c-KIT activation in AML cells, and explore possible mechanisms of poor prognosis of CBF-AML.
Methods: c-KIT N822K mutation status in AML cells was determined by exon 17 sequencing. The level of c-KIT expression was detected by flow cytometry (FCM) and colony formation was assessed after hu-SCF stimulation. After exposure to sunitinib (a kind of tyrosine kinase inhibitor, TKI), cell proliferation inhibition was tested by MTT, cell cycle and apoptosis were measured by FCM, autophagy was assessed by fluorescence microscopy and immunoblotting.
Results: Kasumi-1 cell line was detected to bear c-KIT N822K (T>A) mutation. After hu-SCF stimulation, CD117 expression was decreased and the colony formation efficiency was not altered in Kasumi-1 cells. After sunitinib inhibited the c-KIT activity, the colony formation efficiency was reduced, and the half-maximal inhibitory concentration (IC50) of sunitinib was low (0.44±0.17μM) at 48 hours. Moreover, cells were arrested in G0/G1 phase, corresponding to an increase of apoptosis ratio. Acidic vesicular organelles (AVO) were observed along with an altered expression of autophagy-related proteins in Kasumi-1 cells.
Conclusions: Our data indicated that inhibition of N822K T>A mutation-induced constitutive c-KIT activation in AML cells triggered apoptotic and autophagic pathways leading to death, and c-KIT N822K mutation may have clinical application as a CBF-AML treatment target.
Keywords: c-KIT, function mutation, biological behavior, CBF-AML, treatment target
Introduction
C-KIT is a type III receptor tyrosine kinase that the gene is located on chromosome 4q11~12 and the gene sequence is highly conserved. C-KIT is also a 145kDa monomeric transmembrane glycoprotein (976 amino acids in length) that can be activated by stem cell factor (SCF). It includes an extracellular ligand-binding domain, an intracellular tyrosine kinase domain, and a transmembrane domain that connects the two regions. The intracellular tyrosine kinase domain is subdivided by a kinase insert domain into a proximal kinase domain (TK1) and distal kinase domain (TK2). The activation loop (A-loop) is located in the TK2, which is responsible for catalyzing the transfer of the phosphate group from ATP to the enzyme substrate [1]. C-KIT and its ligand (SCF) play important roles in the survival, migration, proliferation, differentiation, and functional activation of hematopoietic progenitor cells [2]. Moreover, c-KIT plays a critical role in the occurrence and expansion of malignant tumors, including gastrointestinal stromal tumor (GIST) and acute myeloid leukemia (AML) [3,4].
AML carrying the t (8; 21) or inv (16)/t (16; 16) chromosomal abnormality are classified as CBF-AML. Gain-of-function mutations of c-KIT can be detected in 48% of patients with CBF-AML [5, 6]. These mutations mainly cluster in exon 17 (encoding the A-loop of the intracellular tyrosine kinase domain) and exon 8 (encoding the extracellular domain of c-KIT). In addition, the exon 17 A-loop mutations on t (8; 21) in adult or child patients with CBF-AML are all associated with high recurrence rate and low survival rate, as well as other indicators of poor prognosis [7, 8]. Multifactor analysis of long-term follow-up data to determine the prognostic effect of c-KIT mutations to t (8; 21) AML showed no significant difference in complete remission rate between patients with mutated c-KIT (mut c-KIT) and wild-type c-KIT (wt c-KIT), but the 5-year accumulative recurrence rate in patients with mut c-KIT (70%) was nearly twice to that with wt c-KIT (36%) [9]. Among the different c-KIT mutations, the N822K and D816V mutations have significant negative impact on overall survival (OS) in CBF-AML [10]. The frequency of c-KIT N822K and D816V mutations is high in the A-loop region. At the A-loop D816V site of c-KIT, the replacement of aspartic acid by valine or tyrosine leads to changes in structure, kinase activity, and substrate-receptor specificity, hematopoietic cell factor-independent growth, and the occurrence of constitutive c-KIT activation [11]. Previous study indicated that N822K mutation had similar biological function with D816Vmutation [12]. However, our previous study found that there might be other effects except proliferation and apoptosis in c-KIT N822K mutation. Based on the above, it is important to further clarify the biological characteristics of AML cells bearing c-KIT N822K mutation.
As already known, sunitinib is a novel, orally available, and multitargeted TKI against FLT3, c-KIT, PDGFR, and VEGFRs [13]. It has been approved by the U.S. Food and Drug Administration for the treatment of GIST and advanced renal cell carcinoma [14]. GIST-T1 cells bear the activating mutation of c-KIT, and were found to induce growth inhibition and apoptosis by sunitinib via blockage autophosphorylation of c-KIT [15]. In the present study, AML cell lines Kasumi-1, HL-60, and NB4 were screened for c-KIT N822K mutation by exon 17 sequencing initially, and the c-KIT N822Kmutation cell line (Kasumi-1) was selected as study object. C-KIT activity after hu-SCF stimulation was detected by FCM and colony formation assay. Furthermore, we investigated changes in the biological behavior of AML cells in association with inhibition of c-KIT activity by sunitinib, and explored possible mechanisms of poor prognosis of CBF-AML.
