Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(2):264-275. doi:10.7150/ijms.28580 This issue Cite
Research Paper
Platelet Reactivity And Circulating Platelet-Derived Microvesicles Are Differently Affected By P2Y12 Receptor Antagonists
Bernadeta Chyrchel1, Anna Drożdż2, Dorota Długosz3, Ewa Ł. Stępień4*, Andrzej Surdacki1* ![]()
1. Second Department of Cardiology, Jagiellonian University Medical College, Cracow, Poland
2. Małopolska Center of Biotechnology, Jagiellonian University, Cracow, Poland
3. Students' Scientific Group at the Second Department of Cardiology, Jagiellonian University Medical College, Cracow, Poland
4. Department of Medical Physics, Institute of Physics, Faculty of Physics, Astronomy and Applied Computer Science, Jagiellonian University, Cracow, Poland
* joint senior authors on this work.
Received 2018-7-17; Accepted 2018-11-29; Published 2019-1-1
Abstract
Background: Platelet-derived microvesicles (PMVs), shed from platelet surface membranes, constitute the majority of circulating microvesicles and have been implicated in procoagulant, pro-inflammatory and pro-atherosclerotic effects. Our aim was to compare plasma PMVs numbers in relation to platelet reactivity during dual antiplatelet therapy (DAPT) with various P2Y12 adenosine diphosphate (ADP) receptor antagonists.
Methods: In pre-discharge men treated with DAPT for an acute coronary syndrome, plasma PMVs were quantified by flow cytometry on the basis of CD62P (P-selectin) and CD42 (glycoprotein Ib) positivity, putative indices of PMVs release from activated and all platelets, respectively. ADP-induced platelet aggregation was measured by multiple-electrode aggregometry.
Results: Clinical characteristics were similar in patients on clopidogrel (n=16), prasugrel (n=10) and ticagrelor (n=12). Platelet reactivity was comparably reduced on ticagrelor or prasugrel versus clopidogrel (p<0.01). Compared to clopidogrel-treated patients, CD42+/CD62P+ PMVs counts were 3-4-fold lower in subjects receiving ticagrelor (p=0.001) or prasugrel (p<0.05), while CD42+ PMVs were significantly reduced on ticagrelor (by about 6-fold, p<0.001), but not prasugrel (p=0.3). CD42+/CD62P+ PMVs numbers correlated positively to the ADP-induced aggregation on clopidogrel (p<0.01) or prasugrel (p<0.05), which was absent in ticagrelor users (p=0.8). CD42+ PMVs counts were unrelated to platelet reactivity (p>0.5).
Conclusions: Higher antiplatelet potency of prasugrel and ticagrelor versus clopidogrel is associated with decreased plasma CD42+/CD62P+ PMVs numbers. However, in contrast to thienopyridines, the association of reduced CD42+/CD62P+ PMVs counts with ticagrelor use appears independent of its anti-aggregatory effect. Despite similar platelet-inhibitory activity of ticagrelor and prasugrel, only the treatment with ticagrelor seems associated with lower total PMVs release. Our preliminary findings may suggest a novel pleiotropic effect of ticagrelor extending beyond pure anti-aggregatory properties of the drug.
Keywords: dual antiplatelet therapy, P2Y12 antagonists, platelets, microvesicles, ticagrelor
Introduction
Dual antiplatelet therapy (DAPT) with low-dose aspirin and a P2Y12 adenosine diphosphate (ADP) receptor antagonist is a standard of care in patients with acute coronary syndromes (ACS) and after coronary stenting. Although clopidogrel, prasugrel and ticagrelor are all treatment options, ticagrelor is a preferential drug in ACS patients treated with percutaneous coronary stent implantation, and those with medically managed ACS without persistent ST-segment elevation [1-3]. The Platelet Inhibition and Patient Outcomes (PLATO) trial demonstrated lower risk of all-cause mortality, cardiovascular (CV) mortality, myocardial infarction and definite stent thrombosis in ACS subjects randomized to ticagrelor compared to clopidogrel for 12 months, regardless of ST-segment elevation and treatment strategy [4]. The clinical benefits of ticagrelor, a non-thienopyridine antagonist of P2Y12 receptors, have traditionally been ascribed to its ability to rapidly block platelet P2Y12 receptors with a more consistent platelet inhibition than that achieved with thienopyridines [5]. Additionally, ticagrelor, in contrast to thienopyridines, does not require previous hepatic metabolic activation and binds reversibly to P2Y12 receptors. Recently, it has been suggested that mechanisms of clinical benefits of ticagrelor may extend beyond pure blockade of platelet P2Y12 receptors [5-7].
Membrane-coated platelet-derived microvesicles (PMVs), irregularly shaped and ranging in size from 0.1 to 1 μm, constitute the majority of circulating microvesicles (MVs), and are released by surface membrane shedding accompanying platelet activation [8-10]. Notably, an ex vivo study demonstrated that about 23-40% of the procoagulant activity of human platelet suspensions appeared associated with PMVs [11]. Besides providing an additional anionic surface for coagulation, including procoagulant activity at a distance from the site of platelet activation, PMVs have been implicated in pro-inflammatory and pro-atherosclerotic effects [10,12-14]. Elevated numbers of plasma PMVs were described in patients with clinical atherosclerotic CV disease or risk factors, which has been linked to chronic platelet activation [10,12]. In ACS, PMVs counts are several-fold elevated and rapidly fall after initiation of clopidogrel-based DAPT, generally following the pattern of platelet activation [15,16]. Previous studies in patients receiving clopidogrel-based DAPT revealed an association between platelet aggregability and PMVs release, i.e. increased pre-discharge PMVs counts in ACS patients with high on-treatment platelet reactivity [17,18].
To the best of our knowledge, circulating PMVs have not been measured in ACS subjects treated with prasugrel or ticagrelor [14]. Very recently, Gąsecka et al. [19] demonstrated the capability of ticagrelor, added to platelet-rich plasma obtained from healthy volunteers, to inhibit in vitro PMVs generation in response to ADP, nevertheless, that study has been focused on the release of PMVs from ADP-stimulated platelets.
Therefore, our aim was to compare the number of plasma PMVs in relation to platelet reactivity in ACS patients on DAPT with ticagrelor and thienopyridine P2Y12 receptor antagonists.
Materials and Methods
Patients
We studied 38 men, aged 45-75 years, admitted to our department for an ACS who underwent invasive management. DAPT was initiated at admission before coronary angiography according to practice recommendations [1-3] and included ticagrelor (loading dose [LD]: 180 mg followed by a maintenance dose [MD] of 90 mg twice daily), prasugrel (LD: 60 mg, MD: 10 mg once daily in the morning) or clopidogrel (LD: 600 mg; MD: 75 mg once daily in the morning), in addition to low-dose aspirin (75-100 mg once daily in the morning). Patients with a history of previous ACS, coronary revascularization or DAPT prior to the index hospitalization, a complicated in-hospital course or left ventricular ejection fraction ≤35% (by echocardiography), significant valvular heart disease, relevant coexistent diseases (except for well-controlled diabetes and hypertension), and those with significant abnormalities in routine blood or urine assays, had been excluded. Besides DAPT, all the study subjects were receiving standard medical therapy, i.e. an angiotensin-converting enzyme inhibitor (ACEI) and a statin.
