Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Adipogenic Markers in Adipose...
Phenotypic Alterations in...
Oxidative stress and ROS...
Role of Na/K-ATPase/Src...
Inflammation in NAFLD
Pharmacological Interventions...
Effectiveness of pNaKtide in...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(14):1591-1599. doi:10.7150/ijms.27953 This issue Cite
Review
Phenotypic Alteration of Hepatocytes in Non-Alcoholic Fatty Liver Disease
Hari Vishal Lakhani1, Dana Sharma1, Michael W. Dodrill1, Athar Nawab1, Nitin Sharma1, Cameron Lee Cottrill1, Joseph I. Shapiro1, Komal Sodhi2 ![]()
1. Department of Internal Medicine, Marshall University Joan C Edwards School of Medicine, Huntington, WV, USA;
2. Department of Surgery, Marshall University Joan C Edwards School of Medicine, Huntington, WV, USA.
Received 2018-6-18; Accepted 2018-8-31; Published 2018-10-20
Abstract
Non-Alcoholic Fatty Liver Disease (NAFLD) has been recognized as the most common liver disorder in developed countries. NAFLD progresses from fat accumulation in hepatocytes to steatohepatitis to further stages of fibrosis and cirrhosis. Simple steatosis, i.e. fat deposition in the liver, is considered benign and gives way to non-alcoholic steatohepatitis (NASH) with a higher probability of progressing to cirrhosis, and liver-related mortality. Evidence has been found that this progression has been associated with marked alterations in hepatocyte histology and a shift in marker expression of healthy hepatocytes including increased expression of peroxisome proliferator-activated receptor gamma (PPARγ), adipocyte protein (aP2), CD36, interleukin-6 (IL-6), interleukin-18 (IL-18) and adiponectin. This progression shares much in common with the obesity phenotype, which involves a transformation of adipocytes from small, healthy cells to large, dysfunctional ones that contribute to redox imbalance and the progression of metabolic syndrome. Further, activation of Src/ERK signaling via the sodium potassium adenosine triphosphatase (Na/K-ATPase) α-1 subunit in impaired hepatocytes may contribute to redox imbalance, exacerbating the progression of NAFLD. This review hypothesizes that an adipogenic transformation of hepatocytes propagates redox imbalance and that the processes occurring in adipogenesis become activated in fat-laden hepatocytes in liver, thereby driving progression to NAFLD. Further, this review discusses therapeutic interventions to reverse NAFLD including the thiazolidinediones (TZDs) and a variety of antioxidant species. The peptide, pNaKtide, which is an antagonist of Na/K-ATPase signaling, is also proposed as a potential pharmacologic option for reducing reactive oxygen species (ROS) and reversing NAFLD by inhibiting the Na/K-ATPase-modulated ROS amplification loop.
Keywords: Non-Alcoholic Fatty Liver Disease, Hepatocytes
Introduction
In recent years, NAFLD has become an increasingly prevalent health problem internationally and has been found to affect approximately 30% of the United States population [1]. NAFLD is a broad culmination of pathological conditions which encompasses simple steatosis, predominantly caused by the excessive lipid accumulation in the liver, to a more severe and progressive steatohepatitis (NASH), involving fibrosis and cirrhosis [2-5]. As NAFLD progresses, a variety of histopathological changes occur in patients' livers, such as fibrosis, hepatocellular damage/dysfunction, lobular inflammation, and hepatocellular ballooning [2]. Since there are no pharmacological therapies approved for NAFLD, interest has spiked in regard to the molecular pathways, which contribute to this condition in order to obtain viable treatment options to improve patient outcomes.
The condition of obesity is closely associated with hepatic steatosis that subsequently progresses to NASH. Obesity is characterized by an excessive accumulation of body fat due to impaired fat storage and metabolism, specifically having a body mass index (BMI) greater than 30 kg/m2 [6]. This condition has become increasingly prevalent worldwide. It is estimated that by 2030 around 38% of the world's population will be overweight and another 20% will be obese [7]. Common molecular markers present at increased levels in adipogenesis leading to obesity include PPARγ, CCAAT/enhancer binding proteins (C/EBPs), aP2, adiponectin, IL-18, IL-6, and tumor necrosis factor alpha (TNF-α) [8-11]. Obesity directly contributes to the abundance of hepatic triglyceride accumulation associated with the progression towards NAFLD. Therefore, it is reasonable to conclude that correlations between phenotypic changes in adipocytes and hepatocytes may be present. Prior research has demonstrated that adipogenic changes characteristic of obesity can occur in hepatocytes and contribute to the development of NAFLD [12]. Notably, PPARγ, a key regulator of adipocyte differentiation and lipid storage, as well as aP2 have been observed to be upregulated in livers of mice fed a high-fat diet suggesting a strong association with hepatocyte differentiation. Further, steatotic primary hepatocytes were also observed to release the pro-inflammatory cytokines IL-6, IL-18, monocyte chemoattractant protein-1 (MCP-1) and TNF-α in a manner characteristic of adipogenesis. In addition, molecular changes contributing to the transformation of hepatocytes away from their normal physiology have been observed. This includes reduction in levels of cell membrane-bound E-cadherin and albumin [12, 13].