Materials and Methods
Cell lines and culture
The Kasumi-1 cell line (acute myelogenous leukemia-M2b) was obtained from the Department of Hematology of the Fifth People's Hospital of Shanghai, Fudan University. The HL-60 cell line (acute myelogenous leukemia) and NB4 cell line (acute promyelocytic leukemia) were obtained from the Institute of Hematology, Affiliated Union Hospital of Fujian Medical University. Cells were maintained in RPMI 1640 medium containing 15% fetal bovine serum (FBS), and cultured at 37°C in a 5% CO2 humidified atmosphere.
Cell Sequencing
The target gene was amplified with the following primers: c-KIT-F 5'-TAGTGTATTCACAGAGACTTGGC-3'; c-KIT-R 5'-TTTGACTGCTAAAATGTGTGATA-3'. The PCR conditions were as follows: initial denaturation at 95°C for 3 minutes, 35 cycles of denaturation at 94°C for 30 seconds, annealing at 54°C for 35 seconds, extension at 72°C for 40 seconds, and a final extension at 72°C for 5 minutes. The amplified PCR products were purified using a purification recovery kit (Sangon Biotech, China) and sequenced by a 3730 DNA sequencing analyzer (Sangon Biotech, China).
C-KIT detection by FCM
These three cell lines were cultured in separate tubes overnight inRPMI-1640 medium without FBS, treated with 1μL of hu-SCF (BioVision, USA) for 0, 6, and 12 minutes, and then incubated withanti-CD117-PE (an antibody to a c-KIT immunological marker, Ebioscience, USA). Flow cytometry (FACS calibur, BD Bioscience, USA) was used to quantify the level of c-KIT expression.
Colony formation assay
Hu-SCF treatment: Some cells were treated (others were not treated) with 20ng hu-SCF every other day from first day. There were two types of strategies in sunitinib (Pfizer Inc., USA) treatment. For Kasumi-1 cells, the concentrations of sunitinib were 0, 0.08, 0.16, and 0.32 μM. For HL-60 and NB4 cells, the concentrations of sunitinib were 0, 1.0, 2.0, and 4.0 μM. Cells were seeded in 24-well plates containing methylcellulose (200 cells per well; final concentration of methylcellulose, 0.8%), and incubated at 37°C in a 5% CO2 atmosphere for 3 weeks. The number of colonies was counted and the efficiency of colony formation was calculated. Each assay was repeated for three times.
Cell proliferation inhibition assay by MTT
Cells (Kasumi-1, HL-60, and NB4) were seeded in 200 μL of medium per well in 96-well plates at a density of 1.0×l05 cells/ml, 2.0×l05 cells/ml, and 0.8×l05 cells/ml, respectively. Then the Kasumi-1 cells were incubated with 0.04, 0.08, 0.16, 0.32, and 0.64 μM sunitinib, and HL-60 and NB4 cells were incubated with 0.5, 1.0, 2.0, 4.0, and 8.0 μM sunitinib for 48 hours at 37°C. Proliferation was assessed by the MTT assay and the absorbance at 490nm and 630nm from amicroplate reader (Stat Fax2100, Awareness Technology, USA). SPSS17.0 software was used to calculate the IC50. The experiment was repeated for three times.
Analysis of cell cycle by FCM
Cells at a density of 1.0×106/ml were treated with 0, 0.08, and 0.80 μM sunitinib for 24 and 48 hours, respectively. Cell cycle analysis was performed by using a Cell Cycle Detection Kit (KeyGEN Biotech, China) according to manufacturer's instructions. Flow cytometry (FACS calibur, BD Bioscience, USA) was used to detect the cell cycle distribution.
Analysis of apoptosis by FCM
Cells at a density of 5.0×105/ml were treated with 0, 0.08, and 0.80 μM sunitinib for 24 hours, labeled using an Annexin V-PE/7AAD Apoptosis Detection Kit (BD Bioscience, USA), and counted by FCM (C6, BD Bioscience, USA) to determine the percentage of apoptotic cells.
Detection of acidic vesicular organelles (AVO) by fluorescence microscopy
Cells were incubated with 0, 0.04, 0.16, and 0.64μM sunitinib for 24 and 48 hours, respectively. Then the cells were stained with AO dye (Sigma, USA) according to manufacturer's recommendations, and the AVO was observed under a fluorescence microscope (IX71, Olympus, Japan). The assay was repeated for three times.
