International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(3):384-393. doi:10.7150/ijms.30084 This issue Cite
Research Paper
PM2.5 Upregulates MicroRNA-146a-3p and Induces M1 Polarization in RAW264.7 Cells by Targeting Sirtuin1
Yijue Zhong, Jiping Liao, Yan Hu, Yunxia Wang, Chao Sun, Cheng Zhang, Guangfa Wang ![]()
Department of Respiratory and Critical Care Medicine, Peking University First Hospital, Beijing, China, 100034.
Received 2018-9-20; Accepted 2018-12-31; Published 2019-1-29
Citation:
Zhong Y, Liao J, Hu Y, Wang Y, Sun C, Zhang C, Wang G. PM2.5 Upregulates MicroRNA-146a-3p and Induces M1 Polarization in RAW264.7 Cells by Targeting Sirtuin1. Int J Med Sci 2019; 16(3):384-393. doi:10.7150/ijms.30084. https://www.medsci.org/v16p0384.htm
Other stylesAbstract
Background: Fine particulate matter (PM2.5) exposure is proved to be associated with illnesses, but the mechanism is not clear. Potential effects of PM2.5 on innate immunity have become a hotspot recently. Confronting PM2.5, macrophages are able to be activated and induce inflammatory responses. Whether PM2.5 exposure affects macrophage polarization and associated mechanisms remains to be further explored. Afterwards, whether Sirtuin1 (SIRT1) an important intermediate regulator in various physiological processes takes part in the macrophage polarization induced by PM2.5 is unknown. MiRNAs are acknowledged as key regulator in posttranscriptional modification and our previous study found that miR-146a is a novel biomarker of PM2.5 exposure. Thus, we propose a hypothesis, PM2.5 exposure induces M1 polarization and miR-146a-3p is a potential upstream regulator by targeting SIRT1.
Methods: RAW264.7 cells were treated with different concentrations of PM2.5 for 24h. The expressions of cytokines and key molecular markers were detected by qRT-PCR, Western blotting and ELISA. The activation degree of TLRs and NF-κB was assessed by Western blotting. The specific agonist and antagonist of SIRT1 were used to explore the potential role of SIRT1 in M1 polarization induced by PM2.5. MiR-146a-3p mimic and inhibitor were pre-transfected into RAW264.7 cells and the effects on M1 polarization induced by PM2.5 were evaluated. Luciferase analysis was used to identify the binding site of miR-146a-3p and SIRT1.
Results: PM2.5 increased the mRNA and protein expression of M1 markers including interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α) and inducible nitric oxide synthase (iNOS) in RAW264.7 cells. The protein level of TLR4 was significantly increased and the ratio of phosphorylated NF-κB p65 versus p65 subunit was also elevated in PM2.5 group. PM2.5 decreased the protein level of SIRT1 but not the mRNA expression in vitro and in vivo experiments. Pre-treatment with SIRT1 agonist SRT1720 rescued the PM2.5 induced M1 response. Whereas, SIRT1 antagonist EX527 augment the effect. MiR-146a-3p was upregulated in PM2.5 treated RAW264.7 cells. Luciferase experiments reported that SIRT1 was directly targeted by miR-146a-3p. Overexpression of miR-146a-3p downregulated the expression of SIRT1 protein in untreated RAW264.7 cells. Importantly, inhibition of miR-146a-3p upregulated SIRT1 protein and suppressed M1 polarization in PM2.5 treated RAW264.7 cells.
Conclusions: These results suggested that PM2.5 induces the inflammatory M1 polarization and TLR4/NF-κB signal transduction pathway might be involved in the process. MiR-146a-3p is a novel regulator of PM2.5 exerted M1 polarization by targeting SIRT1.
Keywords: PM2.5, macrophages, polarization, sirtuin1, miR-146a-3p
Introduction
Fine particulate matter (PM2.5) is the particulate matter with diameter equal to or less than 2.5 μm and has become a serious threat to human health as a number of epidemiological studies have demonstrated marked association between PM2.5 exposure and increased incidence and aggravation of respiratory and cardiovascular diseases [1, 2]. Once inhaled, PM2.5 deposits in lung tissues and diffuses in blood inducing lung and systematic injuries [3, 4]. Although the intrinsic molecular mechanisms are not well understood, inflammatory responses and oxidative stress have been proposed as fundamental mechanisms underlying the process [5, 6].
As the first defense line, macrophage is one of the most important parts of innate immune system and is a cross-link between innate immunity and adaptive immunity. In general, macrophages can be polarized into two distinct phenotypes: the classically activated macrophages (M1) and alternatively activated macrophages (M2). M1 macrophages which are mainly induced by lipopolysaccharide (LPS) are considered to have higher antigen-presenting capacity and release a lot of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6). On the contrary, M2 macrophages mainly induced by interleukin-4 (IL-4) act as anti-inflammatory ones and take part in regulating angiogenesis, tissue remodeling and wound healing [7-10]. The imbalance of M1 and M2 macrophages causes damage to the body and poses threat to human health. Toll-like receptor (TLR) can bound with LPS or other pathogens and promote the downstream events consequently. TLR/nuclear factor kappa B (NF-κB) is a classical signal pathway which is implicated in various diseases especially inflammatory responses [11-13]. Nowadays, whether PM2.5 induces macrophage polarization directly and the signal transduction pathway has not been fully elucidated.
Sirtuin1 (SIRT1), a type III histone deacetylase, belongs to the silent information regulator 2 (Sir2) family and regulates a variety of physiological processes including oxidative stress, inflammation, cellular senescence, proliferation, apoptosis, and DNA damage response due to its ability to deacetylate various intracellular signaling molecules and chromatin histones [14-17]. Recent studies also indicate that SIRT1 plays an important role in the regulation of immune responses. Zhang et al reported that SIRT1 is an anti-inflammation factor and leads to amelioration of macrophage function [18]. Whether SIRT1 is a potential regulator of macrophage polarization induced by PM2.5 needs to be further explored.