Blood for PMVs assay and the ADP-induced platelet aggregation was drawn from an antecubital vein on the last day of hospitalization, after an overnight fast before administration of the morning dose of drugs, at routine pre-discharge blood sampling. We used a 20-gauge needle, and a light tourniquet was released promptly after the appearance of freely flowing blood in the plastic collection tube.
Platelet pre-activation was prevented by discarding the first 2-3 ml of blood. Blood for PMVs assay was collected to plastic tubes with potassium ethylenediaminetetraacetic acid (EDTA) (final concentration: 1.6 mg EDTA per mL of blood), mixed gently and centrifuged at room temperature for 10 min at 2700 g (Thermo Scientific Heraeus Labofuge 200, Newtown, CT, USA), with a similar delay (30 min) between blood sampling and the centrifugation step. Then plasma was separated, frozen at -80oC and stored until assayed. The university ethics committee had approved the study (KBET/277/B/2013) and patients gave written informed consent.
ADP-induced platelet aggregation
Multiple electrode aggregometry was used to estimate platelet reactivity (Multiplate analyzer, Dynabyte, Munich, Germany). This method is based on elevated electrical impedance owing to platelet adhesion to the surface of a sensor wire [20]. As described previously [21,22], immediately after blood sampling, thrombin inhibitor-anticoagulated blood was diluted with 0.9% NaCl (1:2) and stirred at 37oC for 3 min. Then, we added ADP in a final concentration of 6.4 mmol/L. ADP-induced platelet aggregation was quantified as the area under the curve (in arbitrary units [AU] x min), representing increases in electrical impedance in the function of time during a 6-min recording [20].
Enumeration of PMVs
PMVs were enumerated in stored plasma samples. Before analysis, samples were thawed at 37oC in a water bath to avoid cryoprecipitation and then elaborated and prepared for MVs assay as described previously [16]. In particular, after thawing, samples were centrifuged once for 15 min at 2500 g, to remove residual platelets. All analyses were performed by means of the Apogee A50 micro flow cytometer (Apogee Flow Systems Ltd., Northwood, UK) (Figure 1) for 180 s with a flow rate of 1.5 µL/min; the sample volume was set at 150 µL and the sheath pressure at 150 hPa. As the first, the average events count was measured and appropriate dilutions were performed in order to obtain 5000 events/s. PMV staining was performed in 20 µL of a plasma sample by the incubation with previously chosen antibodies against platelet surface markers CD42(b)/CD62P: a mixture of 1 μL phycoerythrin (PE) anti-human CD42(b) Antibody (BioLegend, San Diego, CA, USA) and 1 μL of Alexa Fluor® 647 anti-human CD62P antibody (BioLegend) (Figure 1C), and isotype control (1 µL of Alexa Fluor® 647 Mouse IgG1, κ Isotype Ctrl (FC) Antibody) (BioLegend) (Figure 1D). To count entire numbers of MVs, a portion (20 µL) of each sample was stained with 1 µL of annexin V, binding to membrane phosphatidylserine (PS), expressed on the surface of MVs irrespective of their origin (fluorescein isothiocyanate [FITC] Annexin V; Annexin V Binding Buffer, BioLegend) (Figure 1B), After 30 minutes of incubation, samples were diluted in phosphate-buffered saline to obtain a final volume of 200 µL. The trigger was set on a middle angle light scattering (MALS) detector with a voltage of 415 V. For daily calibration, we used the reference beads (Figure 1A) (ApogeeMix, cat#1493, Apogee Flow Systems Ltd.) composed of a mixture of 180 nm, 240 nm, 300 nm, 590 nm, 880 nm, and 1300 nm silica vesicles with a refractive index of 1.43, and 110-nm and 500-nm green fluorescent (excited by blue laser) latex beads with a refractive index of 1.59. Final results were corrected by a dilution factor.
Flow cytometry analysis of platelet-derived microvesicles (PMVs) and microvesicles (MVs) with the Apogee A50-Micro flow cytometer. Data were collected on middle angle light scattering. Dot plots present: (A) calibration beads; (B) MVs stained with annexin V-FITC; (C) PMVs stained with anti-CD42-PE and anti-CD62P Alexa Fluor 647; (D) isotype control for anti-CD62P. FITC: fluorescein isothiocyanate; PE: phycoerythrin.
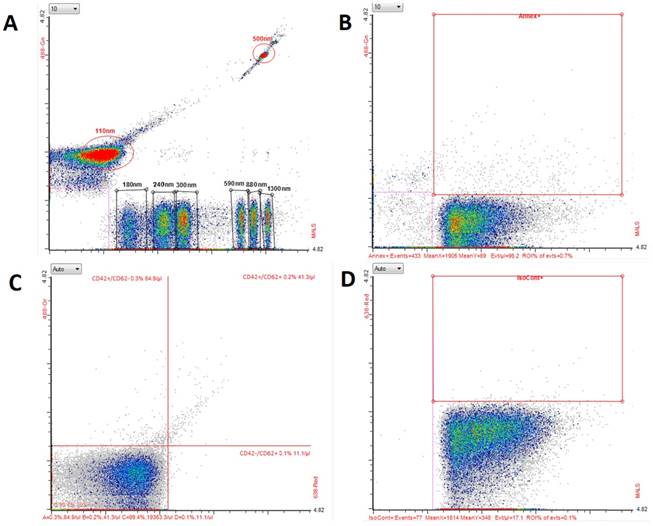
Statistical analysis
Patients' characteristics are presented as means and SD or n (%). Due to a non-Gaussian distribution, MVs/PMVs counts and platelet reactivity were shown as median and interquartile or absolute range. The significance of intergroup differences was assessed by analysis of variance (ANOVA) or Kruskal-Wallis ANOVA (followed by Mann-Whitney U test) for normally or non-normally distributed continuous data, respectively, and the chi-squared test for categorical characteristics. Bivariate relations of platelet aggregability to plasma counts of PMVs and MVs were estimated by Pearson's correlation coefficients (r) after logarithmic transformation to obtain a normal distribution. Mann-Whitney U test was applied to compare numbers of PMVs or MVs in patients with an above-median ADP-induced platelet aggregation relative to those with platelet reactivity lower than the respective median, calculated separately for patients treated with clopidogrel, prasugrel and ticagrelor. A p-value below 0.05 was assumed significant.
Results
Patients' clinical and biochemical characteristics were similar in patients treated with clopidogrel, prasugrel and ticagrelor (Table 1).