Western blot analysis
Cells incubated with 0, 0.04, 0.16, and 0.64 μM sunitinib for 24 hours were subjected to western blot analysis following a standard protocol. Total proteins were extracted from Kasumi-1, HL-60, and NB4 cells, respectively. Blots were probed with primary antibodies directed against Beclin-1 (Cell Signaling Technology, USA) and LC3B (Beyotime Biotech, China) according to manufacturer's recommendations. The internal reference, β-actin, was detected using β-actin antibody (Bioworld Technology, USA). All western blot analyses were repeated at least twice.
Statistical analysis
SPSS 17.0 was used to perform the statistical analysis, and data were expressed as mean ± SD. Comparison of difference between two groups was evaluated by Students' t-test. The difference between more than two groups was determined by one-way analysis of variance (ANOVA). The least significant difference (LSD) test was used in multiple comparisons. P<0.05 was considered as statistically significant.
Results
N822K T>A mutation leads to activation of c-KIT
We identified a homozygous T>A mutation (N822K) in exon 17 of the C-KIT gene inKasumi-1 cell line, but not inHL-60 and NB4 cell lines (Figure 1A). We thus chose HL-60 and NB4 cells as wt c-KIT controls.
We further assessed the level of CD117 (an immunological marker of c-KIT activation) in these three cell lines with or without hu-SCF stimulation. In the absence of hu-SCF, the intensity of CD117 expression was estimated to be 368.98, 19.41, and 14.74 in Kasumi-1, HL-60, and NB4 cells, respectively. After 6 minutes of hu-SCF stimulation, CD117 expression decreased to 317.88in Kasumi-1 cells, increased to 31.24 in HL-60 cells, and did not change in NB4 cells. After 12 minutes of hu-SCF stimulation, these data were 359.64, 25.92, and26.66, respectively (Figure 1B), indicating that hu-SCF could stimulate CD117 expression in HL-60 and NB4 cells in a short time but decreased expression in Kasumi-1 cells in relative longer time (i.e., though CD117 expression was higher at 12 minutes than 6 minutes, it was still lower at 12 minutes than 0 minute).
We further evaluated whether hu-SCF stimulation could affect cell proliferation. The colony formation efficiencies of stimulated HL-60 and NB4 cells were 25.17±2.25% and 78.00±5.22%, significantly higher than that of un-stimulated cells (P=0.033 and P=0.001, Figure 1C), whereas the colony formation efficiencies of stimulated (43.67±2.89%) and un-stimulated (41.17±3.01%) Kasumi-1 cells were statistically similar (P=0.358, Figure 1C). These results demonstrated that hu-SCF could significantly stimulate the colony formation of HL-60 and NB4 cells, but not Kasumi-1 cells.
N822K T>A mutation-induced c-KIT activation increases sensitivity to sunitinib
Intriguingly, treatment with different concentrations of sunitinib decreased the colony formation efficiency of Kasumi-1 cells from 41.17±3.01% to 1.53±1.33% (P<0.001, Figure 2A), HL-60 cells from 20.17±1.53% to 0.00±0.00% (P<0.001, Figure 2B), and NB4 cells from 46.67±3.06% to 1.17±0.76% (P<0.001, Figure 2B). Both the number of colonies and cells per colony were reduced (data not shown). These results suggested that sunitinib could reduce the colony formation efficiency of these three cell lines in a concentration-dependent manner. Notably, the drug concentration required to suppress Kasumi-1 cells colony-forming efficiency was only one tenth of that required to suppress HL-60 and NB4 cells colony-forming efficiency.
To determine whether the cells with c-KIT N822K mutation were more sensitive to sunitinib, we used MTT to assess the IC50 of sunitinib in these three cell lines. At 48 hours, the IC50 of sunitinib in Kasumi-1, HL-60, and NB4 cells was 0.44±0.17μM, 4.62±0.63μM, and 3.04±0.57μM, respectively (Figure 2C-E). The IC50 of sunitinib was about ten-fold higher in HL-60 and NB4 cells than that of Kasumi-1 cells. Such changes were consistent with the results of colony-forming assay.
Inhibiting c-KIT activity induces G0/G1 phase arrest and apoptosis in AML cells with c-KIT N822K mutation
FCM analysis was performed to evaluate the effect of c-KIT activation on cell cycle and apoptosis. With the sunitinib concentration from 0 to 0.80 μM, onlyKasumi-1 cells showed increasing proportion of G0/G1 phase from 55.29±1.87% to 79.28±3.49% (P<0.001, Figure 3A) at 24 hours, and from 54.16±2.16% to 78.68±0.65% (P<0.001, Figure 3B) at 48 hours. The proportion of G0/G1 phase in HL-60 and NB4 cells did not alter significantly over time under the same conditions.