MicroRNAs (miRNAs) are one member of endogenous noncoding RNAs family which participates in regulation of cell development, proliferation, differentiation and death. It has been suggested that the changes in their expression and their posttranscriptional regulator function are associated with many human diseases [19, 20]. Researchers have shown that air pollutants including PM2.5 can alter miRNA expression in recent years. Serena et al found an association between exposure to ambient PM2.5 and downregulation of several miRNAs in elderly men [21]. Our pervious study showed that miR-146, miR-139 and miR-340 expressions are elevated during acute exposure to PM2.5 in mice [22]. However, the role of these miRNAs especially in regulating the macrophage polarization caused by PM2.5 is not clear.
In this study, we established the hypothesis that PM2.5 induces M1 polarization in RAW 264.7 cells. Then, we explored related regulatory mechanisms and found that PM2.5 regulates the expression of miR-146a-3p and augments M1 polarization by inhibiting SIRT1.
Materials and Methods
PM2.5 collection and preparation
PM2.5 samples were collected persistently by using high volume sampler system (Staplex® NO PM-2.5 SSI, USA) at the roof of a ward building in Peking University First Hospital which located in the central of downtown Beijing from October to December 2016. PM2.5 samples on the glass fiber filters were extracted in accordance with the method described previously [23]. Final extracted PM2.5 samples were stored in the ultra-low temperature freezer. Different dosages of PM2.5 were weighted, re-suspended in culture medium with 1% fetal bovine serum or saline and sonicated for complete dissolution.
Cell culture and treatment
The mouse macrophage cell line RAW264.7 was cultured in Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, USA), 100U/ml penicillin and 100μg/ml streptomycin (Gibco, USA) and maintained at 37°C in a saturated humidity atmosphere containing 95 % air and 5 % CO2. Cells were treated with different concentrations of PM2.5 (0, 5, 10, 25, 50, 100μg/ml) for 24h or pretreated with SRT1720 (1μM, Selleck Biochem, USA) or EX527 (10μM, Selleck Biochem, USA) for 6h followed by PM2.5 (25μg/ml) for 24h. MiR-146a-3p mimic and its negative control, anti-miR-146a-3p and anti-miR-146a-3p negative control were purchased from RIBOBIO Company (Guangzhou, China). MiR-146a-3p, anti-miR-146a-3p and corresponding control microRNAs were complexed respectively with ribo FECT ™ CP Transfection Kit (RIBOBIO, Guangzhou, China) according to the manufacturer's instructions. RAW264.7 cells were transfected with miR-146a-3p mimic (50μM), miR-146a-3p inhibitor (100μM) and corresponding negative control for 24h followed by PM2.5 (25μg/ml) for 24h.
Cell viability assay
RAW264.7 Cells were cultured in the 96-well plate and there were six wells for each group. After treatment with different concentrations of PM2.5 (0, 5, 10, 25, 50, 100μg/ml) for 24h, the mediums were discarded and the fresh mediums containing 10μl CCK-8 solution were added in each well according to the instruction of EnoGene Cell Counting Kit-8 (CCK-8, DOJINDO, Japan). The plate was put in the incubator for about 3 hours. The wavelength of each well at 450nm was measured by microplate reader when the time was up.
Animals and endotracheal instillation of PM2.5
Twelve adult Balb/c mice (female, 9weeks old) were purchased from SPF Biotechnology Company (Beijing, China) and were housed in the specific pathogen-free environment with room temperature (23°C-25°C), relative humidity (40%-70%) and 12h light/dark cycles. The study conformed to the animal welfare and was approved by Laboratory Animal Ethics Committee of Peking University First Hospital (NO.201706).The mice were randomly divided into control and PM2.5 treated groups. Mice were anesthetized and endotracheal instillation of 50μl saline or PM2.5 (5mg/kg) dissolved in the equal volume of saline was performed in the control group or PM2.5 treated group respectively once a week for consecutive eight weeks. The mice were sacrificed for further experiments on the 7th day from the last instillation.
Quantitative assessment of mRNA and miRNA Expression
TRIZOL reagent (Invitrogen Life Technologies, USA) was used to extract total RNA. 1μg total RNA quantified by Nanodrop 2000 (Thermo Fisher Scientific, USA) from each sample was reversely transcribed to 20 μl complementary DNA (cDNA) according to the instruction of RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, USA). For miRNAs, reverse transcription of 600ng total RNA was conducted by using Mir-XTM miRNA First Strand Synthesis Kit (Clontech Laboratories, Inc. USA). The primers of mRNA and miRNA specific primers were purchased from Sango Biotech Company (Shanghai, China). The primers used for qRT-PCR were as follows: SIRT1 (sense: CGGCTACCGAGGTCCATATAC, antisense: ACAATCTGCCACAGCGTCAT);
IL-6 (sense: CCCCAATTTCCAATGCTCTCCT, antisense: CATAACGCACTAGGTTTGCCG);
iNOS (sense: GTTCTCAGCCCAACAATACAAGA, antisense: GTGGACGGGTCGATGTCAC);
TNF-α (sense: CATCTTCTCAAAATTCGAGTGACAA, antisense: TGGGAGTAGACAAGGTACAACCC);
Arg-1 (sense: AGCACTGAGGAAAGCTGGTC, antisense: CAGACCGTGGGTTCTTCACA);
CD206 (sense: GGCTGATTACGAGCAGTGGA, antisense: CATCACTCCAGGTGAACCCC);
β-actin (sense: GCTTCTTTGCAGCTCCTTCGT, antisense: AGCGCAGCGATATCGTCATC);
miR-146a-3p (5'-3': CCTGTGAAATTCAGTTCTTCAG).The forward and reverse primer of U6 were included in the Mir-XTM miRNA First Strand Synthesis Kit. qRT-PCR was carried out in a 20μl reaction system containing 1μl cDNA and 10μl POWER SYBR® Green Master Mix (Thermo Fisher Scientific, USA) on Step One Plus Real-Time PCR System (7500, Applied Biosystems, USA). The relative expression of mRNA or miRNA was calculated by 2-ΔΔCT method as described elsewhere and normalized to the expression of β-actin or U6 respectively.
