Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(13):1573-1581. doi:10.7150/ijms.27426 This issue Cite
Research Paper
Pin1 Is Involved in HDAC6-mediated Cancer Cell Motility
Hsiang-Hao Chuang1*, Ming-Shyan Huang2,3*, Pei-Hui Wang1, Yu-Peng Liu4, Michael Hsiao5, Chih-Jen Yang1,6,7 ![]()
1. Division of Pulmonary and Critical Care Medicine and Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
2. Department of Internal Medicine, E-DA Cancer Hospital, Kaohsiung, Taiwan
3. School of Medicine, I-Shou University, Kaohsiung, Taiwan
4. Graduate Institute of Clinical Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
5. Genomics Research Center, Academia Sinica, Taipei, Taiwan
6. Department of Internal Medicine, Kaohsiung Municipal Ta-Tung Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
7. Faculty of Medicine, Department of Respiratory Therapy, College of Medicine, Kaohsiung Medical University, Taiwan
*These authors contributed equally to this work.
Received 2018-5-23; Accepted 2018-8-29; Published 2018-10-20
Abstract
Histone deacetylase 6 (HDAC6), a member of the HDAC enzymes, has been reported to play substantial roles in many cellular processes. Evidence shows that deregulation of HDAC6 may be involved in the progression of some cancers, neurodegenerative diseases, and inflammatory disorders. However, little is known regarding the effect of post-translational modification of HDAC6 on cellular localization and biological functions. In the present study, we identified four phosphorylation sites on HDAC6 under normal conditions by mass spectrometry analysis. Two phosphorylation sites, pSer22 and pSer412, are recognized as Pin1 (peptidyl-prolyl cis/trans isomerase NIMA-interacting 1) substrates. Pin1 can interact with HDAC6 and be involved in HDAC6-mediated cell motility. Pin1 depletion abrogates HDAC6-induced cell migration and invasion in H1299 lung cancer cells. The findings of this study suggest that Pin1 might regulate HDAC6-mediated cell motility through alteration of protein conformation and function. Our results indicate the complexity of activity regulation between HDAC6 and Pin1, expanding knowledge regarding the multifunctional roles of Pin1 in tumorigenesis and cancer progression.
Keywords: HDAC6, invasion, migration, Pin1.
Introduction
Histone deacetylase 6 (HDAC6) is a class II deacetylase that belongs to the HDAC superfamily. This deacetylase is unique in the HDAC superfamily because it consists of two catalytic domains (DAC1 and DAC2) [1, 2] responsible for deacetylation and a C-terminal zinc-finger motif (HUB domain) that interacts with ubiquitin and regulates protein ubiquitination [3]. HDAC6 is mostly retained in the cytoplasm, because two nuclear export signals and one Ser-Glu-containing tetrapeptide (SE14) domain make HDAC6 absent from the nucleus [4]. Cytoplasmic HDAC6 is able to catalyze the deacetylation of α-tubulin, β-catenin, cortactin, heat shock protein 90, Ku70, peroxiredoxins, survivin, and Tat. After enzymatic post-translational modifications (PTMs), its substrates commit to given cellular events, such as angiogenesis, cell motility, cell survival, inflammation, protein degradation, and transcription [5]. In addition to cytoplasmic substrates, HDAC6 can catalyze nuclear histones and transcription factors to abrogate the synaptic connection as a pathogenic character of Alzheimer's disease [6]. Clinically, HDAC6-modified nuclear histones and transcription are considered biomarkers for the prognostic estimation of lung cancer [7]. Several reports have documented that protein kinases, such as Aurora A, CK2, EGFR, ERK, GRK2, GSK3β, and PKCɑ, can regulate the enzymatic activity of HDAC6 [8-14]. Indeed, the catalytic activity of HDAC6 can be posttranslationally regulated by these kinases. This PTM is not only a chemical change of protein but also activates catalytic activity and motivates cytological compartmentalization.