Patients' characteristics by the use of ticagrelor and thienopyridine P2Y12 antagonists.
| Characteristic | P2Y12 receptor antagonist | ||
|---|---|---|---|
| Clopidogrel n=16 | Prasugrel n=10 | Ticagrelor n=12 | |
| Age, years | 63 ± 10 | 63 ± 10 | 64 ± 9 |
| Body-mass index, kg/m2 | 28.5 ± 5.1 | 26.3 ± 4.5 | 29.6 ± 3.5 |
| Diabetes, n (%) | 3 (19%) | 2 (20%) | 2 (17%) |
| Hypertension, n (%) | 13 (81%) | 7 (70%) | 8 (67%) |
| GFR, mL/min per 1.73 m2 | 77 ± 18 | 80 ± 17 | 77 ± 22 |
| LDL cholesterol, mmol/L | 2.6 ± 0.8 | 2.7 ± 0.9 | 2.6 ± 0.8 |
| HDL cholesterol, mmol/L | 1.0 ± 0.3 | 1.1 ± 0.3 | 1.0 ± 0.4 |
| Triglycerides, mmol/L | 1.6 ± 0.6 | 1.7 ± 0.6 | 1.6 ± 0.6 |
| Ejection fraction, % | 50 ± 12 | 48 ± 8 | 51 ± 12 |
| Platelets, 103/uL | 245 ± 97 | 205 ± 57 | 201 ± 65 |
| Erythrocytes, 106/uL | 4.5 ± 0.6 | 4.4 ± 0.5 | 4.3 ± 0.6 |
| Leukocytes, 103/uL | 9.5 ± 2.5 | 9.0 ± 2.4 | 8.4 ± 1.9 |
| Hemoglobin, g/dL | 13.5 ± 1.7 | 14.0 ± 1.3 | 13.3 ± 2.0 |
| Medication, n (%) (besides DAPT+ACEI+statin) | |||
| β-blocker | 13 (81%) | 10 (100%) | 11 (92%) |
| Diuretic | 6 (38%) | 4 (40%) | 4 (33%) |
| Calcium channel blocker | 2 (13%) | 1 (10%) | 2 (17%) |
| Hospitalization length, days | 5 ± 2 | 4 ± 2 | 5 ± 2 |
Data are shown as mean and SD or n (%).
Abbreviations: ACEI: angiotensin-converting enzyme inhibitor; DAPT: dual antiplatelet therapy; GFR: glomerular filtration rate; HDL: high-density lipoproteins; LDL: low-density lipoproteins.
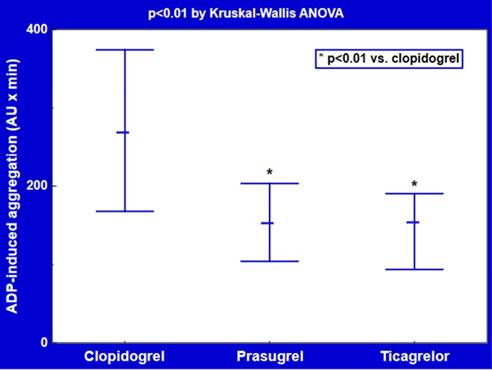
Ex vivo platelet reactivity in response to ADP was reduced to a similar degree in patients treated with ticagrelor (median [interquartile range]: 152 [94-190] AU x min, p<0.01) or prasugrel (151 [104-203] AU x min, p<0.01) versus clopidogrel (266 [168-374] AU x min) (Figure 2).
Platelet reactivity on ticagrelor and thienopyridine P2Y12 antagonists. Data are shown as median and interquartile range. ADP: adenosine diphosphate; AU: arbitrary units.
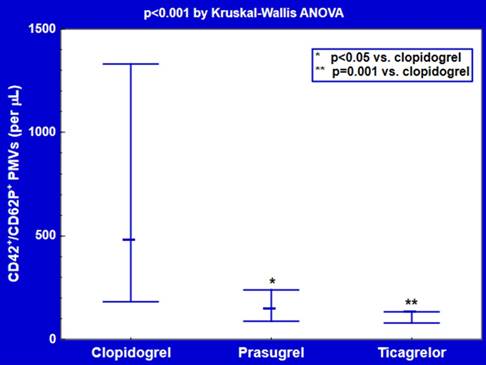
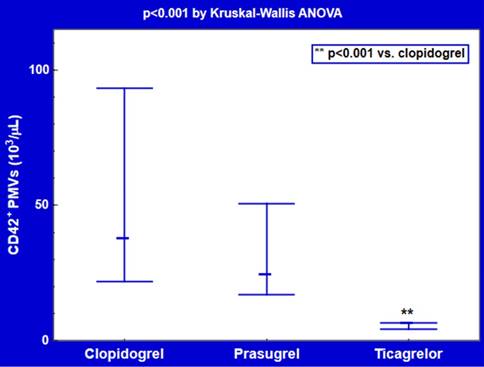
Plasma counts of CD42/CD62P double-positive PMVs were about 3-4-fold lower in subjects receiving ticagrelor (128 [78-133] per μL, p=0.001) or prasugrel (142 [87-238] per μL, p<0.05) compared with clopidogrel users (474 [181-1330] per μL) (Figure 3). CD42-positive PMVs were significantly decreased in patients treated with ticagrelor (5.7 [4.2-6.6] 103/μL, p<0.001), but not prasugrel (23.8 [16.9-50.6] 103/μL, p=0.3), in comparison with those on clopidogrel (37.2 [21.8-93.3] 103/μL) (Figure 4).
Plasma counts of CD42+/CD62P+ platelet-derived microvesicles (PMVs) on ticagrelor and thienopyridine P2Y12 antagonists. Data are shown as median and interquartile range.
Plasma counts of CD42+ platelet-derived microvesicles (PMVs) on ticagrelor and thienopyridine P2Y12 antagonists. Data are shown as median and interquartile range.
Among CD42+ PMVs, the proportion of CD42+/CD62P+ PMVs was elevated (p<0.05) in the ticagrelor group (2.0 [1.3-2.7] %) versus prasugrel (0.8 [0.5-1.3] %) or clopidogrel (1.3 [0.7-1.8] %), mainly due to over 6-fold reductions of CD42+ PMVs on ticagrelor.
CD42+/CD62P+ PMVs numbers correlated positively to the ADP-induced platelet aggregation in patients on either clopidogrel (r = 0.60, p<0.01) or prasugrel (r = 0.65, p<0.05), which was absent on ticagrelor (r = 0.1, p=0.8) (for log-transformed values). Accordingly, CD42+/CD62P+ PMVs counts were significantly higher (p<0.05; p=0.01) in patients with above-median platelet aggregability only in those receiving thienopyridine P2Y12 receptor antagonists (Table 2). Platelet reactivity was unrelated to either CD42+ PMVs or total MVs numbers (p>0.5).
Plasma platelet-derived microvesicles (PMVs) counts by platelet reactivity on ticagrelor and thienopyridine P2Y12 antagonists.
| P2Y12 receptor antagonist | ADP-induced platelet aggregation | |
|---|---|---|
| below-median | above-median | |
| CD42+/CD62P+ PMVs [per μL] | ||
| Clopidogrel | 181 [78-516] | 1329 [256-4556]** |
| Prasugrel | 87 [33-238] | 167 [133-489]* |
| Ticagrelor | 117 [30-244] | 128 [67-133] |
| CD42+ PMVs [103/μL] | ||
| Clopidogrel | 27.0 [6.9-132.6] | 58.2 [18.9-614.8] |
| Prasugrel | 16.9 [2.6-34.5] | 26.0 [9.2-85.1] |
| Ticagrelor | 5.4 [3.0-7.7] | 6.0 [2.9-7.1] |
Data are shown as median and range.
**p=0.01, *p<0.05 versus patients with below-median ADP-induced platelet aggregation.
Median ADP-induced platelet aggregation was 248, 151 and 152 AU x min in patients receiving clopidogrel, prasugrel and ticagrelor, respectively.
Abbreviations: ADP: adenosine diphosphate; AU: arbitrary units.