Luciferase analysis
According to the binding site on SIRT1 mRNA 3′-untranslated region (3′-UTR), a wild-type (wt) SIRT1-3′-UTR gene or a mutated (mut) SIRT1-3′-UTR gene was constructed and cloned into the pMIR-REPORT miRNA expression reporter vector (Obio Technology Corp, Shanghai, China). The HEK293T cells (National Infrastructure of Cell Line Resource, Shanghai, China) were transfected with empty vector, SIRT1-3′-UTR-wt vector and SIRT1-3′-UTR-mut vector with miR-146a-3p mimic or scramble control. After 48 h, the transfected cells were analyzed by Dual-Luciferase Reporter Assay System (Promega Corporation, Fitchburg, WI, USA).
Western blotting
The total protein was fractionated from the whole Cell lysate using RIPA buffer and cocktail (Sigma-Aldrich, USA). Protein concentrations were quantified by BCA method (Beyotime, Shanghai, China). Equivalent amounts of extracted protein were resolved on SDS-PAGE and transferred onto NC membrane. After blocking the background staining with 5% non-fat milk in TBST, the membranes were incubated in primary antibodies against SIRT1 (1:500, Abcam), iNOS (1:1000, Abcam), TLR4 (1:500, Abcam), TLR2 (1:500, Abcam), NF-κB p65 (1:1000, CST), phospho-NF-κB p65 (1:1000, CST), β-actin (1:2000, ZSGB-BIO, China) overnight at 4°C. Immuno-reactive proteins were detected using HRP conjugated secondary antibodies and ECL kit (Merck Millipore, USA) according to the manufacturer's instructions. The bands were quantitated by ImageJ v1.28 system and the fold expression was indicated as the relative protein level.
Enzyme-linked immunosorbent assays (ELISA)
After treatment, the culture supernatants were isolated, centrifuged and stored at -80°C until measured. The commercially available ELISA kits (NOVUS, China) were used for detection of TNF-α and IL-6 according to the manufacturer's instructions. The absorbance at 450 nm was measured and corrected at 570nm in a microplate reader.
Figure 1
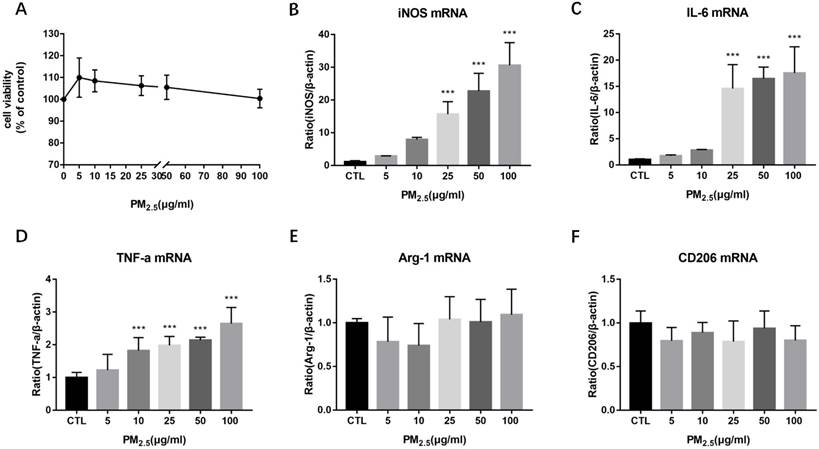
Effect of PM2.5 exposure on macrophage polarization. RAW264.7 cells were treated with 0, 5, 10, 25, 50, 100μg/ml PM2.5 for 24h. (A) Effect on cell viability detected by CCK-8 kit. (B-D) Effect on the mRNA expression of M1 markers quantified by qRT-PCR. (E-F) Effect on the mRNA expression of M2 markers quantified by qRT-PCR. ***p<0.001 versus control group.
Statistical analysis
Statistical analysis of the data was performed using the SPSS14.0 system (SPSS Inc, USA) by two tailed Student's t-test for comparison of two groups or analysis of variance (ANOVA) and appropriate post hoc analysis for comparison of more than two groups. Bar graphs were protracted using Prism (GraphPad Software Ltd, version 5.0 USA). All data in the figures were expressed as means ± standard deviation (SD). Values of p< 0.05 were considered to be statistically significant.
Results
Effect of PM2.5 exposure on macrophage polarization
To evaluate the effect of PM2.5 exposure on the cell viability of macrophages, RAW 264.7 cells were exposed to 0, 5, 10, 25, 50 and 100μg/ml PM2.5 for 24h and the cell viability was detected by CCK-8 assay. It showed that the cell viability was reduced slightly compared with the control group but there is no significant difference (Figure 1A). To determine the impact of PM2.5 exposure on macrophage polarization, RAW 264.7 cells were treated with different dosages of PM2.5 (0, 5, 10, 25, 50 and 100μg/ml) for 24h. The mRNA expressions of M1 and M2 markers were quantified by qRT-PCR. The results showed that the mRNA expressions of M1 markers were gradually increased with the increase of PM2.5 concentration but not that of M2 markers. The mRNA expressions of iNOS and IL-6 were elevated significantly by 25, 50 and 100 μg/ml PM2.5 treatment and TNF-α mRNA expressions were significantly higher in 10, 25, 50 and 100μg/ml PM2.5 group compared with the control group (Figure 1B-1F). Consistent with the results of qRT-PCR, Western blotting analysis showed that compared with the control group, the protein levels of iNOS were significantly increased by 25, 50 and 100μg/ml PM2.5 treatment (Figure 2A, Figure 2B). The concentrations of IL-6 and TNF-α in the supernatants were also significantly increased compared with the control group by using Commercialized ELISA kits (Figure 2C, Figure 2D). To explore whether TLR/NF-κB signal pathway was related with the PM2.5 inducing inflammatory responses, the activation degree of TLRs and NF-κB was assessed by Western blotting. Our data revealed that the protein levels of TLR4 but not those of TLR2 were significantly increased after 25, 50 and 100μg/ml PM2.5 treatment. Furthermore, the ratios of phosphorylated NF-κB p65 versus p65 subunit were also elevated in PM2.5 groups compared with the control group (Figure 2E, Figure 2F). Taken together, these results demonstrated that PM2.5 can induce M1 polarization directly and TLR4/NF-κB signal transduction pathway might be involved in the process.