In addition to phosphorylation, the cis/trans isomerization of proline imidic peptide bonds is a conformational switch. Peptidyl-prolyl cis/trans isomerase, NIMA-interacting 1 (Pin1) catalyzes the cis/trans isomerization of peptidyl-prolyl peptide bonds. This catalytic action as a molecular switch controls diverse biological processes by causing changes in the protein conformation [15-18]. For example, Pin1 controls cyclin D1 level through its enzymatic action as a configuration switch, resulting in the transcriptional upregulation and posttranslational stabilization [19-21]. This leads to a rise in the proliferation rate. Mounting evidence shows that Pin1 expression level is higher in human cancers. This is thought to be a prognostic marker for unfavorable outcomes [22-26]. In addition to tumorigenesis, Pin1 plays a crucial role in the pathogenic development of Alzheimer's disease [18, 27, 28].
In this study, we found more than two Pin1 recognition sites on HDAC6. We biochemically characterized the interaction of Pin1 with HDAC6, and this interaction involves HDAC6-modulating migratory activity and invasion of non-small cell lung cancer cells. The regulation of the biological activity of HDAC6 by Pin1 might underlie a novel mechanism of malignant tumorigenesis.
Materials and methods
Materials. HeLa cells were provided by Dr. Hui-Chun Wang in Kaohsiung Medical University. Detailed materials information is listed in the key resources table.
Plasmid construction and site-directed mutagenesis. The GFP-HDAC6 plasmid was constructed by amplifying the cDNA of human HDAC6 from the plasmid pcDNA3.1(+)-flag-HDAC6 and cloning into the pEGFP C1 vector with HindIII/SalI restriction enzyme sites. Specific mutations were individually introduced to wildtype GFP-HDAC6 by following the manual of a QuikChange site-directed mutagenesis kit (Agilent Technologies,. La Jolla, CA, USA). All mutants were verified through full-length sequencing.
Immunoprecipitation analysis. GFP-HDAC6-expressing cells were harvested in an immunoprecipitation buffer (50 mM Tris/HCl, pH 7.4; 150 mM NaCl; 1 mM EDTA; 0.5 mM EGTA; 1% Triton X-100; 0.1% sodium deoxycholate; 0.1% SDS; 1 mM PMSF; protease inhibitor cocktail; and phosphatase inhibitor cocktail). Lysates were incubated with anti-GFP antibody conjugated agarose beads (GFP-Trap) at 4 ◦C for 90 min and subjected to SDS-PAGE.
In-gel digestion and liquid chromatography tandem-mass spectrometry (LC-MS/MS). The protein band corresponding to GFP-HDAC6 was taken for trypsin digestion. The peptides were extracted with 0.1% formic acid and loaded on to a reverse-phase column. The desalted peptides were subjected to LC-MS/MS by using an Orbitrap Elite Hybrid Ion Trap-Orbitrap tandem-mass spectrometer interfaced with an 1D-LC (RP), Dionex Ultimate 3000 RSLCnano system (TOOLS Biotech Company). The raw MS/MS spectra data analysis was conducted by using Proteome Discoverer software (version 1.4, Thermo Fisher Scientific). The MS/MS spectra were searched against the UniProt database (released on March 16, 2016, extracted for Homo sapiens, 20,199 sequences) by using the Mascot search engine (version 2.5, Matrix Science, London, UK) for peptides identification.
Proliferation assay. The cell survival rate was determined by using the MTT colorimetric cell proliferation assay. Briefly, 3, 000 cells per well were seeded in 96-well plates and incubated at 37 °C for 2 days. After incubation, the culture medium was removed, and 100 μL of preformulated MTT mixed reagent (MTT reagent: PBS = 1:5) was added and incubated at 37 °C for 3 h. After 3 h of reaction, the mixed reagent was removed and replaced with 100 μL of DMSO and incubated at room temperature for 10 min. The absorbance of the samples will be measured with a spectrophotometer at a wavelength of 570 nm.