The development and progression to NAFLD is contributed by the accumulation of ROS as well as inflammation resulting from the upregulated release of pro-inflammatory cytokines from hepatocytes, including IL-6 and TNF-α [14]. In hepatocytes, an imbalance between oxidant and antioxidant agents results in ROS accumulation and oxidative stress that increases lipid peroxidation and fat accumulation [15-17]. One mechanism by which oxidative stress is exacerbated is the Na/K-ATPase/Src signaling cascade. Recent studies by our group have demonstrated the role of Na/K-ATPase/Src signaling in the amplification of ROS, whereby excessive systemic ROS causes carbonylation of the α-1 subunit of the Na/K-ATPase pump. In turn, this activates the Src signaling cascade, ultimately leading to further activation of downstream signaling pathways and ROS generation [18]. Studies have shown that the peptide, pNaKtide, specifically inhibits Src activation, therefore demonstrating a potential method to attenuate Na/K-ATPase-modulated ROS amplification in hepatocytes [19]. Although there is the potential of pNaKtide for reducing oxidative stress and attenuating NAFLD, currently available therapeutic options to treat NAFLD are limited. Some of the existing therapeutic strategies to treat NAFLD include possible treatment with TZDs, a class of drugs that reduce insulin resistance and has potential for reducing the levels of fatty accumulation in hepatocytes [20, 21]. Further, a variety of antioxidants have displayed benefits in the treatment of NAFLD, such as vitamin D, superoxide dismutase (SOD), N-Acetylsysteine (NAC), and heme oxygenase-1 (HO-1) [22-25].
Understanding the molecular mechanisms and similarities between hepatocyte transformation leading to NAFLD and adipogenesis is essential in order to formulate new treatment options for patients susceptible to these conditions. This review hypothesizes that adipogenic transformation of hepatocyte leads to its phenotypic alterations that propagates redox imbalance in order to further the literature in this area and contribute to the discovery of new treatment options, such as pNaKtide, which can lead to improved patient outcomes and decreased healthcare costs.
Adipogenic Markers in Adipose Tissue
Adipocytes are characterized by specific morphological and functional characteristics. These characteristics are adapted through the process of adipogenesis which involves the differentiation of fibroblast like pre-adipocytes into mature, lipid laden, insulin-responsive adipocytes [26]. The successful transformation into mature adipocytes is achieved by the marked changes in morphology and gene expression of pre-adipocytes. Although many of the molecular details of adipogenesis are still unknown, numerous factors involved in this process have been identified. Some stimulators include PPARγ, C/EBPα, signal transducers and activators of transcription (STATs), the transcriptional factor, sterol-regulatory-element-binding-protein-1 (SREBP1), insulin like growth factor-1 (IGF-l), macrophage colony stimulating factor, fatty acids, prostaglandins and glucocorticoids. Briefly, the C/EBPα upregulate the expression of PPARy, which in turn heterodimerizes with retinoid X receptor (RXR) and regulates the adipogenic processes that are required to promote fat cell differentiation [27]. This process further induces a positive feedback loop thereby maintaining the differentiated cell state [28]. Studies show that PPARγ is necessary for the differentiation of white fat adipocytes [29]. Overexpressed PPaRγ can rescue differentiation in C/EBPα deficient cells. Recent studies have also identified multiple inhibitors including glycoproteins, transforming growth factor (TGF), inflammatory cytokines, and growth hormones.
In the state of obesity, adipocytes undergo molecular changes that contribute to the pathophysiology of the disease and other comorbidities. An increase in visceral fat is often associated with increased inflammation, dyslipidemia, and insulin resistance [30]. A variety of cytokines such as TNF-α, IL-6, IL-18, and C-reactive protein have been observed to be upregulated in adipogenesis and contribute to inflammation [31]. Together, these molecular markers contribute to adipocyte transformation in the disease state of obesity.
Phenotypic Alterations in Hepatocytes
Prior research suggests that hepatocytes in the diseased state of NAFLD mirror adipogenic markers that are observed in the adipocytes [12]. Using the high-fat diet mouse model of hepatic steatosis, evidence suggests that the pathogenesis of NAFLD could likely involve adipogenic changes in hepatocytes (Table 1). The development and progression of NAFLD is mediated by increased lipid accumulation, suggesting increased triglycerides levels. Even though, the hepatocytes demonstrate typical morphological changes, the acquisition of adipocyte markers and adipose-secreted factors by hepatocytes, induces phenotypical changes as a part of pathophysiology of NAFLD. The phenotypical alterations are further assisted by the excessive oxidative stress and inflammation [11, 12, 15, 32, 33], which in turn exacerbates the diseased condition and impairs metabolic parameters. The secretory markers from adipose tissues promote adipogenesis, causing steatotic hepatocytes to undergo adipogenic alterations and predominantly express adipocyte markers.
Phenotypical and molecular findings in the high fat diet model of NAFLD. This table summarized findings for various in vivo models demonstrating the diet induced molecular alterations of hepatic and adipogenic markers in the pathogenesis of NAFLD.
| Reference | Finding | |
|---|---|---|
| [12] | Species: Mouse Strain: C57BL/6 Diet: HFD for 24 weeks | Induced expression of adipogenic genes: aP2 and PPARγ Increased cytoplasmic aP2 and albumin protein Reduction in membrane bound E-cadherin and albumin expression Impaired adiponectin activity: upregulation of CD36, decreased hepatic adiponectin, decreased adiponectin receptor-2 |
| [34] | Species: Mouse Strain: C57Bl/6Ncrj Diet: HFD for 2-12 weeks | Lipid accumulation at 2 weeks Fatty liver phenotype at 12 weeks Upregulation of genes of lipid metabolism: PPARγ and CD36 Suppression of CREBP |
| [89] | Species: Mouse Strain: C57BL/6 Diet: HFD for 15 weeks | Induced expression of following genes: hepatic fat accumulation: PPARγ, CD36, FABP4 macrophage markers: F4/80, CD68 inflammatory proteins: TNFα, MCP-1 and CCR2 |
| [90] | Species: Mouse Strain: Fat aussie foz/foz Diet: HFD for 300 days | Severe steatohepatitis with hepatocyte ballooning, inflammation, and fibrosis Increased expression of the lipogenic gene SREBP1c Decreased expression of mitochondrial UCP2 Decreased hepatic lipid disposal gene expression of PPARα Increased adipogenic gene expression of aP2 |
| [91] | Species: Rat Strain: Sprague—Dawley Diet: HFD with 5% cellulose for 7 weeks | Induction of lipogenic genes PPARγ, SREBP1c, FAS and FABP4 |
Abbreviations: aP2 - adipocyte protein 2; PPARγ - peroxisome proliferator activated receptor gamma; CREBP - cAMP response element binding protein; FABP4 - fatty acid binding protein 2; TNFα - tumor necrosis factor alpha; MCP-1 - monocyte chemoattractant protein 1; CCR2 - C-C chemokine receptor type 2; SREBP-1c - sterol regulatory element binding protein 1c; UCP2 - uncoupling protein 2; PPARα - peroxisome proliferator activated receptor alpha; FAS - fatty acid synthase.
