Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(2):324-330. doi:10.7150/ijms.27757 This issue Cite
Research Paper
A Pilot Study of Aberrant CpG Island Hypermethylation of SPRED1 in Acute Myeloloid Leukemia
Jingwen Sun, Jinjing Zhang, Yue Wang, Yan Li, Rui Zhang ![]()
Department of Hematology, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning Province, 110001, China
Received 2018-6-8; Accepted 2018-10-13; Published 2019-1-1
Abstract
Background: Epigenetic silencing of tumor suppressor genes plays important role in acute myeloid leukemia (AML). Recently, SPRED1, a negative regulator of the RAS MAPK pathway, is identified as a tumour suppressor downregulated in AML. However, little is known regarding its underlying dysregulation in AML. In this study, we investigated methylation status of SPRED1 promoters and their association with mRNA levels in AML.
Methods: Methylation level were measured in four regions of SPRED1 (#1: 310 bp ~ 723 bp, #2: 810 bp ~ 1299 bp, #3: 1280 bp ~ 1742 bp and #4: 1715 bp ~ 2059 bp) in a total of 16 patients with de novonon-acute promyelocytic leukemia (non-APL) and three patients who got complete remission after induction treatment using the Sequenom MassARRAY platform. Quantitative real-time polymerase chain reaction (q-RT PCR) was used to analyze SPRED1 mRNA levels.
Results: AML patients had a significantly higher average methylation level than controls at regions of #1_CpG_1 (p= 0.04) and #1_CpG_11 (p =0.002). The methylation values for #1_CpG_11 were negatively correlated with mRNA levels (r= -0.558, p=0.013) but there was no significant association between #1_CpG_1 methylation status and mRNA levels (r=-0.103, p=0.675) in AML patients. There was no significant difference in the methylation level when comparing with clinical biochemical parameters and treatment response (p>0.05). Mutations of epigenetic regulation genes such as DNMT3A, TET2 and IDH1/2 were most frequently observed in patients with higher methylation levels. Decreased methylation levels were revealed in three patients who got complete remission.
Conclusions: Aberrant methylation statuses of the SPRED1 promoter regions are associated with the downregulation of gene transcription in AML. The methylation level is probably associated with the treatment response of AML. Mutations of epigenetic regulation genes might be involved in the epigenetic aberration of SPRED1.
Keywords: SPRED1, DNA methylation, AML, Tumor suppressor gene
Introduction
Acute myeloid leukemia (AML) is a malignant clonal disease of hemopoietic stem cells characterized by the inhibition of differentiation and subsequent accumulation of cells at various stages of immaturity, and also by the decreased production of normal hemopoietic ingredients. Various abnormal signaling pathways are involved in adult AML. The Ras/MAPK signaling pathway has already been shown to be involved in the pathogenesis of AML, including distinct prognostic genes, such as NF1 [1], NRAS, and KRAS [2]. SPRED1 is an emerging regulator of Ras/MAPK signaling pathway aberrant in AML [3-4].
Human sprout-related EVH1 domain-containing 1 (SPRED1) gene is found in the rat osteoclast cDNA library, as shown by Yoshimura and his colleagues in as early as 2001 [5]. It is located in 15q13.2 containing seven exons. Human SPRED1 protein consists of 444 amino acids and 3 domains: EVH1 domain at the N-terminal, c-kit domain in the middle, and SPRY-related domain at the C-terminal, belonging to the SPRED family together with SPRED2 and SPRED3 [5]. The expression of human SPRED1 have been discovered dominantly in lung, brain, spinal cord, and kidney, while it is low in liver, pancreas, prostate, thyroid, muscle, skeleton, and bone marrow [6-7]. SPRED1, interacting with the NF1 protein‒neurofibromin, downregulates the Ras/MAPK signaling pathway [8-10]. Ras/MAPK pathway and cell malignancy transformation were found to be inhibited by SPRED1 in hepatocellular carcinoma, prostate cancer, and lymphoma cell lines. Also, SPRED1 was identified as a tumor suppressor of the Ras/MAPK pathway and downregulated in pediatric AML [4]. However, the underlying cause of SPRED1 downregulation was not clear. Although the mutation of SPRED1 has a predisposition to leukemia [11], subsequent studies indicated that the mutation and deletion of SPRED1 were not common in AML [4,12].