Wound healing assay. The cells were suspended in growth media (5 × 105 cells per mL) in 12-well plates and incubated at 37 °Cand 5% CO2 in a humidified atmosphere overnight. The wound was generated by tip scratching and then transferred into an incubation chamber settled on an inverted microscope (Leica DMI6000 B) for time-lapse imaging.
Invasion assay. Cell invasion wase performed by using Matrigel (CORNING) coated filter inserts (8-μm pore size) fitted into a 24-well plate (Merck Millipore, Billerica, MA, USA). After 16 h incubation, the filter inserts were fixed with 4% formaldehyde. The fixed cells were permeabilized by using 0.5% Triton X-100 and stained with 10% Giemsa solution. The filter membrane were washed twice with PBS, and the cells on the top surface of the filter were removed using cotton swabs. The cell number was counted under a fluorescence microscope (Nikon ECLIPSE Ti).
Western blot analysis. The cells or tissues were harvested and lysed in 1x RIPA buffer containing protease inhibitors and phosphatase inhibitors. The protein concentration was determined using a Bio-Rad DC protein assay kit. Subsequently, 30 μg of total protein was loaded onto SDS-PAGE and transferred to a PVDF membrane. The protein was identified by incubating the membrane with the primary antibodies, followed by horseradish peroxidase conjugated secondary antibodies and development with an enhanced chemiluminescence solution.
Lentiviral production and infection. Lentiviruses encoding luciferase, HDAC6, or PIN1 short hairpin RNA (shRNA) were obtained from the National RNAi Core Facility of Academia Sinica, Taiwan. Production and infection of lentiviruses were performed by following the guidelines of the National RNAi Core Facility.
Statistical analysis. Student's t-test was used for statistical analyses. The data in this study are presented as the mean ± SD of at least three independent experiments. *, P < 0.05 indicated significant differences among the experimental groups.
Results
Simulating subcellular localization of endogenous HDAC6 by using GFP-HDAC6
PTM is a regulation mechanism that joins protein functional activity to cellular events. To investigate the effect of PTM on the subcellular localization and enzymatic action of HDAC6, overexpression of N-terminal GFP-tagged HDAC6 in HEK293 cells was established as a systematic visualization of HDAC6 dynamics. The subcellular localization of GFP-HDAC6 in HEK293 cells was recorded. The N-terminal GFP tag in GFP-HDAC6 with N-terminal GFP tag has similar distributions to endogenous HDAC6 and do not mislocalize in HEK293.
Pharmacological inhibition of the CRM1/exportin 1 function with leptomycin B (LMB) does not lead to absence of human HDAC6 in nucleus, but LMB administration retains the mutant human HDAC6 bearing deleted SE14 domain in nucleus (Fig. S1). This result indicates that the SE14 domain is a critical element responsible for human HDAC6 remaining in the cytoplasm. Otherwise, the cytoplasmic localization of murine and human HDAC6 proteins is differentially regulated, becuase the SE14 domain is not evolutionally preserved in murine HDAC6. This results of the pharmacological assay suggested that a GFP-HDAC6 construct was shuttling between the nucleus and cytoplasm. The endogenous HDAC6 could be shuttling protein cycling back and forth between nucleus and cytoplasm.
Identification of phosphorylation sites on HDAC6
Because GFP-HDAC6 and endogenous HDAC6 have similar distributions in HEK293 cells, GFP-HDAC6 is useful for identification of PTM sites in HDAC6. Into further identify PTM sites in HDAC6, GFP-Trap beads-based immunoprecipitation-MS/MS analysis was applied, which revealed that the overexpression of GFP-HDAC6 in HEK293 was the protein bait to sediment GFP-HDAC6 and its associated proteins. Subsequently, the GFP-Trap beads-captured GFP-HDAC6 proteins were separated by SDS-PAGE, and the desired protein band corresponding to the molecular weight of GFP-HDAC6 was collected and digested with trypsin. This protein fraction was subjected to LC-MS/MS analysis (Fig. 1A). Through proteomic analysis, four HDAC6-related phosphopeptides were identified (Fig. 1B). Those were Ser22, Ser43, Ser146, and Ser412. Among these four phosphorylation sites, Ser22 and Ser43 were located in the NLS domain, and the other two (Ser146 and Ser412) were in DAC1 domain (Fig. 1B).