PPARγ has been found to be upregulated in the livers of high-fat diet mice [12, 13]. PPARγ isoforms 1 and 2 have been expressed principally in mature, white and brown adipocytes, and at lower levels in other tissues such as the spleen, liver, skeletal muscle, and the heart [13]. A high-fat diet can increase mRNA expression of both PPARγ-1 and -2 in mouse adipocytes and in steatotic liver [13]. Livers of several murine models of obesity and diabetes express enhanced levels of PPARγ, including ob/ob, A-ZIP, aP2/DTA, and KKAy [34]. aP2, the upstream activator of PPARγ, was also up-regulated in high-fat diet mice. aP2 is involved in terminal adipocyte differentiation [35]. aP2, along with the RXR, forms a heterodimeric DNA binding complex which mediates the effects of the PPAR family including PPARγ [13]. Steatotic livers of lipoapotrophic AZIP mice expressed elevated levels of aP2 and adipose differentiation-related protein (ADRP) [35]. A recent study has demonstrated the upregulated expression of PPARγ and aP2 in steatotic hepatocytes. This implies acquisition of adipocyte like phenotype by the hepatocytes, potentially contributing to the pathogenesis of NAFLD [12]. Lipoapotrophic mice with lack of liver PPARγ (AZIP LKO) manifested reduced mRNA expression of both ADRP and aP2, indicating that liver PPARγ would contribute to the regulation of hepatocyte lipid transport and storage [35].
A majority of these are genes that participate in adipogenic differentiation or lipid metabolism suggesting that the observed lipid accumulation in hepatocytes reflects transformation of hepatocytes toward adipocytes. Recent studies have shown the contribution of alpha fetoprotein (AFP) in the liver regeneration that is associated with hepatic steatosis, NAFLD, and hepatocellular carcinoma [36-38]. While the pathogenesis of upregulated AFP expression remains largely unknown, a significant correlation has been demonstrated in patients with central obesity, elevated triglyceride (TG) levels, upregulated free fatty acid (FFA), high BMI and abdominal fat, with subsequent development of metabolic syndrome (MS) [39]. An underlying mechanism may be ascertained between the development of hepatic steatosis and upregulation of AFP considering that obesity-induced IR affects hepatic metabolic pathways and causes the release of FFA from adipose tissues. This further leads to excessive lipid accumulation, oxidative stress and inflammation causing metabolic imbalance leading to the release of AFP in phenotypically altered hepatocytes.
The presence of the diseased condition and hepatic steatosis also implicates the reduction of hepatocyte function. The adipogenic alteration of hepatocytes has been evidently confirmed by a recent study that demonstrates the reduction in the secretory function of hepatocytes. The extended findings significantly showed decreased levels of hepatocyte markers, albumin, and E-cadherin, in a high fat diet mice model [12]. The decreased expression of these hepatocyte markers implicitly suggests the development of steatotic hepatocytes with impaired epithelial phenotype, further assisting in acquiring adipocyte markers and function. The fact that albumin contributes in the inhibition of adipogenesis [40], the decreased levels suggests the restored adipogenic processes which will be conducive to the pathogenesis of NAFLD.
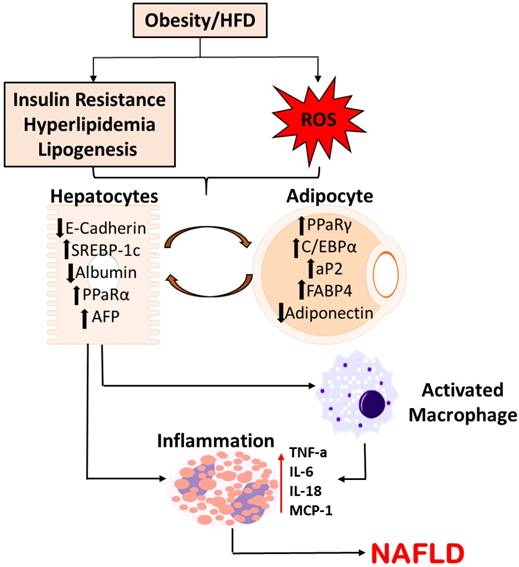
Several other studies have noted upregulated expression of adipocyte markers, adipsin and caveolin-1, that were demonstrated to be elevated in the hepatocytes of steatotic livers [41, 42]. Adipocyte markers, aP2 and PPARγ, increased in the livers of mice at both mRNA and protein levels. Further, it was also observed that hepatocytes release pro-inflammatory cytokines characteristic of adipogenesis including TNF-α, IL-6, and IL-18 [43-45]. These conclusions support the claim that hepatocytes in the disease state of NAFLD undergo molecular changes of adipocytes. Figure 1 summarizes the possible pathway of phenotypic alterations in hepatocytes suggesting possible pathogenesis towards NAFLD.
Schematic Representation of Molecular Alterations in Hepatocytes in the pathogenesis of NAFLD. Diet-induced metabolic complications and systemic ROS causes the molecular alterations in hepatocytes, subsequently activating macrophages and releasing pro-inflammatory cytokines. This pathogenesis potentially plays a crucial role in the disease progression to NAFLD.