Discussion
We found about 3-4-fold lower pre-discharge numbers of plasma CD42/CD62P double-positive PMVs, in CAD patients receiving DAPT comprising of ticagrelor or prasugrel in addition to aspirin, compared with their counterparts on the clopidogrel-based DAPT. However, despite similar inhibition of the ADP-induced ex vivo platelet aggregation on prasugrel and ticagrelor, a positive relationship between platelet reactivity and counts of CD42+/CD62P+ PMVs, originating presumably from activated platelets, was observed only in subjects treated with clopidogrel or prasugrel, being absent in ticagrelor users. Notably, plasma CD42-positive PMVs numbers, putatively reflecting total PMVs release, were significantly reduced only on ticagrelor (by about 6-fold), but not on prasugrel.
Mechanisms of PMVs formation and their procoagulant activity
PMVs, described for the first time in 1967 by Peter Wolf [23] and termed “platelet dust” has re-gained attention on the basis of accumulating evidence. First, Sims et al. [24] demonstrated that an impaired capacity to generate PMVs upon activation may underlie defective platelet function in Scott syndrome, a congenital bleeding disorder. Second, Tans et al. [11] described the 18-28% contribution of PMVs to both procoagulant and anticoagulant activity of human platelets stimulated with various platelet agonists. Furthermore, the percentage was even higher in unstimulated platelets (28-40%) [11]. Formation of PMVs is temporally related to the translocation of aminophospholipids (PS and phosphatidyletanolamine, preferentially localized to the inner leaflet of the membrane bilayer in normal conditions) into the outer leaflet. Platelet vesiculation requires not only the disruption of the asymmetric distribution of aminophospholipids, but also reorganization of the cytoskeleton, both of which are mediated by elevations in intraplatelet Ca2+ activating scramblase and cytosolic calpain, respectively [10,12].
Importantly, owing to exposure of negatively charged PS, PMVs possess high-affinity binding sites for coagulation factors, including factor Va, VIIIa, IXa [12] with a 13-fold higher surface density of factor Xa compared to activated platelets [25], enabling the assembly of intrinsic tenase (factor IXa/VIIIa) and prothrombinase (factor Xa/Va) complexes, thereby amplifying both tissue factor (TF)-independent and TF-dependent coagulation pathways [12]. Additionally, surface densities of β3-integrin (CD61, a component of the GPIIb/IIIa complex) and P-selectin (CD62P) were about 8-fold and 4-fold higher for PMVs than for activated platelets, which accounted for 50-100-fold greater procoagulant activity of PMVs, when computed per unit of surface area [25]. Finally, compared to platelets, the procoagulant activity of PMVs lasts longer and may be exerted distantly from the site of platelet activation [12].
Intraplatelet Ca2+ concentrations, governing platelet vesiculation, are increased by various platelet agonists binding to their respective receptors. Higher Ca2+ levels induce platelet shape changes, trigger the secretion of alpha and delta (dense) granules, and thromboxane A2 (TXA2) synthesis. ADP, released from dense granules, and TXA2 are secondary mediators of aggregation. ADP amplifies activation and aggregation acting on both P2Y1 and P2Y12 receptors. P2Y1 receptors are linked to intraplatelet Ca2+ mobilization via phospholipase C activation. In contrast, P2Y12 signaling results in the inhibition of adenylate cyclase and activation of phosphatidylinositol 3-kinase (PI3K) [26]. These pathways, together with Ca2+ elevations, further potentiate granules secretion and conformational changes in fibrinogen receptor (GPIIb/IIIa), which enables binding of fibrinogen with platelet cross-linking and irreversible aggregation. Additionally, procoagulant activity is also enhanced with consequent thrombin generation, formation of fibrin and thrombus stabilization. Of note, the P2Y12-mediated pathway indirectly contributes to Ca2+ mobilization not only through potentiated release of ADP and its binding to P2Y1, but also via PI3K activation and lowering of intraplatelet cyclic adenosine 3',5'-monophosphate (cAMP), thereby augmenting P2Y1-dependent effects [26].
Accordingly, the joint effects of P2Y1 and P2Y12-dependent signaling control the initiation and amplification of the ADP-induced aggregation, respectively, and also accelerate aggregation and thrombus formation induced by other agonists via multiple positive feed-forward loops, including potentiation of procoagulant activity by PMVs generation.
Comparison with previous studies
The observed association between the numbers of plasma CD42+/CD62P+ PMVs and the ADP-mediated platelet aggregation in patients treated with clopidogrel or prasugrel was not unexpected because surface expression of CD62P, stored in alpha granules, appears on activated platelets. However, it is noteworthy that circulating PMVs counts and the ADP-induced platelet aggregation were correlated only in subjects on a thienopyridine P2Y12 receptor antagonist, being yet unrelated in ticagrelor users. The latter finding appears counterintuitive because positive associations between ex vivo platelet aggregability and in vivo blood PMVs counts could be suspected from previous studies, reporting elevated pre-discharge PMVs numbers in ACS patients with higher on-treatment platelet reactivity on DAPT comprising of aspirin and clopidogrel [17,18]. In addition, a negative correlation between PMVs numbers and plasma concentrations of clopidogrel in stable CAD patients receiving clopidogrel and aspirin was also consistent with that concept [27].
On the other hand, circulating PMVs in subjects treated with prasugrel or ticagrelor have not been estimated so far, to the best of our knowledge [14]. Moreover, in vivo comparisons of blood PMVs counts between patients on various P2Y12 antagonists have not been reported yet. Admittedly, Behan et al. [28] observed that cangrelor not only more effectively decreased platelet aggregation, but also yielded more extensive inhibition of intraplatelet Ca2+ elevations and PMVs formation in response to thrombin receptor-activating peptide in vitro, which contrasted with only partial inhibition of these effects estimated ex vivo in platelet-rich plasma isolated from patients on clopidogrel and aspirin. Additionally, similar to the prasugrel active metabolite, cangrelor also potently blocked phenomena associated with the agonist-induced platelet activation in vitro, i.e. Ca2+ mobilization, procoagulant activity (reflected by PS exposure) and PMVs generation [29]. Very recently, in an vitro study, Gąsecka et al. [19] described an ability of ticagrelor to induce interrelated inhibition of the ADP-induced platelet aggregation and the ADP-mediated release of PMVs from activated platelets (defined as CD61(GPIIIa)/CD62P/PS triple-positive MVs). However, all those studies [19,28,29] were focused on PMVs generation in response to platelet agonists, not on resting PMVs counts in plasma. So, although stronger P2Y12 blockers more efficiently counteract agonists-induced platelet aggregation and PMVs generation in vitro, this notion may not necessarily imply analogous results with regard to circulating PMVs.