Identification of the phosphorylation modification sites of HDAC6. (A) GFP and GFP-HDAC6 overexpressed in HEK293 cells were immunoprecipitated with anti-GFP agarose beads (GFP-Trap) and subjected to SDS-PAGE for Coomassie Blue staining. Molecular masses are indicated in kDa. For LC-MS/MS analysis, the protein band corresponding to GFP-HDAC6 was excised for in-gel digestion. (B) The tandem mass spectra of phospho-peptides. Each peptide has a single phosphorylation and the sequence and locations of modified amino acid residues is shown on the top.
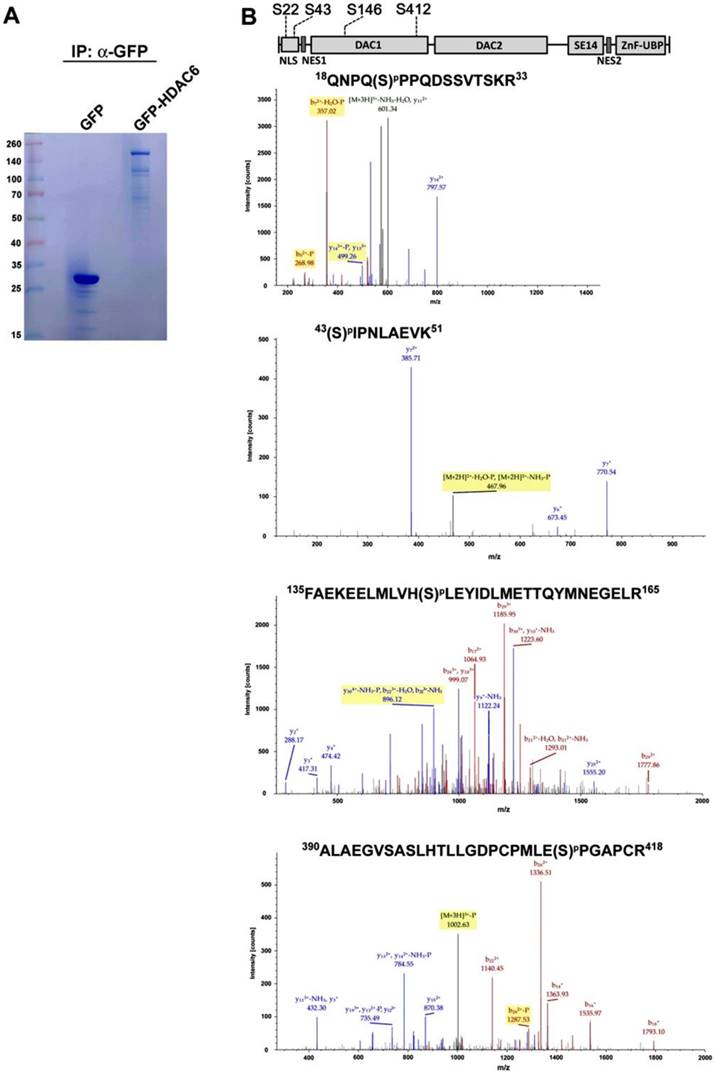
Characterization of the phosphorylation sites on HDAC6
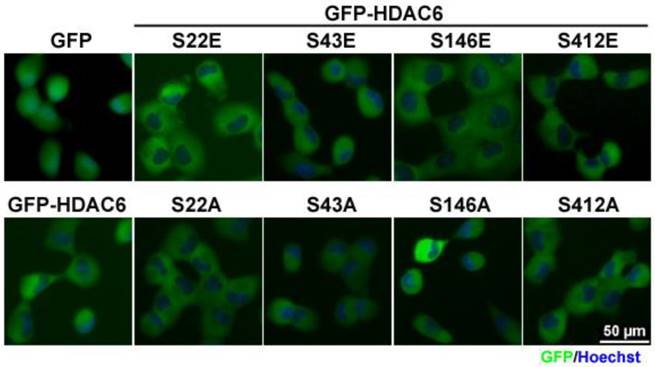
Following the identification of the four phosphorylation sites in HDAC6, the given phosphorylation of HDAC6 destined for its subcellular localization or switched to its catalytic activity was investigated. In this context, the serine-to-glutamic acid substitution as phospho-mimetic constructs and the serine-to-alanine substitution as phospho-defective mutants were designed by using site-directed mutagenesis. In total, eight mutants were constructed, namely HDAC6 (S22E), (S22A), (S43E), (S43A), (S146E), (S146A), (S412E), and (S412A). Then, the N-terminal GFP-tagged HDAC6 mutants were ectopically expressed in H1299 (a non-small cell lung cancer cell line) cells. Visualizing the subcellular distribution of individual HDAC6 mutants revealed that they were mainly present in the cytoplasm (Fig. 2). Mutants expressed in PC9 and HeLa cells had the identical patterns in subcellular localization (Fig. S2A and S2B). Specifically, PTMs exhibited no obvious changes in their subcellular localization.
Post-translational modifications have little effect on cellular localization of HDAC6. H1299 cells expressing GFP, GFP-HDAC6 wild-type and various indicated mutants were stained with Hoechst and monitored for the cellular localization of GFP-fusion proteins by using fluorescence microscopy.