Figure 1
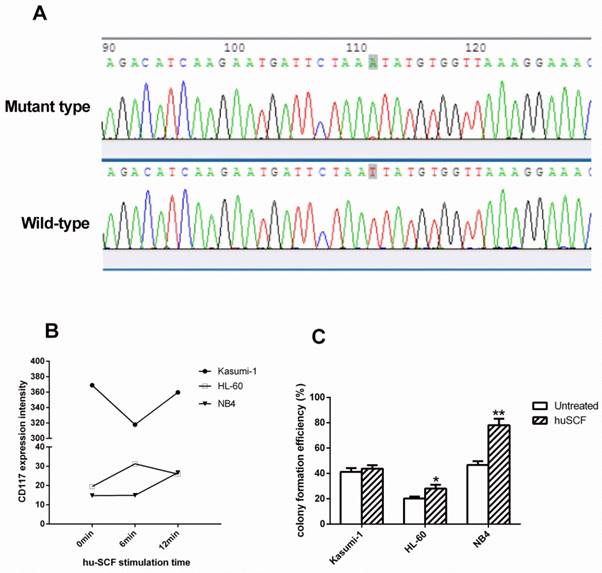
N822K T>A mutation leads to activation of c-KIT. (A) Sequence map of exon 17 showed a typical T>A mutation in codon 822 of the C-KIT gene inKasumi-1 cells. (B) After the three cell lines were starved overnight, the CD117 expression intensity was measured by FCM in cells stimulated for 0, 6, and 12 minutes with hu-SCF. (C) Cell colonies containing ≥40 cells were counted on day 21 using a microscope (×200). (*P<0.050, **P<0.010, vs. non-treated cells).
Figure 2
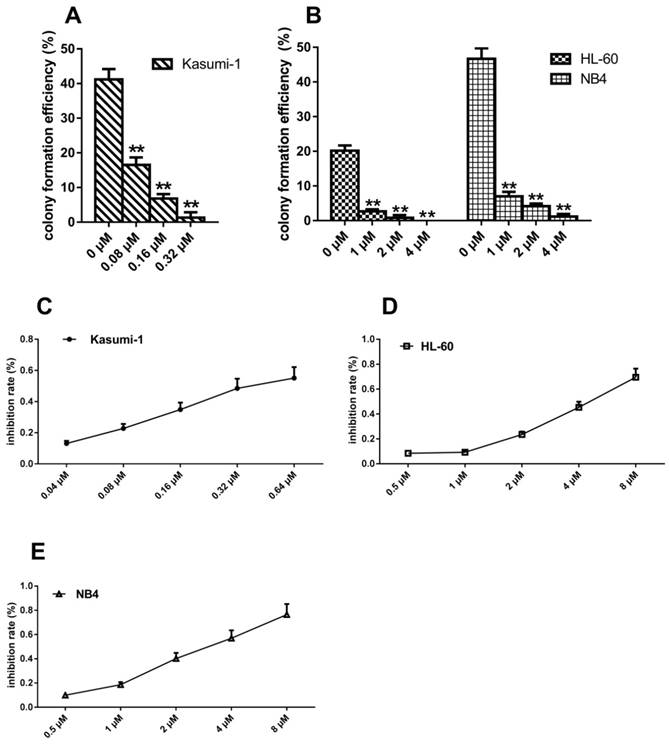
N822KT>A mutation-induced c-KIT activation increases sensitivity to sunitinib. (A, B) Cell colonies containing ≥40 cells were counted on day 21 using a microscope (×200). (C, D, E)Cell proliferation inhibition ratio (%) = [1-(average OD of the treated group-average OD of the blank group) / (average OD of the untreated group-average OD of the blank)] × 100%. The half-maximal inhibition concentration (IC50) was calculated using SPSS17.0 software. (**P<0.010, vs. non-treated cells).
An increase of sunitinib concentration from 0 to 0.80 μM caused an increase of apoptosis ratio in Kasumi-1 cells from 4.71±0.56% to 18.38±1.48% (P<0.001, Figure 3C) after treatment for 24 hours. The apoptosis ratio in HL-60 and NB4 cells after exposure to 0.80 μM sunitinib were 49.78±4.92% and 27.36±1.76%, respectively. These ratios were significantly higher than that of Kasumi-1 cells (P<0.001, Figure 3C). However, the apoptosis ratio of Kasumi-1 cells was lower than expected; we speculated that some Kasumi-1 cells used another death pathway other than the apoptotic one.
Inhibition of c-KIT activity induces autophagy in AML cells with c-KIT N822K mutation
To determine whether sunitinib induced autophagy in Kasumi-1 cells, we assessed the formation of AVO (a morphological characteristic of autophagy) by fluorescence microscopy. At 48 hours, red punctate fluorescence in the cytoplasm of Kasumi-1 cells tended to increase in a sunitinib concentration-dependent manner (Figure 4A). In the cytoplasm of HL-60 and NB4 cells, the red punctuate fluorescence appeared only as a weak red halo, indicating the absence of AVO formation (figures not shown).
To further confirm the existence of autophagy in Kasumi-1 cells at 24 hours following sunitinib-treated, we measured the expression of autophagy-related proteins including Beclin-1 and LC3B. Our data manifested that sunitinib caused significant up-regulation of Beclin-1 and LC3B (P=0.000, Figure 4C&D) in a concentration-dependent manner in Kasumi-1 cells but not in HL-60 and NB4 cells, suggesting the existence of autophagy in Kasumi-1 cells.