Hypothetical mechanisms of different relations between PMVs counts and platelet reactivity in patients receiving ticagrelor and thienopyridine P2Y12 receptor antagonists
This finding can result from differences in pharmacokinetics and pharmacodynamics between ticagrelor and thienopyridine P2Y12 receptor antagonists. Ticagrelor, contrary to thienopyridines whose active metabolites are detected in the systemic blood only for about 4 h after prodrug intake, allows a constant plasma exposure of an active compound over 24 h [5]. Therefore, newly-generated immature platelets, known to be associated with CV risk [30], are rapidly inhibited in ticagrelor users. In contrast, in patients on a thienopyridine P2Y12 receptor antagonist, juvenile platelets remain active until the next prodrug intake, if they have been formed after the elimination of the active metabolite [5]. At later time points after clopidogrel intake, but not after ticagrelor, Kuijpers et al. [31] demonstrated, in an experimental study, a gradually emerging subpopulation of uninhibited newly-formed platelets which preferentially contributed to augmented thrombus formation on collagen under high shear-stress conditions. More recently, a 2-fold lower inhibition of immature platelets in prasugrel-treated than ticagrelor-treated ACS patients was shown late after the last drug intake, i.e. 1 h before the next dose [32]. Because the consequent differences in anticoagulant potency between clopidogrel and ticagrelor were not detected by conventional platelet aggregation assays [31], this juvenile platelets-mediated effect was presumably also masked in our patients, whose blood was sampled before the morning drug dose. Therefore, it can by hypothesized that a stronger ability of ticagrelor than prasugrel to inhibit juvenile platelets in vivo might have translated into lower circulating PMVs, but was missed by the ADP-induced platelet aggregation, thereby obscuring the association of PMVs and platelet reactivity in patients on ticagrelor.
Second, under a strong and constant P2Y12 blockade, as in patients receiving ticagrelor, P2Y12-dependent platelet responses to ADP are presumably profoundly inhibited, including the ability to potentiate P2Y1-dependent Ca2+ elevations [26], and amplification of platelet activation/aggregation evoked by other stimuli. Increases in intracellular Ca2+ levels are also pivotal for the release of PMVs from platelets stimulated by soluble agonists [33,34] and high shear-stress [35,36]. In contrast, shedding of PMVs from unstimulated platelets is governed by Ca2+-independent pathways [37]. Indeed, PMVs formation in suspensions of unstimulated platelets was not dependent on intracellular Ca2+ elevation or calpain activation, and appeared to be mediated by so-called “outside-in” GPIIb/IIIa signaling [37]. So, Ca2+-independent pathways of PMVs release may be relatively more important for plasma PMVs counts in patients on ticagrelor, thereby explaining a lack of correlation between blood PMVs and platelet reactivity to ADP in our ticagrelor-treated CAD patients. Additionally, only about 20% of PMVs released by unstimulated platelets exposed PS, mediating phospholipid-dependent procoagulant activity of PMVs [38]. This observation is consistent with a minimal level of activation of unstimulated platelets generating PMVs in stored platelet-rich plasma, with a 5-8% expression of PS and activated GPIIb/IIIa, unlike ADP-activated platelets expressing almost 100% of these surface markers [37]. Thus., the predominance of non-activated platelets in ticagrelor users could contribute to the lack of correlation between in vitro ADP-induced platelet aggregation and in vivo PMVs counts on ticagrelor. The suggested hypothetical explanation for the dissociation of blood PMVs counts and platelet reactivity is in analogy to that proposed for a lack of association between solid cerebral microembolic signals on transcranial Doppler ultrasonography and blood PMVs, coagulation parameters or platelet activity in patients with a mechanical heart valve on oral anticoagulation [39]. The authors suggested that even if a non-significant increase in PMVs might have reflected enhanced platelet activation, it probably did not reach the irreversible phase of platelet aggregation [39].
Third, a dissociation of plasma PMVs counts and ADP-induced aggregation on ticagrelor, but not on thienopyridines, could result from the potentiation of the anti-platelet effect of ticagrelor by endogenous mediators such as nitric oxide (NO), which was reflected by circulating PMVs counts (a measure of in vivo platelet activation and associated PMVs release), but not by ex vivo testing of platelet reactivity. A similar concept was proposed by Chan et al. [40] to explain the lack of prognostic benefits from ex vivo monitoring of platelet aggregability and subsequent tailoring of DAPT intensity. P2Y12 receptor blockade strongly increases the ability of endogenous nitric oxide (NO) and prostacyclin (PGI2) to attenuate platelet reactivity [41,42], and NO and PGI2 further synergistically inhibit platelet activation via interactions between their respective second messengers, cyclic guanosine 3',5'-monophosphate (cGMP) and cAMP [43]. On the other hand, although improvement of endothelial function was reported after ticagrelor in comparison with thienopyridines in stable post-ACS patients [44,45], this finding was yet not confirmed in a recent study by Ariotti et al. [46].
As to adenosine, ticagrelor, in addition to binding to P2Y12 receptors, reduces cellular uptake od adenosine and increases its extracellular concentrations in humans [47], owing to the drug-induced inhibition of equilibrative nucleoside transporter-1 [6]. Adenosine - via platelet A2A and A2B receptors - activates adenylate cyclase and down-regulates P2Y1 receptor expression [48], inhibiting platelet aggregation and attenuating Ca2+ elevations both directly, acting on P2Y1 [26], and indirectly, modulating the P2Y12-mediated amplification of P2Y1-dependent effects [26]. Intraplatelet Ca2+ governs PMVs shedding from activated platelets [33,34]. Therefore, the ticagrelor-induced accumulation of adenosine, a cAMP “elevator”, may strengthen the above presented hypothesis of synergistic interactions between P2Y12 antagonism, intraplatelet cAMP and cGMP [40,43] in circulating platelets. These interactions might be an explanation for the dissociation of ex vivo platelet reactivity and circulating CD42+/CD62P+ PMVs counts, integrating also possible in vivo effects of endogenous NO, PGI2 and adenosine on PMVs release in patients receiving ticagrelor, in contrast to thienopyridine P2Y12 antagonists.
Irrespective of the mechanisms involved, the independence of in vivo PMVs release and ex vivo platelet reactivity on ticagrelor may not only reflect the in vivo amplification of the drug-specific inhibition of platelet activation and associated PMVs shedding, but could also add to ticagrelor pleiotropic effects, including prevention of systemic inflammatory activation and, importantly, reduced risk of adverse CV events.
Depressed plasma counts of PMVs - a novel pleiotropic effect of ticagrelor ?
Biological effects of PMVs extend beyond blood coagulation. PMVs promote formation of platelet/PMVs/leukocyte aggregates and facilitate leukocyte tethering to activated endothelia, providing a basis for the contribution of PMVs to pro-inflammatory and pro-atherosclerotic responses [10,12-14]. These effects have been accounted for by a corollary of pathways, e.g. surface expression of GPIb (a counter-receptor for the leukocyte β2-integrin CD11b/CD18, i.e. Mac-1), P-selectin (binding to P-selectin glycoprotein ligand-1 on leukocytes [PSGL-1]) and CD40L, a ligand for leukocyte Mac-1 and CD40 on antigen-presenting dendritic cells, B-cells and endothelial cells. Notably, the P-selectin-PSGL-1 interaction also contributes to the delivery of TF from TF-exposing monocytes into developing platelet thrombus via monocyte-derived MVs bearing both PSGL-1 and TF [12,49]. Other pro-inflammatory pathways include transcellular transfer of arachidonic acid, a substrate for cyclooxygenase-2, with subsequent upregulation of intercellular adhesion molecule-1 (ICAM-1) and β2-integrins on endothelial cells and monocytes, respectively [50], conversion of pentameric C-reactive protein to its highly pro-inflammatory monomeric form [51], and enhancement of endothelial adhesiveness for neutrophils via interleukin-1β exported from activated platelets [52]. Thus, PMVs are likely to contribute to the previously reported ability of activated platelets to induce monocyte-platelet and monocyte-endothelial interactions through secretion of monocyte chemotactic protein-1 by endothelia and monocytes as well as endothelial surface expression of ICAM-1, a counter-receptor for leukocyte Mac-1 [53,54]. In accordance with the this concept, Ueba et al. [55] observed an association of PMVs, identified on the basis of CD42-positivity, with interleukin-6 levels in healthy subjects.