Oxidative stress and ROS production in NAFLD
The trigger for the transition of NAFLD from steatosis to steatohepatitis, while unknown, is thought to be associated with an increase in ROS and subsequent oxidative stress. Disruption of metabolic balance, insulin resistance, and increased ROS production are key to steatohepatitis in lipid-laden hepatocytes [46]. Multiple studies have demonstrated the occurrence of NAFLD in the setting of high fat diet, which promotes redox imbalance, increased FFA, hepatic de novo lipogenesis, and increased fibrosis due to intrahepatic lipid accumulation [47-53]. The transition from steatosis to steatohepatitis involves an overall increase in oxidative stress-related parameters with proteolysis and diminished antioxidant capacity. Oxidative stress in the liver has also been associated with a higher peroxidation and depletion of n-3 long-chain poly unsaturated fatty acids which, in turn, impairs fatty acid oxidation and export of triacylglycerol [43]. The modification of hepatic lipid metabolism gene transcription by dietary fat, achieved via long-chain poly unsaturated fatty acids, has been likely the factor into the pro-oxidant status that could promote the transition of the steatotic to a steatohepatitic liver [43]. A recent study further highlights that hepatic steatosis is exacerbated by oxidative stress in the liver, which was directly related to increased lipotoxicity [54-56].
In hepatocytes, the consequence of excessive ROS production in the setting of excessive FFA is that there is ATP/NAD depletion, membrane damage via lipid peroxidation and pro-inflammatory cytokines release [57]. The primary sources of endogenous intracellular ROS production in liver includes, cellular mitochondria, ER stress via activation of cytochrome P450 enzymes, activation of Ras pathway, excessive production of Angiotensin II and upregulated activity of NADPH oxidase. Activated AMP kinase (AMPK) is also known to aggravate the pathophysiological condition by increasing fatty acid oxidation via stimulation of carnitine palmitoyltransferase-1 (CPT-1), which catalyzes the formation of acylcarnitine (from Acyl-CoA) [58]. The excessive production of ROS is further exacerbated by the diseased physiological condition of hepatocytes that alters the cellular strategies to maintain a balance of oxidant and antioxidant markers. These phenotypic changes in hepatocytes leads to structural and functional damage causing the pathogenesis of NAFLD, by reducing anti-oxidant defenses. The livers of the patients with steatohepatitis revealed that oxidative stress was amplified with induction of the CYP2E1 and by reduction of the protein carbonyl content, hepatic glutathione (GSH) content, SOD, GPx catalase, and ferric reducing ability of plasma compared to controls levels [54].
Role of Na/K-ATPase/Src signaling in NAFLD
Since oxidative stress plays a key role in the development of steatohepatitis, it is imperative to examine potential targets that can restore cellular redox balance. In this regard, Na/K-ATPase, a member of P-type ATPase, has been recently shown to affect cellular signaling and cause oxidative stress, in addition to its distinct pumping function [59]. The Na/K-ATPase α-1 subunit serves a scaffolding function with Src, allowing conformational changes in the NA/K-ATPase to activate membrane bound Src and initiate a signaling cascade [60]. The carbonylation of α-1 subunit is initiated by excessive systemic ROS in a diseased condition. This activates the Src signaling cascade with downstream modulation of extracellular signal regulated kinase 1/2 (ERK 1/2), ultimately leading to further ROS generation [61, 62]. Thus, ROS is not only generated from the Na/K-ATPase signaling cascade, but can also initiate it, creating a feed-forward oxidant amplification loop, which has been recently characterized [63, 64]. In vitro and in vivo studies have provided strong evidence suggesting a key role of Na/K-ATPase signaling in multiple clinical disorders, including, but not limited to, obesity, atherosclerosis, chronic renal failure and experimental uremic cardiomyopathy [19, 33, 57]. Activation of Na/K-ATPase signaling cascade promiscuously leads to the feed forward oxidant amplification loop, that consequently activates Ras-Raf-Nox pathway and inflammatory pathways in a diseased state. The studies elucidated the ability of Na/K-ATPase signaling in the amplification of ROS involved in adipogenesis, while also worsening the insulin sensitivity [19]. Recent studies also implicated the role of Na/K-ATPase signaling in the aging process, which was shown to exacerbate cellular senescence, apoptosis and oxidant stress [65]. These studies in experimental disease models established Na/K-ATPase signaling cascade as a potential therapeutic target to restore redox imbalance and metabolic homeostasis. Given the pathophysiological condition in obesity, the increased FFAs causes induction of excessive ROS generation, leading to the activation of Na/K-ATPase/ROS amplification loop and initiating downstream signaling pathways, contributing to steatosis and NASH. Prior studies have demonstrated that Na/K-ATPase signaling cascade also activates PKCδ, which in turn, phosphorylates Fli-1 leading to the amplification of collagen expression [33]. The deposition of collagen aggravates rapid advancement of fibrosis, causing disease progression from NAFL to NASH.
Inflammation in NAFLD
NAFLD is characterized by hepatic fatty changes with lobular hepatitis, focal necrosis containing inflammatory infiltrates and often Mallory bodies and fibrosis [66, 67]. It is well documented that oxidative stress and hepatic steatosis in hepatocytes cause the release of various inflammatory cytokines. High fat diet increases pro-inflammatory cytokines during initial fibrogenic processes that initiate a crosstalk between injured hepatocytes and hepatic stellate cells (HSCs). The activated HSCs express large amounts of extracellular matrix (ECM), which is further enhanced by the production of tissue inhibitors of metalloproteinases, leading to a gradual disruption of normal liver architecture and fibrosis [68]. Recent study demonstrated that C57BL/6 mice placed on a high-fat diet, for 12 weeks, developed fatty liver phenotype, diabetes, triglyceride accumulation in hepatocytes and increased serum total cholesterol. Interestingly, the livers of more than 70% of these mice displayed scattered inflammatory foci of mononuclear cells with hepatocyte ballooning, hepatocyte necrosis and increased lipid droplets [12]. Further in vitro studies have shown elevated mRNA expression of pro-inflammatory cytokines, including TNF-α, MCP-1, IL-6 and IL-18, in steatotic hepatocytes [12]. The release of inflammatory cytokines also activates Kupffer cells that, in turn, influence hepatocyte lipid metabolism and storage [69]. This further leads to the production of fibrogenic genes, TGFβ-1, causing exacerbation of hepatic fibrosis [32, 70-77].