Epigenetics refers to changes in gene expression but without changes in the DNA sequence itself. Currently, the most widely studied epigenetic modification in humans is DNA methylation, which occurs almost exclusively in the context of CpG dinucleotides that control the transcriptional activity of genes [13] and is observed in various diseases, such as glioblastoma [14], lymphocytic leukemia [15], and AML [16]. Recent years have witnessed a large amount of genomic data; however, it has already been reported that almost 50% of all patients with AML belonging to the intermediate-risk group lack cytogenetic abnormalities [17], and a great proportion of the patients carry unknown AML-associated driver genes [18-19]. It is apparent that epigenetic modifications in AML are more widespread than can be explained by recurrent somatic mutations alone.
The goal of the present study was to explore the epigenetic status of SPRED1 and its influence on AML.
Methods
Patients and controls
Bone marrow samples were obtained prior to treatment from 16 de novo patients with non-acute promyelocytic leukemia (non-APL), classified according to the criteria given by the World Health Organization [20], and three patients who achieved complete remission (CR) after induction treatment. Patients younger than 14 years old were excluded from this study. The median age of patients was 34.5 years old (range, 16 to 67 years). The median percentage of blast cells in bone marrow was 66% (range, 24.5% to 95.2%). The treatment and the response assessment were based on Chinese expert consensus on treatment of AML (2009) [21]. CR was defined as a bone marrow with normal hematopoiesis, blasts in bone marrow less than 5%, granulocyte count≥1.0 × 109/L, platelet count≥100 × 109/L, no myeloid blasts in the peripheral blood and no extramedullary disease. Control samples were taken from seven adult healthy volunteers with median age of 47 years old (range, 24 to 66 years). The study was approved by the Ethics Committee of the First Affiliated Hospital of China Medical University (# AF-SOP-07-1.0-01).
RNA extraction and quantitative reverse transcription polymerase chain reaction
RNA was extracted from mononuclear cells of bone marrow samples using the TRIzol reagent (TaKaRa, Japan) and reverse transcribed using a PrimeScript Reverse Transcription Reagent Kit with gDNA Eraser (Perfect Real Time, TaKaRa) according to the manufacturer's protocol. The integrity of synthesized cDNA was confirmed using β-actin as the endogenous control.
Quantitative reverse transcription polymerase chain reaction (PCR) was performed using 7900 real-time PCR system and SYBR Green (TaKaRa) as a double-stranded DNA-specific dye. Target genes were amplified with primers designed by Invitrogen (Shanghai, China). Specific primer sequences of SPRED1 were as follows: forward: 5'-GATGAGCGAGAGACGGAGAC-3' and reverse: 5'-GTCTCTGAGTCTCTCCACGGA-3'. The following protocol was used for real-time PCR: 1 cycle at 95°C for 30 s, followed by 40 cycles at 95°C for 5 s and 60°C for 34 s, and then 1 cycle at 95°C for 15 s, 60°C for 1 min, 95°C for 15 s, and 60°C for 15 s. A melting curve was generated for every PCR amplicon to check the specificity of the PCR reaction. The relative level of SPRED1 was analyzed using the ABI 7900 Sequence Detection System (Applied Biosystems, CA, USA) and calculated using the 2-ΔΔCt method.
DNA extraction and Quantitative MassARRAY analysis of gene methylation status
DNA was extracted from the mononuclear cells of bone marrow tissue using a QIA amp DNA Mini Kit (Qiagen, Hilden, Germany). Sodium bisulfite modification was performed using the EZ DNA Methylation Kit (Zymo Research, CA, USA), and the quantification of DNA methylation was performed using the MassArray platform (Sequenom, CA, USA). Twenty-three samples were analyzed. The primers were designed using the Sequenom EpiDesigner software (www.epidesigner.com). The sequences for primers are listed in Table 1. The PCR conditions were as follows: 94°C for 4 min (activation), 94°C for 20 s (activation), 64°C for 30 s (annealing), 72°C for 1 min (elongation), and 72°C for 5 min (elongation).