Effect of PM2.5 exposure on SIRT1 expression
To examine the expressions of SIRT1 after PM2.5 exposure, we detected SIRT1 protein and mRNA levels both in vitro and in vivo experiments. SIRT1 protein levels were markedly down-regulated by PM2.5 stimulation in RAW264.7 cells (Figure 3A, Figure 3B). Similarly, compared to the saline treated mice, PM2.5 instillation significantly decreased the expression of SIRT1 protein in the lung (Figure 3D, Figure 3E). However, there was no significant change in SIRT1 mRNA level neither in vitro nor in vivo experiments (Figure 3C, Figure 3F). These results indicated that there might have post-transcriptional regulation of SIRT1 expression after PM2.5 exposure.
Figure 2
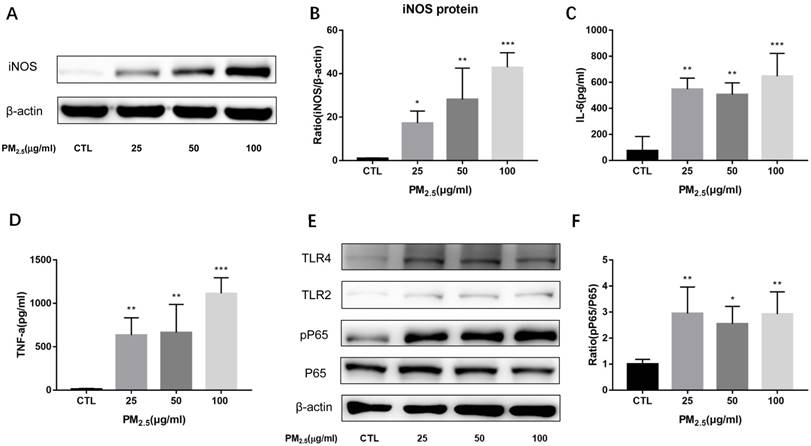
Effect of PM2.5 exposure on macrophage polarization. RAW264.7 cells were treated with 0, 25, 50, 100μg/ml PM2.5 for 24h. (A-B) Effect on the protein expression of iNOS quantified by Western blotting. (C-D) Effect on the release of IL-6 and TNF-α in the supernatant quantified by ELISA. (E-F) Effect on the protein expression of TLR4, TLR2, phosphorylated NF-κB p65 and NF-κB p65 quantified by Western blotting. *p<0.05, **p<0.01, *** p<0.001 versus control group.
The role of SIRT1 in the regulation of M1 polarization induced by PM2.5
To verify whether SIRT1 take part in the regulation of M1 polarization induced by PM2.5, RAW264.7 cells were pretreated with the SIRT1 specific agonist SRT1720 or antagonist EX527 for 6h followed by the PM2.5 (25μg/ml) exposure for 24h. Compared with single PM2.5 exposure group, the mRNA expressions of iNOS and IL-6 were decreased in the PM2.5 plus SRT1720 group. At the meanwhile, the mRNA expressions of iNOS, IL-6 and TNF-α were significantly higher in the PM2.5 plus EX527 group than the single PM2.5 exposure group (Figure 4A-4C). Consistently, the protein level of iNOS was increased in the PM2.5 plus EX527 group and decreased in the PM2.5 plus SRT1720 group compared with the single PM2.5 group (Figure 4E, Figure 4F). The above results showed that SIRT1 might be an upstream regulator of M1 polarization induced by PM2.5.
PM2.5 increased the expression of miR-146a-3p and SIRT1 was a potential target of miR-146a-3p
In order to observe the expression of miR-146a-3p in PM2.5 treated RAW264.7 cells, we detected the expression of miR-146a-3p by qRT-PCR. Compared with control group, the expression of miR-146a-3p showed a significant elevation in 25, 50, 100μg/ml PM2.5 exposure group, though there is not explicit dose dependent relationship (Figure 5A). To explore the mechanism of miR-146a-3p in PM2.5 exposed RAW264.7 cells, we found that SIRT1 was a potential regulatory targets of miR-146a-3p predicted in three bioinformatics databases (TargetScan, miRanda and miRWalk). As shown above, the protein expression but not the mRNA expression of SIRT1 was decreased after PM2.5 exposure. It indicated that miR-146a-3p might decrease the expression of SIRT1 through post-transcriptional regulation. The putative target sequence is located in the 494-500nt of the 3'-UTR of murine SIRT1 mRNA. To verify this, the luciferase analysis was used to identify the predicted binding site of miR-146a-3p and SIRT1. The SIRT1 mRNA containing the wild-type (wt) putative binding site or mutated (mut) binding site of miR-146a-3p were constructed (Figure 5D) and cloned into the luciferase expressing pMIR vector. SIRT1-3′-UTR-wt vector or SIRT1-3′-UTR-mut vector was co-transfected with mmu-miR-146a-3p mimic or scrambled control (miR-NC) into HEK293T cells respectively. The results showed that the relative luciferase activity decreased significantly in cells transfected with SIRT1-3′-UTR-wt vector, but luciferase activity showed no significant change in cells transfected with SIRT1-3′-UTR-mut (Figure 5E). This result confirmed that miR-146a-3p directly regulated SIRT1.