The hormone adiponectin is an adipokine secreted from adipose tissue that plays an anti-lipogenic and anti-inflammatory role, regulating glucose and fatty acid metabolism. Adiponectin is a factor distinguishing NASH from simple steatosis [45]. A study of NAFLD patients found that plasma adiponectin levels were decreased with increased waist circumference, body mass index, triglycerides, insulin resistance, and amino-transferase levels [44]. The expression of adiponectin and the adiponectin receptor II was reduced in hepatic biopsies from patients with NASH compared to simple steatosis [45].
Pharmacological Interventions for NAFLD
The prevalence of NAFLD is associated with its poor prognosis, with limited prominent treatment options available. The potent understanding of pathogenic pathways involved in the development and progression of NAFLD may allow for the targeted intervention strategies. The current treatment options include superficial treatment of metabolic derangements associated with NAFLD using antioxidant therapy and multiple drugs to improve biochemical levels and attenuate inflammation.
A popular treatment option comprises the use of TZDs, a class of heterocyclic compounds. Treatment with TZDs allows for the histologic improvement of NASH by acting as a PPAR-γ agonist and improving the efficiency of glucose metabolism [78]. Representatives of TZDs, rosiglitazone and pioglitazone, are often used in combination with other treatment options. In a controlled trial, rosiglitazone treatment of adult patients with biopsy-proven NASH resulted in improvement of hepatic enzymes to near pretreatment levels by 6 months, with 45% no longer meeting NASH criteria by 48 weeks, along with improved insulin sensitivity and mean ALT levels [79]. Patients with daily pioglitazone treatment for 48 weeks resulted in normal serum ALT in 72% of patients, reductions in hepatic fat content and size, improved glucose and free fatty acid sensitivity to insulin and improvement in histological appearance of cellular injury, parenchymal inflammation, mallory bodies and fibrosis [80]. However, there have been several concerns over the safe administration and side effects of TZDs, including rosiglitazone and pioglitazone. While the use of rosiglitazone has been completely eliminated in some countries and highly limited in others, the viability of TZD class of drugs have been questioned due to its potential risks in causing heart failure, macular edema and bladder cancer [81].
Since oxidative stress underlies the etiopathogenesis of diseases including obesity, diabetes, and NAFLD, it is imperative to evaluate the potential application of antioxidant therapy. Multiple randomized controlled trials have demonstrated the effectiveness of Vitamin E in attenuating the extent of NASH pathology. Studies demonstrated that administration of Vitamin E at various doses in pediatric and adult population improved levels of aminotransferase, ALT, hepatocellular ballooning, lobular inflammation and hepatic steatosis [82-85]. However, prolonged intake of Vitamin E has been reported to cause prostate cancer in healthy adults and may increase all-cause mortality [86, 87], therefore, treatment of NAFLD cannot be completely relied on such antioxidants.
Each class of drug has its own benefit and safety profile, however, the systemic complications associated with the administration of these agents may present challenges for clinicians in the intervention strategies for clinical disorder. Based on these observations, it is important to adapt a targeted intervention strategy to ameliorate the consequences associated with NAFLD and avoid contraindications.
Effectiveness of pNaKtide in attenuating NAFLD
A cell permeable peptide, pNaKtide, has emerged to play a paramount role in attenuating multiple pathophysiological diseased conditions. The development of pNaKtide was detailed in 2009 [88]. Briefly, pNaKtide acts as an antagonist of the scaffolding function of Na/K-ATPase/Src signaling that blocks the formation of the receptor Na/K-ATPase-Src complex. The CTS induced activation of Na/K-ATPase signaling cascade is negated by the pNaKtide. Our group has extensively demonstrated the effectiveness of pNaKtide, in C57BL/6 mice fed a western diet for 2 months. The study demonstrated that pNaKtide effectively improved the fatty acid oxidation genes in adipose tissues (CPT-1, PGC-1α and UCP 1/2), hepatic lipogenesis and attenuated expression for markers of macrophage infiltration and inflammation (F40/80, MCP-1, IL-6) [33]. Additionally, treatment with pNaKtide in our murine model of high fat diet established robust evidence exhibiting reduction in oxidative stress and hepatic fibrosis (assessed by collagen synthesis in liver and mRNA expression of hepatic fibrogenic genes, fibronectin, MMP9 and MMP13. The potency of pNaKtide has been previously compared with similar Src inhibitors, like PP2 [18, 88]. However, unlike other Src inhibitors, pNaKtide has demonstrated exclusivity to Na/K-ATPase by evidently impeding the formation of Na/K-ATPase/Src receptor complex, further inhibiting the activation of downstream pathways, including ERK 1/2 [88]. pNaKtide specifically localizes to the membrane component of the cell that inhibits the association with membrane bound Src. The pNaKtide superficially seems to mimic antioxidants, as it has been effective in ameliorating oxidative stress, however the specific mode of action of pNaKtide is what makes it novel for further implications. Collectively, pNaKtide has been shown to ameliorate obesity, insulin resistance, dyslipidemia, metabolic syndrome, hepatic steatosis and NASH phenotype by mediating the blockage of Na/K-ATPase/ROS amplification loop [33].