Notably, Mause et al. [56] demonstrated that the expression of both GPIb (CD42(b)) and P-selectin (CD62P) on the surface of PMVs was required for PMVs interactions with activated endothelial cells, including PMVs rolling and adhesion, deposition of PMVs-derived RANTES (Regulated on Activation, Normal T cell Expressed and Secreted) on endothelial surface and the PMVs-dependent monocyte recruitment to activated endothelium. In our CAD patients receiving ticagrelor, 3-6-lower plasma counts of CD42/CD62P-double positive and CD42-positive PMVs were observed compared to subjects on clopidogrel, regardless of platelet aggregability.
So, it does not seem implausible to assume that markedly depressed counts of PMVs in patients on ticagrelor might be not only an epiphenomenon, accompanying strong and constant platelet inhibition in vivo, but - more importantly - can also represent a novel mechanism of the anti-inflammatory effect of the drug, with a possible contribution of endogenous adenosine accumulation [6,47]. Ticagrelor, inhibiting cellular adenosine uptake, was shown to augment vascular and platelet effects of exogenous adenosine, including its vasodilatory [57] and anti-aggregatory [58] effects. Notably, some of pleiotropic effects of ticagrelor may be at least partially ascribed to endogenous adenosine, such as enhancement of cardiac reactive hyperemia [59], anti-inflammatory activity (at high adenosine levels, contrary to pro-inflammatory effects of low adenosine [60]), as well as better outcome in patients with pulmonary infection [61], prevention of cardiac ischemia/reperfusion injury [62], and improvement of endothelial function [44].
Whether the ticagrelor-associated reductions of PMVs generation might contribute to proposed anti-inflammatory and anti-atherosclerotic effects of the drug with prognostic implications in CAD patients, remains to be investigated in a randomized prospective study.
Limitations of the study
A small number of study subjects is a major limitation of our preliminary observational report. However, there were no intergroup differences in clinical characteristics, medication or hospitalization length. In particular, all the patients were on statins, known to affect PMVs counts [63]. Additionally, in order to reduce the heterogeneity of the study patients, we had excluded subjects with a history of previous ACS, coronary revascularization or DAPT, a complicated clinical course, coexistent diseases or relevant abnormalities in routine blood and urine assays.
Second, blood samples were centrifuged once at 2700 g for 10 min and then frozen until PMVs enumeration, while double centrifugation (2 x 15 min at 2500 g) is recommended to remove residual platelets which persist after a single centrifugation and may release MVs or fragment during a freeze-thaw cycle [64,65]. In our hands, to remove residual platelets from stored plasma, a second centrifugation (15 min at 2500 g) was carried out after thawing the sample, before flow cytometry analysis. Keeping in mind the importance of pre-analytical steps for PMVs assay [64-66], an additional analysis was done in order to compare the effects of inter-protocol differences on the concentration of residual platelets. We were able to demonstrate that the content of residual platelets in the plasma prepared using the recommended protocol [64,65] and that obtained by our procedure was similar under the condition that a second centrifugation was performed after thawing the frozen samples, as in our protocol (Supplementary Table S1).
Third, we used CD42(b) (GPIb) to define PMPs by flow cytometry, similar to previous studies enumerating PMVs exposing CD42(b) and/or CD42(a) (GPIX) [16,18,27,29,55,67]. However, other criteria for PMVs identification were also reported, including the positivity for CD41 (GPIIb) [64] or CD61 (GPIIIa) [15,17,19]. Additionally, Rank et al. [68] even suggested that a considerable portion of CD41-positive MVs, in contrast to CD62P-positive MVs, probably originates from megakaryocytes, not from platelets, which could also occur for GPIb [69]. Nevertheless, that we observed associations of ex vivo platelet reactivity only with plasma counts of CD42+/CD62P+ MVs, but not CD42+ or annexin V+ MVs, strongly suggests activated platelets as the predominant source of CD42+/CD62P+ MVs. Accordingly, the lack of association between CD42+/CD62P+ counts and the ADP-induced platelet aggregation in patients on ticagrelor, but not on thienopyridine P2Y12 ADP receptor antagonists, argues in favor of a ticagrelor-specific effect, i.e. a dissociation of the drug-induced inhibition of in vivo PMVs release and ex vivo ADP-induced platelet aggregation. That platelet reactivity was attenuated to a similar degree in subjects treated with prasugrel and ticagrelor, also supports the proposed hypothetical concept.
Fourth, we observed much higher numbers of CD42-positive than CD42/CD62P double-positive PMVs, which was rather unexpected keeping in mind the notion of PMVs generation during platelet activation, associated with surface CD62P expression. Nonetheless, Kafian et al. [18] reported CD62P-positivity on less than a half of plasma PMVs, defined on the basis of surface GPIX (CD42(a)) expression, which points into a relevant contribution of MVs shed from non-activated platelets to total PMVs release in vivo. Moreover, in agreement with our finding, Gąsecka et al. [19], studying in vitro platelet suspensions, described about 15-fold higher release of CD61+/CD62P-/PS- PMVs than CD61+/CD62P+/PS+ PMVs from suspensions of resting platelets, putatively reflecting MVs generation by non-activated and activated platelets, respectively. Intriguingly, unlike Gąsecka et al. [19], who reported the ability of ticagrelor (1 μmol/L) to inhibit the release of CD62P-positive, but not CD62P-negative PMVs in response to ADP, we observed lower plasma counts of PMVs in patients receiving ticagrelor, irrespective of CD62P-positivity. Nevertheless, comparisons between these reports are profoundly hampered by differences in study design and distinct criteria for the identification of PMVs by flow cytometry.
Conclusions
Higher antiplatelet potency of prasugrel and ticagrelor versus clopidogrel is associated with decreased plasma counts of CD42+/CD62P+ PMVs, originating presumably from activated platelets. However, in contrast to thienopyridines, the association of reduced CD42+/CD62P+ PMVs numbers with ticagrelor use appears independent of its anti-aggregatory effect. Notably, despite similar antiplatelet activity of ticagrelor and prasugrel, only the treatment with ticagrelor seems to be associated with lower counts of CD42+ PMVs, putatively reflecting total PMVs release. Thus, inasmuch as PMVs can contribute to CV risk, our preliminary findings may suggest a novel pleiotropic effect of ticagrelor extending beyond pure anti-aggregatory properties of the drug.
Abbreviations
ACEI: angiotensin-converting enzyme inhibitor; ACS: acute coronary syndrome; ADP: adenosine diphosphate; AU: arbitrary units; CAD: coronary artery disease; cAMP: cyclic adenosine 3',5'-monophosphate; cGMP: cyclic guanosine 3',5'- monophosphate; CV: cardiovascular; DAPT: dual antiplatelet therapy; EDTA: ethylenediaminetetraacetic acid; FITC: fluorescein isothiocyanate; GFR: glomerular filtration rate; GP: glycoprotein; HDL: high-density lipoproteins; ICAM-1: intercellular adhesion molecule-1; LDL: light-density lipoproteins; MVs: microvesicles; NO: nitric oxide; PE: phycoerythrin; PGI2: prostacyclin; PI3K: phosphatidylinositol 3-kinase; PMVs: platelet-derived microvesicles; PS: phospatidylserine; PSGL-1: P-selectin glycoprotein ligand-1; SD: standard deviation; TF: tissue factor; TXA2: thromboxane A2.