Primer sequences, position, product length, and CpG unit used for MassArray quantitative methylation analysis
| Regimen | Primer | Position (bp) | Product Length (bp) | CpG units |
|---|---|---|---|---|
| #1 | AGGATAATGTTGTTGTTGAGGTAGG (F)1 | -1690∼-1277 | 414 | 12 |
| CTAAATCCCAAATACTCCCAAATTC (R)2 | ||||
| #2 | GGTTGGGTTGATTTTAGGTTTTAGT (F)1 | -1190∼-701 | 490 | 15 |
| CCTCCTACTATCCCCCTAATTACAC (R)2 | ||||
| #3 | ATTAGGGGGATAGTAGGAGGAGTTT (F)1 | -720∼-258 | 563 | 14 |
| CAAATTTCAAAAAAATAATTCCCTC (R)2 | ||||
| #4 | GAGGAGGGAATTATTTTTTTGAAAT (F)1 | -285∼59 | 345 | 12 |
| AACCAATACCCACCAACAACTC (R)2 |
Note:1tag: aggaagagag and 2tag: cagtaatacgactcactatagggagaaggct were added.
DNA sequencing analysis
PCR-based mutational analysis on FLT3 gene and the next generation sequencing (NGS) based assay were performed at Yuanqi Biomedical technology co. LTD (Shanghai, China). The panel covers hot spots of AML associated genes including FLT3, DMNT3A, NPM1, CEBPA, TET2, IDH1/2 and C-KIT.
Statistical analysis
Data were analyzed using GraphPad Prism (version 5.0; GraphPad Software Inc., CA, USA) and SPSS (version 22; SPSS Inc., IL, USA). The Mann-Whitney test was performed to evaluate the significance of any differences between the patients with AML and controls. The Spearman correlation analysis was performed to evaluate the correlations between methylation statuses and mRNA levels of SPRED1 genes. All statistical analysis was two sided, and a P value <0.05 was considered statistically significant.
Results
Increased methylation level of SPRED1 in patients with non-APL
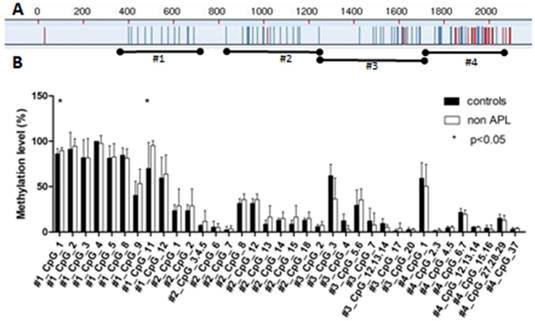
The methylation status of SPRED1 gene was analyzed using the Sequenom MassARRAY platform. Four amplicons in SPRED1 (#1: 310-723 bp, #2: 810-1299 bp, #3: 1280-1742 bp, and #4: 1715-2059 bp) including 53 CpG units were analyzed in bone marrow specimens from 16 patients with de novo non-APL and 7 healthy controls (Fig. 1A). Prior to analysis, strict quality control was performed to remove potentially unreliable measurements as previously described [22]. The CpG units that failed to produce data from more than 30% of samples (unreliable CpG units) and samples missing more than 30% of the data points (unreliable samples) were discarded. All in all, 36 CpG units including 49 CpG sites were evaluated, and each unit included single or multiple CpG sites. The methylation levels was significantly higher in patients with non-APL at units of #1_CpG_1 (89.56% ± 3.41%) and #1_CpG_11 (95.19% ± 5.41%), compared with the control group (86% ± 6.12%, P = 0.04, and 70.17% ± 28.61%, P = 0.002, respectively) (Fig. 1B). No significant difference was observed in the methylation levels for other CpG units.