Figure 3
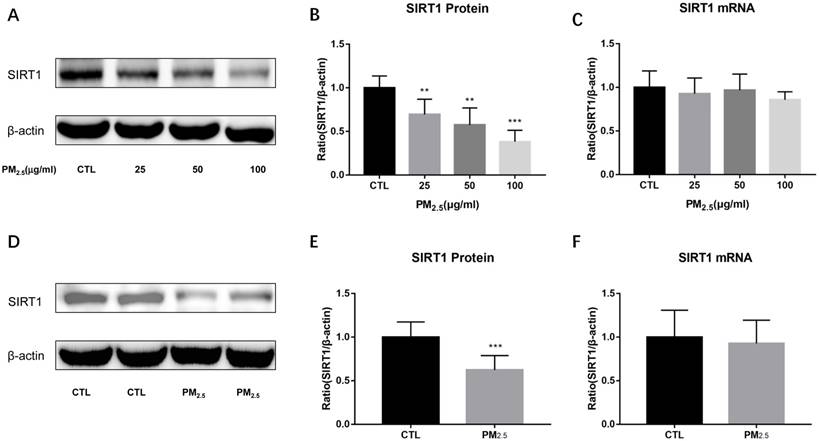
Effect of PM2.5 exposure on SIRT1 expression. (A-C) RAW264.7 cells were treated with 0, 25, 50, 100μg/ml PM2.5 for 24h. The protein and mRNA expressions of SIRT1 were detected by Western blotting and qRT-PCR. (D-F) Endotracheal instillation of saline or PM2.5 (5mg/kg) re-suspended in saline was performed in Balb/c mice once a week for consecutive eight weeks. The protein and mRNA expressions of SIRT1 in lungs were detected by Western blotting and qRT-PCR. **p<0.01 and *** p<0.001 versus control group.
Overexpression of miR-146a-3p directly downregulated the expression of SIRT1 protein
To further explore the potential regulation relationship between miR-146a-3p and SIRT1, untreated RAW264.7 cells were transfected with miR-146a-3p mimic, inhibitor and scrambled controls for 24h. The efficiency of transfection and the expression of SIRT1 mRNA were detected by qRT-PCR (Figure 5B, Figure 5C). The expression of SIRT1 protein was detected using Western blotting analysis (Figure 5F). The SIRT1 mRNA expression showed no significant difference among different treatment. However, SIRT1 protein increased significantly after inhibition of miR-146a-3p and decreased significantly after overexpression of miR-146a-3p. These results showed that Overexpression of miR-146a-3p downregulated the expression of SIRT1 protein in untreated RAW264.7 cells.
Inhibition of miR-146a-3p upregulated SIRT1 protein and suppressed M1 polarization in PM2.5 treated RAW264.7 cells
Lastly, to observe whether miR-146a-3p could regulate M1 macrophage polarization in PM2.5 treated RAW264.7 cells, cells were pre-transfected with miR-146a-3p mimic, inhibitor and scrambled controls for 24h followed by PM2.5 (25μg/ml) treatment. Western blotting results showed the SIRT1 protein expression decreased significantly in cells treated with mimic compared with the control group. Whereas, The SIRT1 protein expression significantly increased in cells treated with inhibitor (Figure 6D, Figure 6E). Overexpression of miR-146a-3p significantly increased the mRNA expression of M1 markers including iNOS, IL-6 and TNF-α in PM2.5 treated RAW264.7 cells. To the contrary, inhibition of miR-146a-3p significantly decreased the mRNA expression of iNOS, IL-6 and TNF-α (Figure 6A-6C). The Western blotting analysis also shown that overexpression of miR-146a-3p significantly increased the protein expression of iNOS. Conversely, inhibition of miR-146a-3p significantly decreased the protein expression of iNOS (Figure 6D, Figure 6F). Above results demonstrated that decreased miR-146a-3p upregulated SIRT1 protein and suppressed M1 polarization in PM2.5 treated RAW264.7 cells.
Figure 4
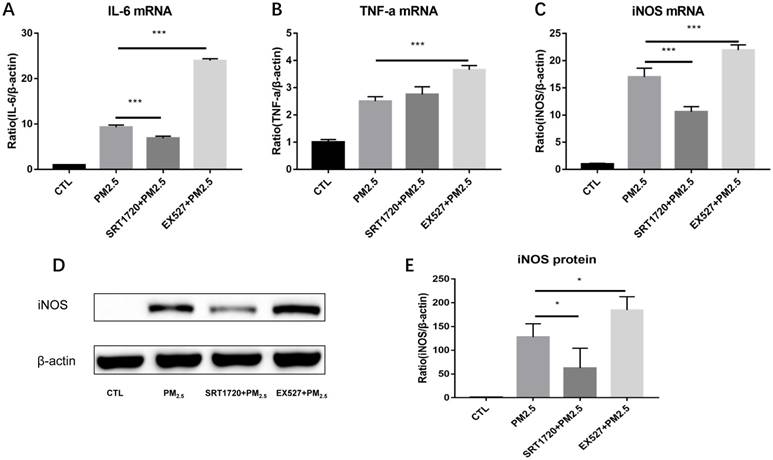
The role of SIRT1 in the regulation of M1 polarization induced by PM2.5. RAW264.7 cells were pretreated with the SIRT1 specific agonist SRT1720 or antagonist EX527 for 6h followed by the PM2.5 (25μg/ml) treatment for 24h. (A-C) The mRNA expressions of IL-6, TNF-α and iNOS were detected by qRT-PCR. (D-E) The protein expression of iNOS was detected by Western blotting. *p<0.05 and *** p<0.001 between indicated groups.
Discussion
China, a developing country which is undergoing a rapid period of urbanization and industrialization suffers from environmental pollution inevitably. One of most serious and widespread environmental pollution is air pollution and it can't be eliminated in short term. PM2.5, a complex mixture of small particles and liquid droplets in the air is one of the most important sources of air pollution. Similar to other studies [24-26], our previous studies indicated that PM2.5 contained high concentration of endotoxin up to 20.8ng/m3 and toxic heavy metals such as Cu, Zn, Al, Mn and Pb [22, 23]. Once inhaled, PM2.5 can cause damage to the whole body. Although the associated mechanism has not yet been elucidated, inflammation and oxidative stress are involved in the process.