Supplementary Material
Supplementary table.
Acknowledgements
This work was supported by a grant from the Jagiellonian University Medical College, Cracow, Poland (K/ZDS/006104). An abstract based on this study has been submitted for poster presentation at the 23rd Cardiology Update Course, Davos, Switzerland, February 16th, 2019.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Levine GN, Bates ER, Bittl JA. et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;68(10):1082-1115
2. Valgimigli M, Bueno H, Byrne RA. et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: The Task Force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. 2018;39(3):213-260
3. Roffi M, Patrono C, Collet JP. et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J. 2016;37(3):267-315
4. Wallentin L, Becker RC, Budaj A. et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057
5. Nylander S, Schulz R. Effects of P2Y12 receptor antagonists beyond platelet inhibition-comparison of ticagrelor with thienopyridines. Br J Pharmacol. 2016;173(7):1163-1178
6. Cattaneo M, Schulz R, Nylander S. Adenosine-mediated effects of ticagrelor: evidence and potential clinical relevance. J Am Coll Cardiol. 2014;63(23):2503-2509
7. Kubisa MJ, Jeżewski MP, Gąsecka A, Siller-Matula JM, Postuła M. Ticagrelor - toward more efficient platelet inhibition and beyond. Ther Clin Risk Manag. 2018;14:129-140
8. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999;94(11):3791-3799
9. Berckmans RJ, Nieuwland R, Böing AN, Romijn FP, Hack CE, Sturk A. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb Haemost. 2001;85(4):639-646
10. Zaldivia MTK, McFadyen JD, Lim B, Wang X, Peter K. Platelet-derived microvesicles in cardiovascular diseases. Front Cardiovasc Med. 2017;4:74
11. Tans G, Rosing J, Thomassen MC, Heeb MJ, Zwaal RF, Griffin JH. Comparison of anticoagulant and procoagulant activities of stimulated platelets and platelet-derived microparticles. Blood. 1991;77(12):2641-2648
12. Nomura S, Ozaki Y, Ikeda Y. Function and role of microparticles in various clinical settings. Thromb Res. 2008;123(1):8-23
13. Badimon L, Suades R, Fuentes E, Palomo I, Padró T. Role of platelet-derived microvesicles as crosstalk mediators in atherothrombosis and future pharmacology targets: A link between inflammation, atherosclerosis, and thrombosis. Front Pharmacol. 2016;7:293
14. Rosińska J, Łukasik M, Kozubski W. The impact of vascular disease treatment on platelet-derived microvesicles. Cardiovasc Drugs Ther. 2017;31(5-6):627-644
15. Skeppholm M, Mobarrez F, Malmqvist K, Wallén H. Platelet-derived microparticles during and after acute coronary syndrome. Thromb Haemost. 2012;107(6):1122-1129
16. Stępień E, Stankiewicz E, Zalewski J, Godlewski J, Żmudka K, Wybrańska I. Number of microparticles generated during acute myocardial infarction and stable angina correlates with platelet activation. Arch Med Res. 2012;43(1):31-35
17. Kalantzi KI, Dimitriou AA, Goudevenos JA, Tselepis AD. The platelet hyporesponsiveness to clopidogrel in acute coronary syndrome patients treated with 75 mg/day clopidogrel may be overcome within 1 month of treatment. Platelets. 2012;23(2):121-131
18. Kafian S, Mobarrez F, Wallén H, Samad B. Association between platelet reactivity and circulating platelet-derived microvesicles in patients with acute coronary syndrome. Platelets. 2015;26(5):467-473
19. Gąsecka A, Nieuwland R, van der Pol E. et al. P2Y12 antagonist ticagrelor inhibits the release of procoagulant extracellular vesicles from activated platelets: Preliminary results. Cardiol J. 2018 Epub ahead of print
20. Sibbing D, Braun S, Jawansky S. et al. Assessment of ADP-induced platelet aggregation with light transmission aggregometry and multiple electrode platelet aggregometry before and after clopidogrel treatment. Thromb Haemost. 2008;99(1):121-126
21. Chyrchel B, Surdacki A, Chyrchel M, Dudek D, Dubiel JS. Separate dosing of clopidogrel and omeprazole may improve platelet inhibition on dual antiplatelet therapy. Int J Cardiol. 2011;149(1):124-125
22. Chyrchel B, Totoń-Żurańska J, Kruszelnicka O. et al. Association of plasma miR-223 and platelet reactivity in patients with coronary artery disease on dual antiplatelet therapy: A preliminary report. Platelets. 2015;26(6):593-597
23. Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol. 1967;13(3):269-288
24. Sims PJ, Wiedmer T, Esmon CT, Weiss HJ, Shattil SJ. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J Biol Chem. 1989;264(29):17049-17057
25. Sinauridze EI, Kireev DA, Popenko NY. et al. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost. 2007;97(3):425-434
26. Hardy AR, Jones ML, Mundell SJ, Poole AW. Reciprocal cross-talk between P2Y1 and P2Y12 receptors at the level of calcium signaling in human platelets. Blood. 2004;104(6):1745-1752
27. França CN, Pinheiro LF, Izar MC. et al. Endothelial progenitor cell mobilization and platelet microparticle release are influenced by clopidogrel plasma levels in stable coronary artery disease. Circ J. 2012;76(3):729-736
28. Behan MW, Fox SC, Heptinstall S, Storey RF. Inhibitory effects of P2Y12 receptor antagonists on TRAP-induced platelet aggregation, procoagulant activity, microparticle formation and intracellular calcium responses in patients with acute coronary syndromes. Platelets. 2005;16(2):73-80
29. Judge HM, Buckland RJ, Sugidachi A, Jakubowski JA, Storey RF. The active metabolite of prasugrel effectively blocks the platelet P2Y12 receptor and inhibits procoagulant and pro-inflammatory platelet responses. Platelets. 2008;19(2):125-133
30. Ibrahim H, Schutt RC, Hannawi B, DeLao T, Barker CM, Kleiman NS. Association of immature platelets with adverse cardiovascular outcomes. J Am Coll Cardiol. 2014;64(20):2122-2129
31. Kuijpers MJ, Megens RT, Nikookhesal E. et al. Role of newly formed platelets in thrombus formation in rat after clopidogrel treatment: comparison to the reversible binding P2Y12 antagonist ticagrelor. Thromb Haemost. 2011;106(6):1179-1188
32. Bernlochner I, Goedel A, Plischke C. et al. Impact of immature platelets on platelet response to ticagrelor and prasugrel in patients with acute coronary syndrome. Eur Heart J. 2015;36(45):3202-3210
33. Dachary-Prigent J, Freyssinet JM, Pasquet JM, Carron JC, Nurden AT. Annexin V as a probe of aminophospholipid exposure and platelet membrane vesiculation: a flow cytometry study showing a role for free sulfhydryl groups. Blood. 1993;81(10):2554-2565
34. Dachary-Prigent J, Pasquet JM, Freyssinet JM, Nurden AT. Calcium involvement in aminophospholipid exposure and microparticle formation during platelet activation: a study using Ca2+-ATPase inhibitors. Biochemistry. 1995;34(36):11625-11634