Comparison of median methylation levels for SPRED1 in the control and patients with AML. (A) Red CpG units could not be detected due to sequential problems; blue CpG units were analyzed for DNA methylation. (B) Average methylation levels for SPRED1 of 7 control and 16 patients with non-APL. All of the values represent the average with SD. *P < 0.05 (Mann-Whitney test).
Relationship between SPRED1 methylation status and mRNA expression levels
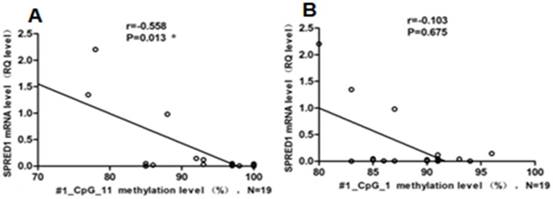
The mRNA expression levels of the SPRED1 gene were determined using qRT-PCR in 16 patients with de novo non-APL and 7 healthy controls. The relative expression levels of SPRED1 mRNA were significantly lower in patients with non-APL (0.034 ± 0.041) compared with controls (1.32 ± 0.46) (P = 0.000008). A correlation analysis was performed in patients with non-APL to identify any relationship between SPRED1 methylation status and respective mRNA levels. The methylation rate of #1_CpG_11 was negatively correlated with the SPRED1 mRNA level (r = -0.558, P = 0.013, Fig. 2A). However, no significant correlation was observed between the methylation rate of #1_CpG_1 and SPRED1 mRNA expression (r = -0.103, P = 0.675, Fig. 2B).
Association of methylation status and mRNA levels for SPRED1 in patients with non-APL. (A) Association of methylation status and mRNA levels for #1_CpG_11 in 19 patients with AML. RQ values represent relative mRNA expression levels for SPRED1 gene. *P < 0.05 (Spearman). (B) Association of methylation status and mRNA levels for #1_CpG_1 in 19 patients with non-APL
Relationship between SPRED1 methylation levels and prognostic parameters
The correlations of #1_CpG_11 methylation level of SPRED1 with a series of prognostic relevant clinical and laboratory parameters of AML, including age, gender, French-American-British (FAB) subtypes, white blood cell count, hemoglobin (Hb), platelet count, blast cells in bone marrow, karyotype, gene mutation, and risk stratification, were investigated. No significant difference was found in the methylation level at a unit of #1_CpG_11 between subgroups of prognostic parameters. It was worth noting that the highest methylation level was observed in patients with DMNT3A mutations (Table 2).
Characteristics and SPRED1 methylation level of 16 cases of non-APLs
| Parameters | Number (ratio) | #1_CpG_11 methylation level of SPRED1 (average±SD, %) |
|---|---|---|
| Age(years) | ||
| ≥ 60 | 1(6.25%) | 86.00±0.00 |
| <60 | 15(93.75) | 95.80±5.02 |
| Gender | ||
| Male | 9(56.25%) | 94.75±4.74 |
| Female | 7(43.75%) | 95.63±5.98 |
| FAB classification, no. (%) | ||
| M2 | 10(62.5%) | 94.50±6.45 |
| M5 | 6(37.5%) | 96.30±2.56 |
| Peripheral blood count (median levels, ranges) | ||
| WBC>10×10⁹/L(38.11,11.48-80.26) | 13(81.25%) | 94.50±4.95 |
| WBC≤10×10⁹/L(5.15,3.76-7.18) | 3(18.75%) | 95.00±7.07 |
| Hb≥80g/L (96,84-136) | 10(62.5%) | 94.50±5.46 |
| Hb<80g/L(61,55-76) | 6(37.5%) | 95.60±5.25 |