Discussion
The Kasumi-1 cell line was established from a AML patient with M2 subtype and has been proved as a very useful tool to study the cellular and molecular profile of AML with t (8;21) [16]. It also provides a typical example of the “two hits” model for the CBF leukemogenesis, for it simultaneously bears the translocation t (8; 21) and the Asn822Lys (N822K) c-KIT mutation [17]. By 17 exon sequencing, we confirmed that the AML cell line Kasumi-1 had the c-KIT N822KT>A mutation, which made it suitable for our study. Since leukemia was highly heterogeneous, we chose two granulocyte lines of AML cell lines including HL-60 and NB4 to be sequenced at the same time. Then HL-60 and NB4 cell lines without the c-KIT N822K T>A mutation were chosen as controls.
Figure 3
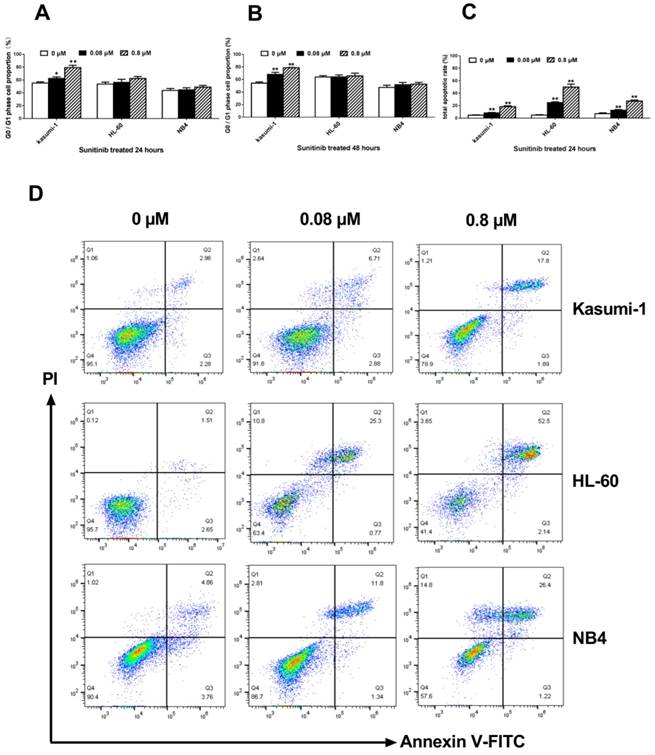
Inhibiting c-KIT activity induces G0/G1 phase arrest and apoptosis in AML cells with c-KIT N822K T>A mutation. (A, B) The proportion of cells in G0/G1 phase after incubation with different concentrations of sunitinib at 24 and 48 hours in Kasumi-1, HL-60and NB4 cells. (C, D)After sunitinib-treated for 24 hours, the apoptosis ratio was measured by FCM analysis. (*P<0.050, **P<0.010, vs. non-treated cells).
Figure 4
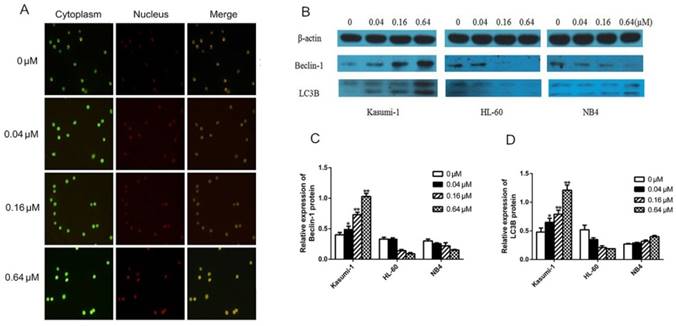
Inhibition of c-KIT activity induces autophagy in AML cells with c-KIT N822K mutation. (A) AO-stained Kasumi-1 cells observed by fluorescence microscopy (×200), the positive cells contained a red fluorescent nucleus, green fluorescent cytoplasm, and red punctate cytoplasmic (AVO). (B) After cells were treated with increasing concentrations of sunitinib for 24 hours, levels of Beclin-1 and LC3B were determined by western blotting using rabbit anti-Beclin-1 (1:1000), rabbit anti-LC3B (1:500) , and mouse anti-β-actin (1:3000, as internal control).The density of (C) Beclin-1 and (D) LC3B bands were normalized to that of β-actin. (*P<0.050, **P<0.010, vs. non-treated cells).