In addition to their subcellular distribution, the effect of the four phosphorylation-modified HDAC6 on cell migration was individually assessed. In this study, wound-healing assay was employed to quantitatively validate migration activity. To neglect the proliferative effect, this assay was settled for 12 h. Our results showed that either the phospho-mimetic residue or phospho-defective mutations in Ser22, an amino acid in the NLS sequence, and mutation with phospho-mimetic substitution at Ser412 residue led to retarded migration. Otherwise, HDAC6 promoted cell motility through phospho-mimetic substitution at Ser43 residue or phospho-defective mutation at Ser412 (Fig. 3A), but the phospho-defective mutant at Ser43 residue reduced cell migration. Regardless, modification at Ser146 does not obviously change cell migration activity.
Post-translational modifications on HDAC6 influence cell motility. (A) H1299 cells expressing GFP, GFP-HDAC6 wild-type and various indicated mutants grew fully in wells. Wounds were generated through tip scratching and cell migration was monitored by microscopy, which was scored according to the distance of migration. The results of relative migration are shown as averages ± SD from three independent experiments compared to WT expressing group. *P < 0.05 and **P < 0.01, based on Student's t-test. (B) H1299 cells expressing GFP, GFP-HDAC6 wild type, and various indicated mutants, were harvested and applied to SDS-PAGE for Western blotting analysis with the indicated antibody. (C) The cell proliferation rate of H1299 cells expressing GFP, GFP-HDAC6 wild type, and various indicated mutants was determined through MTT assay. *P < 0.05 based on Student's t-test.