| PLT≥50×109/L(60,50-62) | 5(31.25%) | 94.20±5.27 |
| PLT<50×109/L(26,14-34) | 11(68.75%) | 95.64±5.42 |
| Bone marrow (median levels, ranges) | ||
| Blast%≥66 (85.2,75.2-95.2) | 8(50%) | 94.00±5.52 |
| Blast%<66 (32,24.5-56.8) | 8(50%) | 96.38±5.02 |
| No. of karyotypic abnormalities | ||
| Normal | 6(37.5%) | 95.67±4.92 |
| 1-2 | 7(43.75%) | 94.50±5.25 |
| complex | 3(18.75%) | 95.50±6.18 |
| No. of Gene mutation | ||
| None | 2(12.25%) | 89.00±4.00 |
| Isolated | 4(62.5%) | 98.00±3.46 |
| More than two | 10(12.5%) | 96.38±5.55 |
| Gene Mutations | ||
| FLT3/C-KIT | 7(43.75%) | 95.86±5.17 |
| CEBPA | 4(25%) | 98.50±1.50 |
| NPM1 | 1(6.25%) | 93.00±0.00 |
| DNMT3A | 3(18.75%) | 99.33±0.94 |
| TET2 | 7(43.75%) | 94.71±5.95 |
| IDH1/2 | 3(18.75%) | 93.00±5.72 |
| Risk stratification* | ||
| Low | 2(12.5%) | 98.50±1.50 |
| Medium | 8(50%) | 92.63±6.36 |
| High | 6(37.5%) | 97.50±2.22 |
* Risk status based on cytogenetic and molecular abnormalities according to national comprehensive cancer network (NCCN) guideline version 3.2017 acute myeloid leukemia.
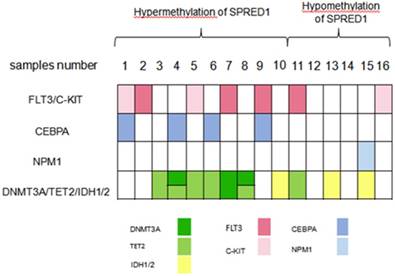
A total of 16 non-APL samples were divided into hypermethylation and hypomethylation of SPRED1 according to the median value (97%) of SPRED1 methylation levels. Mutations in genes including FLT3, c-KIT, CEBPA, NPM1, DNMT3A, TET2, IDH1, IDH2 and NRAS were identified in 15/16 samples. Four patients (25%, samples number 2, 7, 9, 11) had FLT3-ITD mutations. Three patients (18.75%, samples number 1, 5, 16) had mutations in c-KIT. Four patients (25%, samples number 1, 4, 6, 9) carried CEBPA mutation, two of which were biallelic mutation. Three patients had DNMT3A mutation, two of who concurrently carried TET2 mutation. Four patients were presented with single TET2 mutation and three patients had IDH1/2 mutation. The mutation genes were further classified into four subgroups according to their contribution to leukemogenesis: genes inducing proliferation (FLT3 and c-KIT), impairing differentiation (CEBPA), involved in cell cycle (NPM1), and regulating epigenetics (DNMT3A, TET2, and IDH1/2). Overall, mutation in FLT3 or c-KIT was found in 5/10 patients (50%) and 2/6 patients (33.33%) with the hypermethylation and hypomethylation of SPRED1, respectively. All CEBPA mutations were found in patients with hypermethylation of SPRED1 at a ratio of 4/10 (40%). One NPM1 mutation was found in patients with the hypomethylation of SPRED1 (1/6, 16.67%). Mutation in DNMT3A, TET2, or IDH1/2 was found in 7/10 (70%) and 3/6 (50%) patients with the hypermethylation and hypomethylation of SPRED1, respectively. All DNMT3A mutations and all except one TET2 mutation were presented in patients with high methylation level (Figure 3).
Distribution of different gene mutations in patients with AML with the methylation of SPRED1 (n = 16). AML samples were dichotomized according to the median methylation level (97%) of SPRED1 into the low-level or high-level methlyation of SPRED1.