As the first line of defense, the balance of M1 and M2 macrophage is crucial for inflammatory responses in handling PM2.5. Previous studies have indicated that PM from different sources affected pro-inflammatory cytokines secretion of M1 and anti-inflammatory response of M2 [27-29]. In this study, we found that 25μg/ml and higher concentration of PM2.5 collected from Beijing, one of the most polluted cities in China significantly induce the expression and release of pro-inflammatory cytokines of M1. Whereas, different concentrations of PM2.5 didn't affect the expression of M2 macrophage markers. Consistent with previous studies [27, 30], our study demonstrated that PM2.5 exposure induce M1 polarization directly and initiate persistent inflammatory response though the chemical composition of PM2.5 is complicated and the mechanism underlying the effect has not been fully elucidated. Macrophages recognize pathogens via pattern recognition receptors (PRR), such as TLR. TLR2 and TLR4 are the major members of TLR family and have their own ligand and distribution features [31, 32]. Our results indicated that the PM2.5 exposure increased the expression of TLR4 but not that of TLR2. Subsequently, activation of NF-κB was demonstrated. TLR4/NF-κB signal transduction pathway might be involved in the M1 polarization induced by PM2.5.
Figure 5
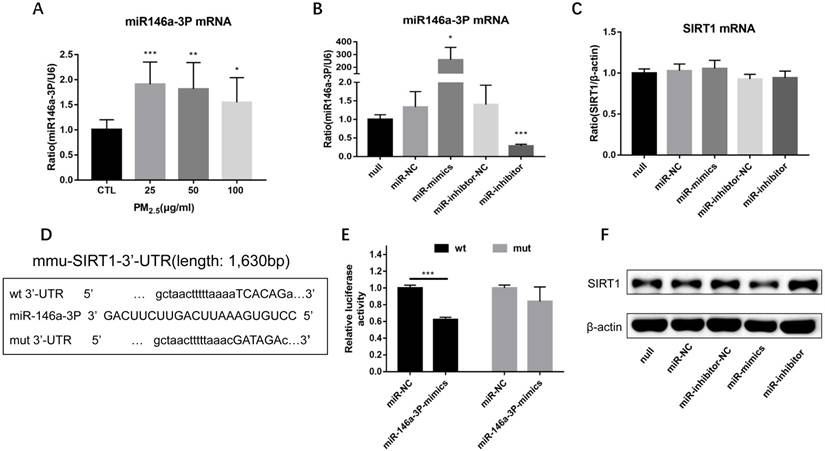
PM2.5 increased the expression of miR-146a-3p and SIRT1 was a potential target of miR-146a-3p. (A) RAW264.7 cells were treated with 0, 25, 50, 100 μg/ml PM2.5 for 24h. The mRNA expression of miR-146a-3p was detected by qRT-PCR. (B-C) Relative expression of miR-146a-3p normalized against the U6 endogenous control and SIRT1 mRNA normalized against the β-actin endogenous control in untreated RAW264.7 transfected with miR-146a-3p mimic, inhibitor or scrambled controls. (D) Wild-type and mutant binding sites of miR-146a-3p in the 3′-UTR of SIRT1. (E) Luciferase analysis. The results showed that miR-146a-3p mimics decreased the fluorescence intensity in cells transfected with SIRT1-3′-UTR-wt but did not change the fluorescence intensity in cells transfected with SIRT1-3′-UTR-mut. (F) Western blotting analysis of SIRT1 in untreated RAW264.7 transfected with miR-146a-3p mimic, inhibitor or scrambled controls. *p<0.05, **p<0.01, ***p<0.001 versus control group or between indicated groups.
Figure 6
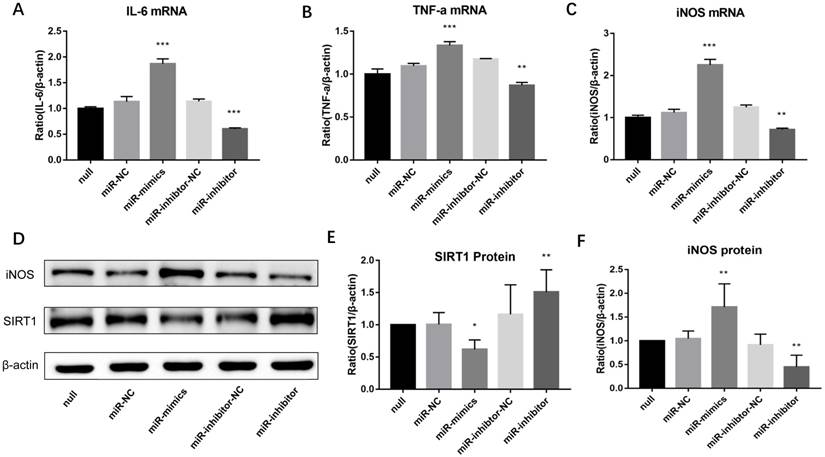
Inhibition of miR-146a-3p upregulated SIRT1 protein and suppressed M1 polarization in PM2.5 treated RAW264.7 cells. RAW264.7 cells were pre-transfected with miR-146a-3p mimic, inhibitor and scrambled controls for 24h followed by PM2.5 (25μg/ml) treatment. (A-C) The mRNA expressions of M1 markers were quantified by qRT-PCR. Inhibition of miR-146a-3p significantly decreased the mRNA expression of IL-6, TNF-α and iNOS in PM2.5 treated RAW264.7 cells. (D-F) Western blotting analysis of iNOS and SIRT1. Inhibition of miR-146a-3p significantly increased the protein expression of SIRT1 and decreased the protein expression of iNOS. *p<0.05, **p<0.01, ***p<0.001 versus control group.
In order to further explore the related regulatory mechanism of M1 polarization induced by PM2.5, the potential role of SIRT1 which was proved to be anti-inflammation factor was detected [33, 34]. Firstly, we found that PM2.5 exposure significantly decreased the protein level of SIRT1 in vitro and in vivo. This is similar to previous studies [35, 36] except that our study showed PM2.5 exposure didn't significantly alter the mRNA expression of SIRT1. Then, we found that pretreatment with SIRT1 agonist SRT1720 significantly decreased the expression of M1 markers in PM2.5 exposed RAW264.7 cells. Conversely, pretreatment with SIRT1 antagonist EX527 further increased the expression of these markers. These results demonstrated a novel notion that SIRT1 is the upstream regulator of M1 polarization induced by PM2.5 and there might have post-transcriptional regulation of SIRT1 expression.