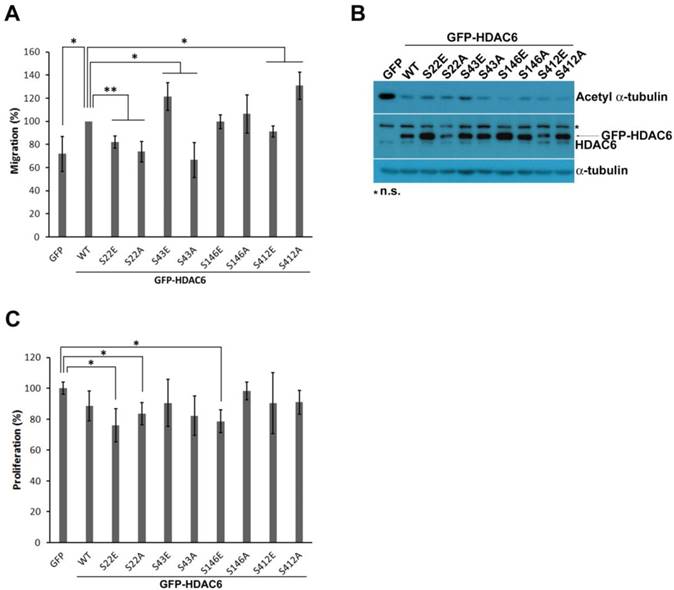
HDAC6 is an enzyme capable of deacetylating ɑ-tubulin. This catalytic reaction on acetylated ɑ-tubulin may also involve regulation of cell migratory activity. In this study, the level of acetylated ɑ-tubulin in H1299 cells bearing the HDAC6 mutants was examined using Western blot analysis. The GFP-HDAC6 overexpression in H1299 also resulted in less acetylated ɑ-tubulin, which implies that HDAC6 enzymatically deacetylates acetylated ɑ-tubulin. During cell migration in the cells bearing GFP-HDAC6 mutants, the acetylated ɑ-tubulin level and cell migration efficiency were not completely concurrent, but lower acetylated ɑ-tubulin level was correlated with increased migration ability.
Regulation of HDAC6 on cell proliferation
The HDAC6-modulated level of c-myc and cyclin D1 has been noted to affect cell proliferative activity [29, 30]. In addition to its role in cell migration, this study confirmed that HDAC6 with variant PTMs is involved in cell proliferation. MTT assay was used to determine proliferative rates. H1299 cells harboring different GFP-HDAC6 mutants had similar growth rates. There appeared to be little change in proliferative activity among the mutants (Fig. 3C).
Interaction of HDAC6 with Pin1
Because the phospho-Ser/Thr-Pro motif is a target site of the Pin1 protein, Pin1 specifically catalyzes the interconversion of peptidyl-prolyl peptide bonds within phosphorylated Pro-directed Ser/Thr (pSer/Thr-Pro) motifs. After cis and trans interconversion, protein functional activity is changed [18]. Pin1 has been found to engage with numerous types of cancer tumorigenesis [22]. Our data showed that mutations in HDAC6 at Ser22 and Ser412, which are recognized by Pin1, had an ambiguous effect on cell proliferative activity. By contrast, HDAC6 mutants at Ser22 and Ser412 in H1299 cells had a clear effect on cell migration. We speculate that the interaction of Pin1 with HDAC6 and the catalytic action of Pin1 on HDAC6 might regulate cell motility. Verdel et al reported that the interaction between HDAC6 and Pin1 promotes Pin1 enzymatic activity [31].
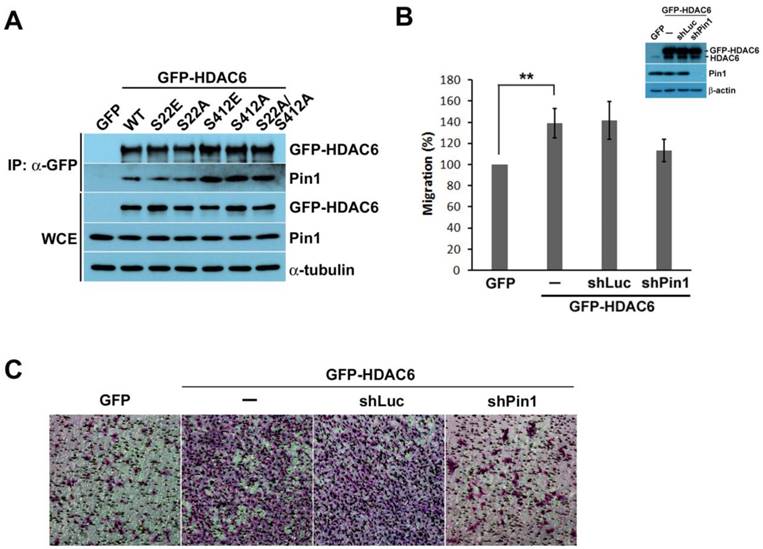
According to the identification of phosphorylation sites on HDAC6, phosphorylated HDAC6 at Ser22 and Ser412 might turn HDAC6 into Pin1 substrates. The interaction of Pin1 with phosphorylated HDAC6 at Ser22 and Ser412 was confirmed by the overexpression of GFP-HDAC6 and the various GFP-HDAC6 mutants in H1299 cells. An antibody specifically recognizing the GFP tag was applied to the immunoprecipitation of GFP-HDAC6 or GFP-HDAC6 mutants and their associated proteins. After immunoprecipitation, the sedimented proteins were analyzed through Western blot. In addition to GFP-HDAC6, other HDAC6 mutants could also interact with Pin1, as noted in the Western blot data (Fig. 4A).