Conclusion
This review hypothesizes that an adipogenic transformation of hepatocytes propagates redox imbalance, inflammation, and fibrosis in the disease state of NAFLD, ultimately leading to the development of NASH. The transformation of hepatocytes during NAFLD progression (summarized in Figure 1), parallels the progression of adipocytes in obesity in which small, healthy adipocytes transform to large, dysfunctional cells. The literature has reported a shift in expression from markers of healthy hepatocytes to an expression of adipogenic markers in conjunction with their NASH phenotypic alterations of ballooning, redox imbalance, and liver damage. These findings are summarized in Table 2. These changes are concurrent with other molecular changes in hepatocytes away from their normal physiology.
Summarized biomarker expression and function in the pathogenesis of NAFLD. This table summarizes the expression of each biomarker and their expression in either hepatocytes or adipocytes during the adipogenic processes and alteration of hepatocyte in NAFLD. The cellular function of each biomarker is summarized and is correlated with their regulation in the cell in the diseased state.
| Biomarkers | Hepatocytes/Adipocytes | Regulation | Function |
|---|---|---|---|
| PPARϒ | Adipocyte | ↑↑ | Adipocyte Differentiation and Lipid Storage |
| aP2 | Adipocyte | ↑↑ | Involvement in terminal adipocyte differentiation |
| AFP | Hepatocyte | ↑↑ | Plays a role in the liver regeneration associated with hepatic steatosis |
| Albumin | Hepatocyte | ↓↓ | Contributes in the inhibition of adipogenesis |
| E-Cadherin | Hepatocyte | ↓↓ | A calcium dependent cell adhesion molecule in the formation and maintenance of hepatocyte. Protects hepatocyte from cell death |
| SREBP-1c | Hepatocyte | ↑↑ | Mediates the activation of lipogenesis: Increases the de novo synthesis of fatty acids |
| Adiponectin | Adipocyte | ↓↓ | Regulates glucose and fatty acid metabolism. Secreted from adipose tissue and plays important role as anti-lipogenic and anti-inflammatory marker. |
| C/EBPα | Adipocyte | ↑↑ | Induces adipogenesis by the mediating the upregulation of PPARϒ |
| PPARα | Hepatocyte | ↑↑ | Central mediator for signaling molecules to maintain lipid homeostasis and regulate genes in cell growth and differentiation. |
Abbreviations: aP2 - adipocyte protein 2; PPARγ - peroxisome proliferator activated receptor gamma; SREBP-1c - sterol regulatory element binding protein 1c; PPARα - peroxisome proliferator activated receptor alpha; FAS - fatty acid synthase; AFP - alpha fetoprotein; C/EBPα - CCAAT enhancer-binding protein alpha.
Although limited pharmacologic treatment for NAFLD currently exists, potential treatment options to reverse the progression of NAFLD include TZDs as well as various antioxidant agents. Notably, the specific Na/K-ATPase antagonist, pNaKtide, has been implicated in the reduction of ROS in hepatocytes thereby reversing fat accumulation and the obesity phenotype. Further research is necessary in order to examine the effects of pNaKtide treatment on reducing expression of adipogenic markers in hepatocytes, hepatocyte transformation, and liver injury. This would implicate both the EFGR/Src pathway in NAFLD progression and establish pNaKtide as a potential pharmacological treatment for NAFLD.
Acknowledgements
This work was supported by National Institutes of Health Grants to JIS (HL109015, HL105649 and HL071556), and by the Brickstreet Foundation (J.I.S.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
References
1. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263-73
2. Haas JT, Francque S, Staels B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu Rev Physiol. 2016;78:181-205
3. Lou Y, Tian GY, Song Y. et al. Characterization of transcriptional modules related to fibrosing-NAFLD progression. Sci Rep. 2017;7:4748
4. Sharma DL, Lakhani HV, Klug RL. et al. Investigating Molecular Connections of Non-alcoholic Fatty Liver Disease with Associated Pathological Conditions in West Virginia for Biomarker Analysis. J Clin Cell Immunol. 2017:8
5. Benedict M, Zhang X. Non-alcoholic fatty liver disease: An expanded review. World journal of hepatology. 2017;9:715-32
6. Kaila B, Raman M. Obesity: a review of pathogenesis and management strategies. Can J Gastroenterol. 2008;22:61-8
7. Hruby A, Hu FB. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics. 2015;33:673-89
8. Neuman MG, Cohen LB, Nanau RM. Biomarkers in nonalcoholic fatty liver disease. Can J Gastroenterol Hepatol. 2014;28:607-18
9. Cawthorn WP, Sethi JK. TNF-alpha and adipocyte biology. FEBS Lett. 2008;582:117-31
10. Kern PA, Ranganathan S, Li C. et al. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2001;280:E745-51
11. Wood IS, Wang B, Jenkins JR. et al. The pro-inflammatory cytokine IL-18 is expressed in human adipose tissue and strongly upregulated by TNFalpha in human adipocytes. Biochem Biophys Res Commun. 2005;337:422-9
12. Pan X, Wang P, Luo J. et al. Adipogenic changes of hepatocytes in a high-fat diet-induced fatty liver mice model and non-alcoholic fatty liver disease patients. Endocrine. 2015;48:834-47
13. Vidal-Puig A, Jimenez-Linan M, Lowell BB. et al. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest. 1996;97:2553-61
14. Ucar F, Sezer S, Erdogan S. et al. The relationship between oxidative stress and nonalcoholic fatty liver disease: Its effects on the development of nonalcoholic steatohepatitis. Redox Rep. 2013;18:127-33
15. Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol. 2014;20:8082-91
16. Morita M, Ishida N, Uchiyama K. et al. Fatty liver induced by free radicals and lipid peroxidation. Free Radic Res. 2012;46:758-65
17. Furukawa S, Fujita T, Shimabukuro M. et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752-61
18. Srikanthan K, Shapiro JI, Sodhi K. The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Obesity and Cardiovascular Disease. Molecules. 2016:21