Relationship between SPRED1 methylation levels and treatment response
A correlation analysis was performed in patients with non-APL to identify any relationship between SPRED1 methylation status and their respective treatment response. For patients with CR and NR (n =10 and 6, respectively) achievement, the average methylation level of #1_CpG_11 was 94.42% ± 5.99% and 93.5% ± 6.13%, respectively. No significant difference was found in the methylation level of #1_CpG_11 compared with the treatment response (P = 0.56).
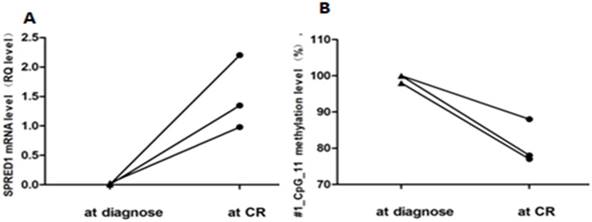
Further, paired methylation rates of unit #1_CpG_11 at diagnosis and at CR were evaluated in three patients. Increased methylation levels at diagnosis (99.33% ± 0.94%) and decreased levels at CR (81% ± 4.97%) are shown in Figure 4A. In addition, paired mRNA expression levels at diagnosis and at CR were evaluated in three patients with non-APLs. Decreased expression levels at diagnosis (0.013 ± 0.011) and increased levels at CR (1.51 ± 0.51) are shown in Figure 4B.
Paired methylation rates of SPRED1 at diagnosis and CR. (A) Methylation levels for #1_CpG_11 at diagnosis and CR; (B) mRNA levels for #1_CpG_11 at diagnosis and CR.
Discussion
In the present study, the quantitative methylation analysis of SPRED1 genes was initially performed in patients with non-APL using the Sequenom MassARRAY platform as a highly accurate, sensitive, and high-throughput method. MassARRAY is an emerging quantitative detection method for DNA methylation in recent years. The MassARRAY system is more efficacious than the bisulfite sequencing PCR (BSP) to reflect the real methylation level of low-methylated regions; it can detect the methylation level as low as 5% [23]. Therefore, it can more accurately determine the correlation with quantitative expression. Significantly increased methylation levels of SPRED1 were found in patients with non-APL, indicating a negative correlation with the methylation rate and SPRED1 transcription level. These findings indicated that changes in the methylation status of SPRED1 might influence gene expression and contribute to the development of AML. Epigenetic control mechanisms are vital in the development and progression of the disease. This includes DNA methylation, histone modification, regulation of mRNA stability, translation by noncoding RNAs, and differential RNA splicing [24]. DNA methylation constitutes an important epigenetic regulation mechanism in many eukaryotes, which has been extensively studied [25]. AML subgroups are defined by diverse DNA methylation patterns, and abnormally methylated genes are identified and related to the clinical outcome [26-28]. It represents a step toward a better understanding of the fact that the tumor suppressor gene SPRED1 is epigenetically silenced in AML. A previous study showed that the expression level of SPRED1 and the incidence rate of tumor invasion and metastasis were negatively correlated [29]. The correlated experiments in vitro and in vivo also showed that the increased expression level of SPRED1 could significantly inhibit tumor cells by suppressing the activity of ERK [30-31]. It was also confirmed that the downregulated expression of SPRED1 correlated with the activation of Ras/MAPK pathway visualized by increased phosphoERK1/2 levels in pediatric AML [4]. Therefore, it was hypothesized that the hypermethylation of SPRED1 led to the decreased SPRED1 expression and activation of Ras/MAPK signaling pathway, facilitating leukemogenesis.