Since previous study reported that the N822K mutation within the A-loop of KIT affected the P-1 residue adjacent to Tyr821 that represented the potential autophosphorylation site, leading to constitutive ligand-independent autophosphorylation, the activation of the KIT [18]. Initially we used hu-SCF, the c-KIT ligand to stimulate the three cell lines, in order to demonstrate c-KIT N822K mutation might induce the activation of c-KIT in AML cells. We monitored CD117 by FCM after hu-SCF stimulation. Our data demonstrated that cells without N822K T>A mutation (HL-60 and NB4) were sensitive to hu-SCF compared to untreated cells, and that their response (increased intensity of CD117 expression) was rapid. Since the kinetics of c-KIT activation was rapid and could be detectable in several minutes after exposure to SCF, binding of SCF induced c-KIT dimerization and intermolecular tyrosine phosphorylation by the kinase domain. That is to say, the c-KIT might be activated by homodimerization and auto-phosphorylation through c-KIT ligand [19]. In contrast, the response of Kasumi-1 cells bearing c-KIT N822K T>A mutation was decreased when compared with untreated cells. A reasonable explanation for these results in Kasumi-1 cells might be that stress caused by the exogenous hu-SCF stimulus triggered negative feedback down-regulation of c-KIT expression on the cell surface. Subsequent colony formation assay results also supported this explanation. Colony formation efficiency was SCF-dependent growth in HL-60 and NB4 cells, whereas SCF-independent growth in Kasumi-1 cells bearing c-KIT N822Kmutation. Such result was consistent with published work of SCF-independent KIT receptor activation in cells with c-KIT N822K mutation [12]. The downstream signaling activation of PI3K/Akt has demonstrated that the expression level of p-PI3K was higher in Kasumi-1 cells with N822K mutation than HL-60 and NB4 cells in our previous work [20]. There was also evidence that the c-KIT D816V mutation was constitutively associated with higher level of PI3K activity than wt c-KIT [21]. In brief, from another angle, the current study provided additional evidence that c-KIT N822K T>A mutation leaded to constitutive activation of c-KIT.
To further study the effect of N822K T>A mutation-induced c-KIT activation on the biological behavior of AML cells, we utilized TKI to interfere with c-KIT activation. Previous data revealed that sunitinib exhibited the higher intracellular accumulation in Kasumi-1 that bearing the c-KIT N822KT>A mutation, indicating that cells were more sensitive to sunitinib [22]. Thus, we used sunitinib as the inhibitor in our study. Takayuki and colleagues found that sunitinib induced growth inhibition and apoptosis of GIST-T1 cells through blockage the activating of c-KIT mutation [15]. We found that sunitinib could inhibit proliferation of the three AML cell lines. The colony formation capacity was reduced and the drug concentration required to inhibit colony formation was higher in HL-60 and NB4 cells than that of Kasumi-1 cells. The IC50 of sunitinib was about ten-fold higher in HL-60 and NB4 cells than Kasumi-1 cells with c-KIT N822KT>A mutation. We focused on the PI3k/Akt pathway to investigate the effect of sunitinib in c-KIT. Our previous study showed that the levels of p-PI3K and p-Akt were reduced in Kasumi-1 cells with a sunitinib concentration-dependent manner, which was sensitive to sunitinib treatment, but not in HL-60 and NB4 cells [20]. Moreover, other TKI mediated c-KIT inhibition could interrupt the association between p85PI3K subunit and c-KIT Tyr721, already described in Kasumi-1 cells [23, 24]. Thus, we believed that the high efficacy of sunitinib in inhibiting colony formation and cell viability might be due to the activation of c-KIT inhibition. Hence, c-KIT N822KT>A mutation could be a potential and effective target for CBF-AML treatment.
Other aspects of the cell cycle and apoptosis were also involved. We found that inhibiting c-KIT activation induced G0/G1 phase arrest in a significantly higher proportion of Kasumi-1 cells than both HL-60 and NB4 cells, which was consistent with others' results [12]. Thus, our result demonstrated that inhibition of c-KIT activity could inhibit proliferation through cell cycle arrest, suggesting that the target of proliferation regulation might involve the cell cycle in Kasumi-1 cells bearing c-KIT N822K T>A mutation. Besides, we also found some new phenomena like the ratio of apoptosis in Kasumi-1 cells was increased slightly, in contrast to HL-60 and NB4 cells at all drug concentrations. Such a slight increase of apoptosis ratio in Kasumi-1 cells was not consistent with the epigenetic results. We proposed that some Kasumi-1 cells died following a different manner after inhibition of c-KIT activity.
The effect of autophagy on the survival of AML cells was double-edged. Autophagy promoted the apoptosis of AML cells, but on the other hand, it could protect AML cells and maintain their survival [25]. Goussetis found that autophagy was the critical mechanism in As2O3-dependent anti-leukemia activity. As2O3-treated could induce autophagy in AML cell line U937, and this phenomenonwas partially reversed by the autophagy inhibitor chloroquine [26]. We further evaluated the role of autophagy in the death of Kasumi-1 cells with c-KIT N822K T>A mutation. As a result, the AVO was observed in Kasumi-1 cells at 48 hours after sunitinib treatment, and the number of AVO increased in a sunitinib concentration-dependent manner. Nevertheless, more specific evidence of autophagy was needed. We detected autophagy specific markers membrane LC3B and regulatory Beclin-1. Our autophagy proteins assay showed significant changes at 24 hours after sunitinib treatment rather than 48 hours, so the changes of LC3B and Beclin-1 expressions occurred before the appearance of AVO. Notably, the autophagy proteins expressed up-regulation in a dose-dependent manner in Kasumi-1 cells. However, no similar phenomenon occurred in HL-60 and NB4 cells under the same conditions.