MiRNAs are acknowledged as key molecules in post-transcriptional modification. They can bind directly to the 3′-UTR of target mRNA leading to mRNA degradation or translation suppression. Due to the stable expression in specific tissues and the conservative role in different species, miRNAs have been considered to have therapeutic potential and became a research focus in recent years [37, 38]. Previous studies have indicated a variety of air pollution exposure associated miRNAs [39-42], but there is still no consensus because the designs of these studies were different. Our previous study indicated that 10 miRNAs (miR-691, miR-181a, miR-146a, miR-146b, miR-21a-5p, miR-129, miR-155, miR-139-5p, miR-21a-3p and miR-340) were upregulated in lung tissues of mice exposed to PM2.5 and involved in regulating Th1/Th2 balance [22]. Among these miRNAs, miR-146a has been shown to regulate pro-inflammatory cytokine production in endotoxin-stimulated human monocytes [43, 44] and has been proved to be associated with enhanced phagocytic activity in patient monocytes [45]. However, the role of miR-146a family particularly miR-146a-3p in PM2.5 exposed macrophage has not been elucidated. In this study, we found that miR-146a-3p directly targeted SIRT1 which is the upstream regulator of macrophage polarization as mentioned above. Our experiments demonstrated that overexpression of miR-146a-3p downregulated SIRT1 protein in untreated RAW264.7 cells. Not only that, inhibition of miR-146a-3p upregulated SIRT1 protein and suppressed M1 polarization in PM2.5 treated RAW264.7 cells. This suggested that miR-146a-3p might be a potential therapeutic target of inflammatory responses induced by PM2.5.
There are some limitations in this study. First, we demonstrated different dosage of PM2.5 exposure for 24h induced M1 polarization directly in RAW264.7 cells, the effect on macrophages from other sources and for other exposure time need to be further explored. Second, we found that SIRT1 is the upstream regulator of M1 polarization induced by PM2.5, but the signal pathways involved was not analyzed in this study. Last, our study illustrated that miR-146a-3p is a very promising therapeutic target for the treatment of PM2.5 induced inflammatory responses. Next, transfected animal studies should be prepared to find a reasonable dose and measurement of treatment.
Conclusions
In a summary, this study proved that PM2.5 upregulates the expression of miR-146a-3p and induces the inflammatory M1 polarization by inhibiting SIRT1 in RAW264.7 cells. TLR4/NF-κB signal transduction pathway might be involved in the process. Overexpression of miR-146a-3p directly downregulated the expression of SIRT1 protein. Inhibition of miR-146a-3p upregulated SIRT1 protein and suppressed M1 polarization in PM2.5 treated RAW264.7 cells. These findings suggest that miR-146a-3p could be a potential therapeutic target for PM2.5 induced inflammatory responses.
Acknowledgements
The work was supported by the grants from the National Natural Science Foundation of China (No. 81500014), the Beijing Municipal Natural Science Foundation (No.7161013 and No.7174357), the Capital Medical Development and Scientific Research Fund (No.2016-1-4071) and National Key Research and Development Plan (No.2017YFC1309500).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liang F, Xiao Q, Gu D. et al. Satellite-based short- and long-term exposure to PM2.5 and adult mortality in urban Beijing, China. Environ Pollut. 2018;242:492-9
2. Rodriguez-Villamizar LA, Rojas-Roa NY, Blanco-Becerra LC, Herrera-Galindo VM, Fernandez-Nino JA. Short-Term Effects of Air Pollution on Respiratory and Circulatory Morbidity in Colombia 2011(-)2014: A Multi-City, Time-Series Analysis. Int J Environ Res Public Health. 2018:15
3. Bernstein JA, Alexis N, Barnes C. et al. Health effects of air pollution. J Allergy Clin Immunol. 2004;114:1116-23
4. Brunekreef B, Holgate ST. Air pollution and health. Lancet. 2002;360:1233-42
5. Donaldson K, Tran CL. Inflammation caused by particles and fibers. Inhal Toxicol. 2002;14:5-27
6. Shukla A, Timblin C, BeruBe K. et al. Inhaled particulate matter causes expression of nuclear factor (NF)-kappaB-related genes and oxidant-dependent NF-kappaB activation in vitro. Am J Respir Cell Mol Biol. 2000;23:182-7
7. Zhu L, Zhao Q, Yang T, Ding W, Zhao Y. Cellular metabolism and macrophage functional polarization. Int Rev Immunol. 2015;34:82-100
8. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958-69
9. Murray PJ, Allen JE, Biswas SK. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14-20
10. Sun C, Sun L, Ma H. et al. The phenotype and functional alterations of macrophages in mice with hyperglycemia for long term. J Cell Physiol. 2012;227:1670-9
11. Kaisho T, Akira S. Toll-like receptors and their signaling mechanism in innate immunity. Acta Odontol Scand. 2001;59:124-30
12. Bhaskar S, Sudhakaran PR, Helen A. Quercetin attenuates atherosclerotic inflammation and adhesion molecule expression by modulating TLR-NF-kappaB signaling pathway. Cell Immunol. 2016;310:131-40
13. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197-216
14. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
15. Rahman I, Kinnula VL, Gorbunova V, Yao H. SIRT1 as a therapeutic target in inflammaging of the pulmonary disease. Prev Med. 2012;54(Suppl):S20-8
16. Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1-13
17. Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins-novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841-53
18. Zhang R, Chen HZ, Liu JJ. et al. SIRT1 suppresses activator protein-1 transcriptional activity and cyclooxygenase-2 expression in macrophages. J Biol Chem. 2010;285:7097-110
19. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421-33
20. Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21-37
21. Fossati S, Baccarelli A, Zanobetti A. et al. Ambient particulate air pollution and microRNAs in elderly men. Epidemiology. 2014;25:68-78
22. Hou T, Liao J, Zhang C, Sun C, Li X, Wang G. Elevated expression of miR-146, miR-139 and miR-340 involved in regulating Th1/Th2 balance with acute exposure of fine particulate matter in mice. Int Immunopharmacol. 2018;54:68-77
23. Zhao C, Liao J, Chu W. et al. Involvement of TLR2 and TLR4 and Th1/Th2 shift in inflammatory responses induced by fine ambient particulate matter in mice. Inhal Toxicol. 2012;24:918-27
24. Ebisu K, Bell ML. Airborne PM2.5 chemical components and low birth weight in the northeastern and mid-Atlantic regions of the United States. Environ Health Perspect. 2012;120:1746-52
25. Zhang Y, Lang J, Cheng S. et al. Chemical composition and sources of PM1 and PM2.5 in Beijing in autumn. Sci Total Environ. 2018;630:72-82
26. Rogula-Kozlowska W, Blaszczak B, Szopa S. et al. PM(2.5) in the central part of Upper Silesia, Poland: concentrations, elemental composition, and mobility of components. Environ Monit Assess. 2013;185:581-601
27. Vogel CF, Garcia J, Wu D. et al. Activation of inflammatory responses in human U937 macrophages by particulate matter collected from dairy farms: an in vitro expression analysis of pro-inflammatory markers. Environ Health. 2012;11:17
28. Jaguin M, Fardel O, Lecureur V. Exposure to diesel exhaust particle extracts (DEPe) impairs some polarization markers and functions of human macrophages through activation of AhR and Nrf2. PLoS One. 2015;10:e0116560
29. Sijan Z, Antkiewicz DS, Heo J. et al. An in vitro alveolar macrophage assay for the assessment of inflammatory cytokine expression induced by atmospheric particulate matter. Environ Toxicol. 2015;30:836-51
30. Zhao Q, Chen H, Yang T. et al. Direct effects of airborne PM2.5 exposure on macrophage polarizations. Biochim Biophys Acta. 2016;1860:2835-43
31. Miyata R, van Eeden SF. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol Appl Pharmacol. 2011;257:209-26
32. Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675-80
33. Yeung F, Hoberg JE, Ramsey CS. et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369-80
34. Wu Z, Liu MC, Liang M, Fu J. Sirt1 protects against thrombomodulin down-regulation and lung coagulation after particulate matter exposure. Blood. 2012;119:2422-9
35. Jin X, Su R, Li R. et al. Amelioration of particulate matter-induced oxidative damage by vitamin c and quercetin in human bronchial epithelial cells. Chemosphere. 2016;144:459-66
36. Yang L, Duan Z, Liu X, Yuan Y. N-acetyl-l-cysteine ameliorates the PM2.5-induced oxidative stress by regulating SIRT-1 in rats. Environ Toxicol Pharmacol. 2018;57:70-5
37. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-22
38. Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
39. Duan J, Yu Y, Li Y. et al. Comprehensive understanding of PM2.5 on gene and microRNA expression patterns in zebrafish (Danio rerio) model. Sci Total Environ. 2017;586:666-74
40. Chen R, Li H, Cai J. et al. Fine Particulate Air Pollution and the Expression of microRNAs and Circulating Cytokines Relevant to Inflammation, Coagulation, and Vasoconstriction. Environ Health Perspect. 2018;126:017007
41. Li X, Ding Z, Zhang C. et al. MicroRNA-1228(*) inhibit apoptosis in A549 cells exposed to fine particulate matter. Environ Sci Pollut Res Int. 2016;23:10103-13
42. Li Y, Duan J, Yang M. et al. Transcriptomic analyses of human bronchial epithelial cells BEAS-2B exposed to atmospheric fine particulate matter PM2.5. Toxicol In Vitro. 2017;42:171-81
43. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481-6
44. El Gazzar M, Church A, Liu T, McCall CE. MicroRNA-146a regulates both transcription silencing and translation disruption of TNF-alpha during TLR4-induced gene reprogramming. J Leukoc Biol. 2011;90:509-19
45. Pauley KM, Stewart CM, Gauna AE. et al. Altered miR-146a expression in Sjogren's syndrome and its functional role in innate immunity. Eur J Immunol. 2011;41:2029-39
Author contact
![]() Corresponding author: Guangfa Wang, M.D., Ph.D., FCCP. Department of Respiratory and Critical Care Medicine, Peking University First Hospital, No.8 Xishiku Street, Xicheng District, Beijing, China, 100034. Email: wangguangfacom.
Corresponding author: Guangfa Wang, M.D., Ph.D., FCCP. Department of Respiratory and Critical Care Medicine, Peking University First Hospital, No.8 Xishiku Street, Xicheng District, Beijing, China, 100034. Email: wangguangfacom.
Citation styles
APA
Zhong, Y., Liao, J., Hu, Y., Wang, Y., Sun, C., Zhang, C., Wang, G. (2019). PM2.5 Upregulates MicroRNA-146a-3p and Induces M1 Polarization in RAW264.7 Cells by Targeting Sirtuin1. International Journal of Medical Sciences, 16(3), 384-393. https://doi.org/10.7150/ijms.30084.
ACS
Zhong, Y.; Liao, J.; Hu, Y.; Wang, Y.; Sun, C.; Zhang, C.; Wang, G. PM2.5 Upregulates MicroRNA-146a-3p and Induces M1 Polarization in RAW264.7 Cells by Targeting Sirtuin1. Int. J. Med. Sci. 2019, 16 (3), 384-393. DOI: 10.7150/ijms.30084.
NLM
Zhong Y, Liao J, Hu Y, Wang Y, Sun C, Zhang C, Wang G. PM2.5 Upregulates MicroRNA-146a-3p and Induces M1 Polarization in RAW264.7 Cells by Targeting Sirtuin1. Int J Med Sci 2019; 16(3):384-393. doi:10.7150/ijms.30084. https://www.medsci.org/v16p0384.htm
CSE
Zhong Y, Liao J, Hu Y, Wang Y, Sun C, Zhang C, Wang G. 2019. PM2.5 Upregulates MicroRNA-146a-3p and Induces M1 Polarization in RAW264.7 Cells by Targeting Sirtuin1. Int J Med Sci. 16(3):384-393.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.