19. Sodhi K, Maxwell K, Yan Y. et al. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci Adv. 2015;1:e1500781
20. Chang E, Park CY, Park SW. Role of thiazolidinediones, insulin sensitizers, in non-alcoholic fatty liver disease. J Diabetes Investig. 2013;4:517-24
21. Zhou G, Myers R, Li Y. et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167-74
22. Barchetta I, Cimini FA, Cavallo MG. Vitamin D Supplementation and Non-Alcoholic Fatty Liver Disease: Present and Future. Nutrients. 2017:9
23. Liu W, Baker SS, Baker RD. et al. Antioxidant Mechanisms in Nonalcoholic Fatty Liver Disease. Curr Drug Targets. 2015;16:1301-14
24. Khoshbaten M, Aliasgarzadeh A, Masnadi K. et al. N-acetylcysteine improves liver function in patients with non-alcoholic Fatty liver disease. Hepat Mon. 2010;10:12-6
25. Hinds TD Jr, Sodhi K, Meadows C. et al. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity (Silver Spring). 2014;22:705-12
26. Lefterova MI, Lazar MA. New developments in adipogenesis. Trends in endocrinology and metabolism: TEM. 2009;20:107-14
27. Brey CW, Nelder MP, Hailemariam T. et al. Kruppel-like family of transcription factors: an emerging new frontier in fat biology. International journal of biological sciences. 2009;5:622-36
28. Rosen ED, Hsu CH, Wang X. et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes & development. 2002;16:22-6
29. Farmer SR. Transcriptional control of adipocyte formation. Cell metabolism. 2006;4:263-73
30. Peterson SJ, Drummond G, Kim DH. et al. L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. Journal of lipid research. 2008;49:1658-69
31. Li M, Kim DH, Tsenovoy PL. et al. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57:1526-35
32. Tang T, Sui Y, Lian M. et al. Pro-inflammatory activated Kupffer cells by lipids induce hepatic NKT cells deficiency through activation-induced cell death. PloS one. 2013;8:e81949
33. Sodhi K, Srikanthan K, Goguet-Rubio P. et al. pNaKtide Attenuates Steatohepatitis and Atherosclerosis by Blocking Na/K-ATPase/ROS Amplification in C57Bl6 and ApoE Knockout Mice Fed a Western Diet. Scientific reports. 2017;7:193
34. Inoue M, Ohtake T, Motomura W. et al. Increased expression of PPARγ in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005;336:215-22
35. Gavrilova O, Haluzik M, Matsusue K. et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. The Journal of biological chemistry. 2003;278:34268-76
36. Babali A, Cakal E, Purnak T. et al. Serum alpha-fetoprotein levels in liver steatosis. Hepatology international. 2009;3:551-5
37. Wu JT. Serum alpha-fetoprotein and its lectin reactivity in liver diseases: a review. Annals of clinical and laboratory science. 1990;20:98-105
38. Qin LX, Tang ZY. Recent progress in predictive biomarkers for metastatic recurrence of human hepatocellular carcinoma: a review of the literature. Journal of cancer research and clinical oncology. 2004;130:497-513
39. Chen Y, Zhao Y, Feng L. et al. Association between alpha-fetoprotein and metabolic syndrome in a Chinese asymptomatic population: a cross-sectional study. Lipids in health and disease. 2016;15:85
40. Schlesinger JB, van Harmelen V, Alberti-Huber CE. et al. Albumin inhibits adipogenesis and stimulates cytokine release from human adipocytes. American journal of physiology Cell physiology. 2006;291:C27-33
41. Razani B, Combs TP, Wang XB. et al. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. The Journal of biological chemistry. 2002;277:8635-47
42. Cook KS, Min HY, Johnson D. et al. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science. 1987;237:402-5
43. Videla LA, Rodrigo R, Araya J. et al. Oxidative stress and depletion of hepatic long-chain polyunsaturated fatty acids may contribute to nonalcoholic fatty liver disease. Free radical biology & medicine. 2004;37:1499-507
44. Bugianesi E, Pagotto U, Manini R. et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. The Journal of clinical endocrinology and metabolism. 2005;90:3498-504
45. Kaser S, Moschen A, Cayon A. et al. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut. 2005;54:117-21
46. Sodhi K, Puri N, Favero G. et al. Fructose Mediated Non-Alcoholic Fatty Liver Is Attenuated by HO-1-SIRT1 Module in Murine Hepatocytes and Mice Fed a High Fructose Diet. PloS one. 2015;10:e0128648
47. Lim JS, Mietus-Snyder M, Valente A. et al. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nature reviews Gastroenterology & hepatology. 2010;7:251-64
48. Nomura K, Yamanouchi T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. The Journal of nutritional biochemistry. 2012;23:203-8
49. Basaranoglu M, Basaranoglu G, Sabuncu T. et al. Fructose as a key player in the development of fatty liver disease. World journal of gastroenterology. 2013;19:1166-72
50. Longato L. Non-alcoholic fatty liver disease (NAFLD): a tale of fat and sugar? Fibrogenesis & tissue repair. 2013;6:14
51. Rebollo A, Roglans N, Alegret M. et al. Way back for fructose and liver metabolism: bench side to molecular insights. World journal of gastroenterology. 2012;18:6552-9
52. Huang D, Dhawan T, Young S. et al. Fructose impairs glucose-induced hepatic triglyceride synthesis. Lipids in health and disease. 2011;10:20
53. Ackerman Z, Oron-Herman M, Grozovski M. et al. Fructose-induced fatty liver disease: hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension. 2005;45:1012-8
54. Videla LA, Rodrigo R, Orellana M. et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clinical science. 2004;106:261-8
55. Patterson RE, Kalavalapalli S, Williams CM. et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. American journal of physiology Endocrinology and metabolism. 2016;310:E484-94
56. Masarone M, Rosato V, Dallio M. et al. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxidative medicine and cellular longevity. 2018;2018:9547613