It was inspiring to confirm the occurrence of autophagy in Kasumi-1 cells bearing c-KIT N822K mutation. Here we attributed the aberrant apoptosis ratio in Kasumi-1 cells to autophagy. Thus, inhibiting constitutive c-KIT activation that triggered by the N822K T>A mutation induced AML cells death via apoptosis- and autophagy-dependent manners. Thus we speculated that constitutive c-KIT activation due to the N822K mutation might be an important factor to maintain the malignant characteristics of AML cells and affect tumor burden in CBF-AML. These promising data suggested the potential of c-KIT N822K mutation as a therapeutic target for CBF-AML.
In conclusion, these results indicated that c-KIT N822K mutation induced to constitutive activation of c-KIT would affect the biological behavior of AML cells. In vitro results of our present study served to illuminate the unfavorable prognosis of CBF-AML bearing c-KIT N822K T>A mutation. Although the autophagy phenotype is potentially interesting, further investigation is needed to confirm that it is caused by c-KIT inhibition. It is important to investigate whether the autophagy can be initiated by c-KIT shRNA.
Abbreviations
CBF-AML: core binding factor acute myeloid leukemia; TKI: tyrosine kinase inhibitor; FCM: flow cytometry; PDGFR: platelet-derived growth factor receptor; VEGFR: vascular endothelial growth factor receptor; IC50: half-maximal inhibitory concentration; AVO: acidic vesicular organelles; SCF: stem cell factor; GIST: gastrointestinal stromal tumor; OS: overall survival.
Acknowledgements
The authors would like to thank Prof. Ligen Liu (Department of Hematology of the Fifth People's Hospital of Shanghai, Shanghai, China) for donation of the Kasumi-1cell line.
Funding
This work was financially supported by the National Natural Science Foundation of China (81371879, 81601827) and the Provincial Natural Science Foundation of Fujian (2015J01320, 2016J01770).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Andrã© C, Martin E, Cornu F, Hu WX, Wang XP, Galibert F. Genomic organization of the human c-kit gene: evolution of the receptor tyrosine kinase subclass III. Oncogene. 1992;7:685-691
2. Ashman LK, Griffith R. Therapeutic targeting of c-kit in cancer. Expert Opin Inv Dru. 2013;22:103-115
3. Ali S, Ali S. Role of c-KIT/SCF in cause and treatment of gastrointestinal stromal tumors (GIST). Gene. 2007;401:38-45
4. Heinrich MC, Blanke CD, Druker BJ, Corless CL. Inhibition of kit tyrosine kinase activity: a novel molecular approach to the treatment of kit-positive malignancies. J Clin Oncol. 2002;20:1692-1703
5. Boissel N, Leroy H, Brethon B, Philippe N, De BS, Auvrignon A. et al. Incidence and prognostic impact of c-KIT, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia(CBF-AML). Leukemia. 2006;20:965-970
6. Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ. et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118:5593-5603
7. Sun A, Ji S, Chen S, Zhang T, Wu D. Study on the pathogenesis and clinical significance of FLT3 and C-KIT mutations in core-binding factor AML. Blood. 2016;128:5236
8. Tokumasu M, Murata C, Shimada A, Ohki K, Hayashi Y, Saito AM. et al. Adverse prognostic impact of KIT mutations in childhood CBF-AML: the results of the Japanese pediatric leukemia/lymphoma study group AML-05 trial. Leukemia. 2015;29:2438-2441
9. Paschka P, Marcucci G, Ruppert AS, Mrózek K, Chen H, Kittles RA. et al. Adverse prognostic significance of kit mutations in adult acute myeloid leukemia with inv (16) and t (8; 21): a Cancer and Leukemia Group B study. J Clin Oncol. 2006;24:3904-3911
10. Wang D, Qiao C, Xiao M, Geng Z, Shang Z, He J. et al. Integrative analysis of prognostic factors in Chinese core binding factor leukemia. Biochem Bioph Res Co. 2012;428:411-415
11. Voisset E, Lopez S, Dubreuil P, De SP. The tyrosine kinase FES is an essential effector of KIT D816V proliferation signal. Blood. 2007;110:2593-2599
12. Omori I, Yamaguchi H, Miyake K, Miyake N, Kitano T, Inokuchi K. D816V mutation in the kit gene activation loop has greater cell-proliferative and anti-apoptotic ability than N822K mutation in core-binding factor acute myeloid leukemia. Exp Hematol. 2017;52:56-64
13. Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J. et al. Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl) amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46:1116-1119
14. Atkins M, Jones CA, Kirkpatrick P. Sunitinib maleate. Nat Rev Drug Discov. 2006;5:279-280
15. Ikezoe T, Yang Y, Nishioka C, Bandobashi K, Nakatani H, Taguchi T. et al. Effect of SU11248 on gastrointestinal stromal tumor-T1 cells: enhancement of growth inhibition via inhibition of 3-kinase/Akt/mammalian target of rapamycin signaling. Cancer Sci. 2006;97:945-951
16. Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8; 21 chromosome translocation. Blood. 1991;77:2031-2036
17. Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genom Hum G. 2002;3:179-198
18. Serve H, Yee NS, Stella G, Sepp-Lorenzino L, Tan JC, Besmer P. Differential roles of PI3K-kinase and Kit tyrosine 821 in Kit receptor-mediated proliferation, survival and cell adhesion in mast cells. EMBO J. 1995;14:473-483
19. Broudy VC, Lin NL, Bühring HJ, Komatsu N, Kavanagh TJ. Analysis of c-kit receptor dimerization by fluorescence resonance energy transfer. Blood. 1998;91:898-906
20. Xu J, Li G, Wu W, Fu X, Zheng J, Li D. et al. Effect of PI3K/Akt/ mTOR signal pathway-mediated N822K mutation on apoptosis induced by TKI sunitinib in AML cells. Chin J Immunol. 2018;34:376-380
21. Chian R, Young S, Danilkovitch-Miagkova A, Rönnstrand L, Leonard E, Ferrao P. et al. Phosphatidylinositol 3 kinase contributes to the transformation of hematopoietic cells by the D816V c-Kit mutant. Blood. 2001;98:1365-1373
22. Hu S, Niu H, Minkin P, Orwick S, Shimada A, Inaba H. et al. Comparison of antitumor effects of multitargeted tyrosine kinase inhibitors in acute myelogenous leukemia. Mol Cancer Ther. 2008;7:1110-1120
23. Larizza L, Magnani I, Beghini A. The Kasumi-1 cell line: a t (8;21)-kit mutant model for acute myeloid leukemia. Leukemia Lymphoma. 2005;46:247-255
24. Mpakou VE, Kontsioti F, Papageorgiou S, Spathis A, Kottaridi C, Girkas K. et al. Dasatinib inhibits proliferation and induces apoptosis in the kasumi-1 cell line bearing the t(8;21)(q22;q22) and the N822K c-kit mutation. Leukemia Res. 2013;37:175-182
25. Torgersen ML, Simonsen A. Autophagy: friend or foe in the treatment of fusion protein-associated leukemias? Autophagy. 2013;9:2175-2177
26. Goussetis DJ, Altman JK, Glaser H, Mcneer JL, Tallman MS, Platanias LC. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J Biol Chem. 2010;285:29989-29997
Author contact
![]() Corresponding author: Prof. Donghong Lin, Associate dean of School of Medical Technology and Engineerin, Fujian Medical University. Tel: +86-591-8356-9212; FAX: +86-591-8356-9250; E-mail:lindh65edu.cn
Corresponding author: Prof. Donghong Lin, Associate dean of School of Medical Technology and Engineerin, Fujian Medical University. Tel: +86-591-8356-9212; FAX: +86-591-8356-9250; E-mail:lindh65edu.cn
Citation styles
APA
Xu, J., Zheng, J., Fu, X., Wu, W., Tao, L., Li, D., Lin, D. (2019). Inhibition of N822K T>A mutation-induced constitutive c-KIT activation in AML cells triggers apoptotic and autophagic pathways leading to death. International Journal of Medical Sciences, 16(5), 757-765. https://doi.org/10.7150/ijms.33532.
ACS
Xu, J.; Zheng, J.; Fu, X.; Wu, W.; Tao, L.; Li, D.; Lin, D. Inhibition of N822K T>A mutation-induced constitutive c-KIT activation in AML cells triggers apoptotic and autophagic pathways leading to death. Int. J. Med. Sci. 2019, 16 (5), 757-765. DOI: 10.7150/ijms.33532.
NLM
Xu J, Zheng J, Fu X, Wu W, Tao L, Li D, Lin D. Inhibition of N822K T>A mutation-induced constitutive c-KIT activation in AML cells triggers apoptotic and autophagic pathways leading to death. Int J Med Sci 2019; 16(5):757-765. doi:10.7150/ijms.33532. https://www.medsci.org/v16p0757.htm
CSE
Xu J, Zheng J, Fu X, Wu W, Tao L, Li D, Lin D. 2019. Inhibition of N822K T>A mutation-induced constitutive c-KIT activation in AML cells triggers apoptotic and autophagic pathways leading to death. Int J Med Sci. 16(5):757-765.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.