The development of AML is associated with methylation-regulated genetic mutations, including DNMT3A, TET2, and IDH. DNA methylation is performed by DNA methyltransferases (DNMTs), and DNMT3A mutations appear to be associated with poor prognosis [18,32]. Patients with TET2 mutations display overlapping hypermethylation signatures [32]. In this study, although no statistical difference on spectrum of mutations between different methylation levels, the mutations involving epigenetics regulating group, especially. DNMT3A and TET2, was found in patients with higher methylation level of SPRED1. Therefore, mutations in DNMT3A and TET2 might be implicated in the aberrant methylation of SPRED1. Their association with SPRED1 aberrant methylation status is needed to be further clarified. FLT3, NPM1, CEBPA, and c-KIT are related to the risk stratification of AML. Previous reports indicated a significant association between the low expression rates of SPRED1 and FLT3 mutation [4]. However, the present study did not find significant association of SPRED1 hypermethylation and FLT3 mutation. The CEBPA gene encodes a critical transcription factor of hematopoietic cell differentiation [33]. Mutations in double alleles of CEBPA are associated with a favorable clinical prognosis [20]. There seemed an overt distribution of DNA methylation signature in SPRED1 promoter region between the presence and absence of mutations in CEBPA genes in this study. Figueroa et al.found that AML with CEBPA mutation (mostly biallelic) can be divided into two distinct subtypes with different methylation signatures, hypermethylation or hypomethylation. The clinical outcome of subtype in marked hypermethylation status was even better than the known favorable risk core-binding factor leukemias, i.e., t(18;21) and inv(16). Therefore, whether hypermethylation of SPRED1 in subtype of AML with CEBPA mutation can be assigned to a risk stratified system and provide a specific therapy target for hypomethylating agents were worth further investigating.
Further studies with a larger number of samples are warranted to confirm the finding that aberrant methylation of SPRED1 contributes to the development of AML. Likewise, the prognostic correlation between the methylation status of SPRED1 and the treatment response should be confirmed by large-scale studies in the future. A detailed understanding of how SPRED1 hypermethylation is relevant to the pathogenesis of AML is required for better risk classification and better approaches to therapy.
Conclusions
The present study examined the methylation status of SPRED1 and found significant changes in the methylation levels in the SPRED1 gene promoter region. Also, the aberrant methylation status of SPRED1 was negatively correlated with its corresponding mRNA expression, indicating that the DNA methylation changes in SPRED1 gene contributed to transcriptional regulation in patients with AML and were vital in the pathogenesis of AML. Mutations of epigenetic regulators might be involved in the aberrant hypermethylation of SPRED1.These findings might provide important clues for the development of novel treatments for AML and provide a more thorough understanding of the etiology of leukemia.
Acknowledgements
This work was supported by the National Natural Science Fundation of China (grant number 81600117).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Boudry-Labis E, Roche-Lestienne C, Nibourel O, Boissel N, Terre C, Perot C. et al. French ALFA group. Neurofibromatosis-1 gene deletions and mutations in de novo adult acute myeloid leukemia. Am J Hematol. 2013;88(4):306-11
2. Badar T, Patel KP, Thompson PA, DiNardo C, Takahashi K, Cabrero M. et al. Detectable FLT3-ITD or RAS mutation at the time of transformation from MDS to AML predicts for very poor outcomes. Leuk Res. 2015;39(12):1367-74
3. Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R. et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nature genetics. 2007;39(9):1120-6
4. Pasmant E, Gilbert-Dussardier B, Petit A, de Laval B, Luscan A, Gruber A. et al. SPRED1, a RAS MAPK pathway inhibitor that causes Legius syndrome, is a tumour suppressor downregulated in paediatric acute myeloblastic leukaemia. Oncogene. 2015;34(5):631-38
5. Brems H, Legius E. Legius syndrome, an update. molecular pathology of mutations in spred1. Keio J Med. 2013;62(4):107-12
6. Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21(5):650-9