57. Liu J, Tian J, Chaudhry M. et al. Attenuation of Na/K-ATPase Mediated Oxidant Amplification with pNaKtide Ameliorates Experimental Uremic Cardiomyopathy. Scientific reports. 2016;6:34592
58. Koo SH. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clinical and molecular hepatology. 2013;19:210-5
59. Liang M, Tian J, Liu L. et al. Identification of a pool of non-pumping Na/K-ATPase. The Journal of biological chemistry. 2007;282:10585-93
60. Yan Y, Shapiro JI. The physiological and clinical importance of sodium potassium ATPase in cardiovascular diseases. Current opinion in pharmacology. 2016;27:43-9
61. Yan Y, Shapiro AP, Haller S. et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. The Journal of biological chemistry. 2013;288:34249-58
62. Wang Y, Ye Q, Liu C. et al. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free radical biology & medicine. 2014;71:415-26
63. Liu J, Tian J, Haas M. et al. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. The Journal of biological chemistry. 2000;275:27838-44
64. Xie Z, Kometiani P, Liu J. et al. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. The Journal of biological chemistry. 1999;274:19323-8
65. Sodhi K, Nichols A, Mallick A. et al. The Na/K-ATPase Oxidant Amplification Loop Regulates Aging. Scientific reports. 2018;8:9721
66. Kleiner DE, Brunt EM, Van Natta M. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313-21
67. Ludwig J, Viggiano TR, McGill DB. et al. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clinic proceedings. 1980;55:434-8
68. Elpek GO. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World journal of gastroenterology. 2014;20:7260-76
69. Huang W, Metlakunta A, Dedousis N. et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. 2010;59:347-57
70. Theret N, Lehti K, Musso O. et al. MMP2 activation by collagen I and concanavalin A in cultured human hepatic stellate cells. Hepatology. 1999;30:462-8
71. Kato J, Sato Y, Inui N. et al. Ethanol induces transforming growth factor-alpha expression in hepatocytes, leading to stimulation of collagen synthesis by hepatic stellate cells. Alcoholism, clinical and experimental research. 2003;27:58S-63S
72. Kanuri G, Bergheim I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). International journal of molecular sciences. 2013;14:11963-80
73. Thakur V, Pritchard MT, McMullen MR. et al. Adiponectin normalizes LPS-stimulated TNF-alpha production by rat Kupffer cells after chronic ethanol feeding. American journal of physiology Gastrointestinal and liver physiology. 2006;290:G998-1007
74. Mandal P, Roychowdhury S, Park PH. et al. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. Journal of immunology. 2010;185:4928-37
75. Shi L, Kishore R, McMullen MR. et al. Chronic ethanol increases lipopolysaccharide-stimulated Egr-1 expression in RAW 264.7 macrophages: contribution to enhanced tumor necrosis factor alpha production. The Journal of biological chemistry. 2002;277:14777-85
76. Kishore R, McMullen MR, Cocuzzi E. et al. Lipopolysaccharide-mediated signal transduction: Stabilization of TNF-alpha mRNA contributes to increased lipopolysaccharide-stimulated TNF-alpha production by Kupffer cells after chronic ethanol feeding. Comparative hepatology. 2004;3(Suppl 1):S31
77. Wobser H, Dorn C, Weiss TS. et al. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell research. 2009;19:996-1005
78. Goldberg LR. Simple models or simple processes? Some research on clinical judgments. The American psychologist. 1968;23:483-96
79. Neuschwander-Tetri BA, Brunt EM, Wehmeier KR. et al. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology. 2003;38:1008-17
80. Promrat K, Lutchman G, Uwaifo GI. et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188-96
81. Irons BK, Minze MG. Drug treatment of type 2 diabetes mellitus in patients for whom metformin is contraindicated. Diabetes, metabolic syndrome and obesity: targets and therapy. 2014;7:15-24
82. Lavine JE. Vitamin E treatment of nonalcoholic steatohepatitis in children: a pilot study. The Journal of pediatrics. 2000;136:734-8
83. Sanyal AJ, Chalasani N, Kowdley KV. et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. The New England journal of medicine. 2010;362:1675-85
84. Harrison SA, Torgerson S, Hayashi P. et al. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. The American journal of gastroenterology. 2003;98:2485-90
85. Lavine JE, Schwimmer JB, Van Natta ML. et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. Jama. 2011;305:1659-68
86. Miller ER 3rd, Pastor-Barriuso R, Dalal D. et al. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Annals of internal medicine. 2005;142:37-46
87. Klein EA, Thompson IM Jr, Tangen CM. et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Jama. 2011;306:1549-56
88. Li Z, Cai T, Tian J. et al. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. The Journal of biological chemistry. 2009;284:21066-76
89. Ma Y, Gao M, Liu D. Chlorogenic acid improves high fat diet-induced hepatic steatosis and insulin resistance in mice. Pharm Res. 2015;32:1200-9
90. Arsov T, Larter CZ, Nolan CJ. et al. Adaptive failure to high-fat diet characterizes steatohepatitis in Alms1 mutant mice. Biochemical and biophysical research communications. 2006;342:1152-9
91. Chiu CY, Chan IL, Yang TH. et al. Supplementation of chitosan alleviates high-fat diet-enhanced lipogenesis in rats via adenosine monophosphate (AMP)-activated protein kinase activation and inhibition of lipogenesis-associated genes. Journal of agricultural and food chemistry. 2015;63:2979-88
Author contact
![]() Corresponding author: Komal Sodhi, M.D., Associate Professor of Surgery and Biomedical Sciences, Marshall University Joan C Edwards School of Medicine, WV 25701, Tel: 304 691-1704, Fax: 914 347-4956, E-mail: Sodhiedu
Corresponding author: Komal Sodhi, M.D., Associate Professor of Surgery and Biomedical Sciences, Marshall University Joan C Edwards School of Medicine, WV 25701, Tel: 304 691-1704, Fax: 914 347-4956, E-mail: Sodhiedu