35. Miyazaki Y, Nomura S, Miyake T. et al. High shear stress can initiate both platelet aggregation and shedding of procoagulant containing microparticles. Blood. 1996;88(9):3456-3464
36. Holme PA, Orvim U, Hamers MJ. et al. Shear-induced platelet activation and platelet microparticle formation at blood flow conditions as in arteries with a severe stenosis. Arterioscler Thromb Vasc Biol. 1997;17(4):646-653
37. Cauwenberghs S, Feijge MA, Harper AG, Sage SO, Curvers J, Heemskerk JW. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006;580(22):5313-5320
38. Connor DE, Exner T, Ma DD, Joseph JE. The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thromb Haemost. 2010;103(5):1044-1052
39. Skjelland M, Michelsen A, Brosstad F, Svennevig JL, Brucher R, Russell D. Solid cerebral microemboli and cerebrovascular symptoms in patients with prosthetic heart valves. Stroke. 2008;39(4):1159-1164
40. Chan MV, Knowles RB, Lundberg MH. et al. P2Y12 receptor blockade synergizes strongly with nitric oxide and prostacyclin to inhibit platelet activation. Br J Clin Pharmacol. 2016;81(4):621-633
41. Cattaneo M, Lecchi A. Inhibition of the platelet P2Y12 receptor for adenosine diphosphate potentiates the antiplatelet effect of prostacyclin. J Thromb Haemost. 2007;5(3):577-582
42. Kirkby NS, Lundberg MH, Chan MV. et al. Blockade of the purinergic P2Y12 receptor greatly increases the platelet inhibitory actions of nitric oxide. Proc Natl Acad Sci U S A. 2013;110(39):15782-15787
43. Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. 1987;92(3):639-646
44. Torngren K, Ohman J, Salmi H, Larsson J, Erlinge D. Ticagrelor improves peripheral arterial function in patients with a previous acute coronary syndrome. Cardiology. 2013;124(4):252-258
45. Jeong HS, Hong SJ, Cho SA. et al. Comparison of ticagrelor versus prasugrel for inflammation, vascular function, and circulating endothelial progenitor cells in diabetic patients with non-ST-segment elevation acute coronary syndrome requiring coronary stenting: A prospective, randomized, crossover trial. JACC Cardiovasc Interv. 2017;10(16):1646-1658
46. Ariotti S, van Leeuwen M, Brugaletta S. et al. Effects of ticagrelor, prasugrel, or clopidogrel at steady state on endothelial function. J Am Coll Cardiol. 2018;71(11):1289-1291
47. Bonello L, Laine M, Kipson N. et al. Ticagrelor increases adenosine plasma concentration in patients with an acute coronary syndrome. J Am Coll Cardiol. 2014;63(9):872-877
48. Johnston-Cox HA, Ravid K. Adenosine and blood platelets. Purinergic Signal. 2011;7(3):357-365
49. Falati S, Liu Q, Gross P. et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003;197(11):1585-1598
50. Barry OP, Praticò D, Savani RC, FitzGerald GA. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J Clin Invest. 1998;102(1):136-144
51. Habersberger J, Strang F, Scheichl A. et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc Res. 2012;96(1):64-72
52. Lindemann S, Tolley ND, Dixon DA. et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1β synthesis. J Cell Biol. 2001;154(3):485-490
53. Weyrich AS, Elstad MR, McEver RP. et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest. 1996;97(6):1525-1534
54. Gawaz M, Neumann FJ, Dickfeld T. et al. Activated platelets induce monocyte chemotactic protein-1 secretion and surface expression of intercellular adhesion molecule-1 on endothelial cells. Circulation. 1998;98(12):1164-1171
55. Ueba T, Nomura S, Inami N. et al. Correlation and association of plasma interleukin-6 and plasma platelet-derived microparticles, markers of activated platelets, in healthy individuals. Thromb Res. 2010;125(6):e329-e334
56. Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. 2005;25(7):1512-1518
57. Wittfeldt A, Emanuelsson H, Brandrup-Wognsen G. et al. Ticagrelor enhances adenosine-induced coronary vasodilatory responses in humans. J Am Coll Cardiol. 2013;61(7):723-727
58. Nylander S, Femia EA, Scavone M. et al. Ticagrelor inhibits human platelet aggregation via adenosine in addition to P2Y12 antagonism. J Thromb Haemost. 2013;11(10):1867-1876
59. van Giezen JJ, Sidaway J, Glaves P, Kirk I, Björkman JA. Ticagrelor inhibits adenosine uptake in vitro and enhances adenosine-mediated hyperemia responses in a canine model. J Cardiovasc Pharmacol Ther. 2012;17(2):164-172
60. Barletta KE, Ley K, Mehrad B. Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol. 2012;32(4):856-864
61. Storey RF, James SK, Siegbahn A. et al. Lower mortality following pulmonary adverse events and sepsis with ticagrelor compared to clopidogrel in the PLATO study. Platelets. 2014;25(7):517-525
62. Wang K, Zhou X, Huang Y. et al. Adjunctive treatment with ticagrelor, but not clopidogrel, added to tPA enables sustained coronary artery recanalisation with recovery of myocardium perfusion in a canine coronary thrombosis model. Thromb Haemost. 2010;104(3):609-617
63. Pinheiro LF, França CN, Izar MC. et al. Pharmacokinetic interactions between clopidogrel and rosuvastatin: effects on vascular protection in subjects with coronary heart disease. Int J Cardiol. 2012;158(1):125-129
64. Lacroix R, Judicone C, Poncelet P. et al. Impact of pre-analytical parameters on the measurement of circulating microparticles: towards standardization of protocol. J Thromb Haemost. 2012;10(3):437-446
65. Coumans FAW, Brisson AR, Buzas EI. et al. Methodological guidelines to study extracellular vesicles. Circ Res. 2017;120(10):1632-1648
66. Witwer KW, Buzás EI, Bemis LT. et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2:20360
67. Judge HM, Buckland RJ, Holgate CE, Storey RF. Glycoprotein IIb/IIIa and P2Y12 receptor antagonists yield additive inhibition of platelet aggregation, granule secretion, soluble CD40L release and procoagulant responses. Platelets. 2005;16(7):398-407
68. Rank A, Nieuwland R, Delker R. et al. Cellular origin of platelet-derived microparticles in vivo. Thromb Res. 2010;126(4):e255-e259
69. Lepage A, Leboeuf M, Cazenave JP, de la Salle C, Lanza F, Uzan G. The alpha(IIb)beta(3) integrin and GPIb-V-IX complex identify distinct stages in the maturation of CD34(+) cord blood cells to megakaryocytes. Blood. 2000;96(13):4169-4177
Author contact
![]() Corresponding author: Andrzej Surdacki, MD, PhD. Second Department of Cardiology, Jagiellonian University Medical College, 17 Kopernika Street, PL31-501 Cracow, Poland. Phone/Fax: +48 12 424 7180; E-mail: andrzej.surdackiedu.pl
Corresponding author: Andrzej Surdacki, MD, PhD. Second Department of Cardiology, Jagiellonian University Medical College, 17 Kopernika Street, PL31-501 Cracow, Poland. Phone/Fax: +48 12 424 7180; E-mail: andrzej.surdackiedu.pl