7. Shmueli O, Hornsaban S, Chalifacaspi V, Shmoish M, Ophir R, Benjaminrodrig H. et al. GeneNote: whole genome expression profiles in normal human tissues. CR Biol. 2003;326(10):1067-72
8. Wakioka T, Sasaki A, Kato R, Shouda T, Matsumot A, Miyoshi K. et al. Spred is a Sprouty-related suppressor of Ras signalling. Nature. 2001;412(6847):647-51
9. Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernandez H. et al. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Gene Dev. 2012;26(13):1421-6
10. Bundschu K, Walter U, Schuh K. The VASP-Spred-Sprouty domain puzzle. J Bio Chem. 2006;281(48):36477-81
11. Pasmant E, Ballerini P, Lapillonne H, Perot C, Vidaud D, Leverger G. et al. SPRED1 disorder and predisposition to leukemia in children. Blood. 2009;114(5):1131
12. Zhang R, Lee J Y, Wang X, Xu W, Hu X, Lu X. et al. Identification of novel genomic aberrations in AML-M5 in a level of array CGH. Plos One. 2014;9(4):e87637
13. Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev of Bioch. 2005;74(74):481-514
14. Vaitkiene P, Skiriute D, Skauminas K, Tamasauskas A. GATA4 and DcR1 methylation in glioblastomas. Diagn Patho. 2013;8(1):7-7
15. Rahmatpanah FB, Carstens S, Guo J, Sjahputera O, Taylor KH, Duff D. et al. Differential DNA methylation patterns of small B-cell lymphoma subclasses with different clinical behavior. Leukemia. 2006;20(10):1855-62
16. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427
17. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A. et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-51
18. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J. et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-89
19. Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ. et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118(20):5593-603
20. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM. et al. The 2016 revision to the World Health Organization (WHO) classification of myeloidneoplasms and acute leukemia. Blood. 2016;127:2391-405
21. Chinese Society of Hematology, Chinese Medical Association. Expert consensus on treatment of acute myeloid leukemia (part I). Chin J Hematol. 2009;30(6):429-31
22. Ollikainen M, Smith KR, Joo EJ, Ng HK, Andronikos R, Novakovic B. et al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum Mol Genet. 2010;19(21):4176-88
23. Ohtani K, Dimmeler S. Epigenetic regulation of cardiovascular differentiation. Cardiovas Res. 2011;90(3):404-12
24. Yagi S, Hirabayashi K, Sato S, Li W, Takahashi Y, Hirakawa T. et al. DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) in mouse promoter regions demonstrating tissue-specific gene expression. Genome Res. 2008;18(12):1969-78
25. Li S, Garrettbakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F. et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. 2016;22(7):792-9
26. Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ. et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13-27
27. Luskin MR, Gimotty PA, Smith C, Loren AW, Figueroa ME, Harrison J. et al. A clinical measure of DNA methylation predicts outcome in de novo acute myeloid leukemia. Jci Insight. 2016:1 (9)
28. Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G. et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci USA. 2005;102(44):15785-90
29. Yoshida T, Hisamoto T, Akiba J, Koga H, Nakamura K, Tokunaga Y. et al. Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene. 2006;25(45):6056-66
30. Kato R, Nonami A, Taketomi T, Wakioka T, Kuroiwa A, Matsuda Y. et al. Molecular cloning of mammalian Spred-3 which suppresses tyrosine kinase-mediated Erk activation. Biochem Biophy Res Commun. 2003;302(4):767-72
31. Nobuhisa I, Kato R, Inoue H, Takizawa M, Okita K, Yoshimura A. et al. Spred-2 Suppresses Aorta-Gonad-Mesonephros Hematopoiesis by Inhibiting MAP Kinase Activation. J Exp Med. 2004;199(5):737-42
32. Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2015;127(1):42-52
33. [33] Mueller BU, Pabst T. C/EBPalpha and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol. 2006;13(1):7-14
Author contact
![]() Corresponding author: Rui Zhang, M.D. Ph.D. Department of Hematology, The First Affiliated Hospital of China Medical University, No.155, Nanjing North Street, Shenyang, Liaoning, 110001, China; Tel: +86 024 83282501; Fax: +86 024 22703576; E-mail: hemozerro2008com
Corresponding author: Rui Zhang, M.D. Ph.D. Department of Hematology, The First Affiliated Hospital of China Medical University, No.155, Nanjing North Street, Shenyang, Liaoning, 110001, China; Tel: +86 024 83282501; Fax: +86 024 22703576; E-mail: hemozerro2008com