Pin1 might be involved in HDAC6-mediated cell motility. (A) H1299 cells expressing GFP, GFP-HDAC6 wild type, and various indicated mutants were harvested for immunoprecipitation with anti-GFP agarose beads. The immunoprecipitation products and cell lysates were applied to SDS-PAGE for Western blotting analysis with the indicated antibody. (B) H1299 cells expressing GFP, GFP-HDAC6 or expressing GFP-HDAC6 in the meanwhile depleting Pin1, respectively, grew fully in wells. Wounds were generated through tip scratching. Cell migration was monitored by using microscopy, and the distance of migration was scored. Western blot analysis of the indicated proteins from the cell sublines was shown. (C) The cell invasion activity of H1299 cells expressing GFP, GFP-HDAC6 and GFP-HDAC6 but Pin1 depleted, respectively, was determined by using Matrigel-coated transwell inserts followed by Giemsa staining.
Interaction of HDAC6 with Pin1 mediates cell motility
The interaction between Pin1 and HDAC6 was identified through immunoprecipitation. Subsequently, we determined whether the interaction between Pin1 and HDAC6 involves regulation of cell motility. In comparison to GFP overexpression in H1299 cells, the H1299 cells harboring GFP-HDAC6 exhibited greater migratory activity (Fig. 4B). Moreover, disrupting the HDAC6 and Pin1 complex through Pin1 depletion repressed the migratory ability of H1299 cells. This result suggests that Pin1 not only acts as a proliferative regulator also affects cell migration.
The Pin1 and HDAC6 complex acting on cell migration was identified in H1299 cells harboring GFP-HDAC6, and it could also contribute to tumor invasion. To assess the role of Pin1 andHDAC6 complex in tumor invasion, H1299 cells overexpressing GFP-HDAC6 were appliedsubjected to the transwell invasion assay. The cells were found to have high invasion potential (Fig. 4C). Subsequently, lower invasion capacity was observed in the Pin1 depletion disrupted Pin1 and HDAC6 complex, as these H1299 cells harboring shRNA against Pin1 were determined in the transwell invasion assay. This indicates that Pin1 plays a crucial role in HDAC6-modulating cell motility and tumor invasion.
Discussion
Dysregulation of HDAC6 is involved in many different diseases such as kidney diseases [32], neurological diseases [33, 34], and cancers [35, 36]. In addition to regulation of cell motility and tumor metastasis, HDAC6 plays a critical role in tumorigenesis and cancer cell stemness [37]. HDAC6 is required for Ras- and ErbB2-induced transformation in fibroblast cells [38]. Furthermore, HDAC6 promotes chemoresistance and radioresistance through activating EGFR signaling and DNA repair pathways [39-41]. Therefore, HDAC6 is considered an important therapeutic target for these diseases.
PTM is a regulation mechanism that affects enzymatic activity and determines protein compartmentalization or protein-protein interaction. In the present study, we identified four phosphorylation sites on HDAC6. It is known that Aurora A, CK2, ERK, GRK2, GSK3β, and PKCɑ respectively, are known to phosphorylate HDAC6, and such associated PTMs can increase HDAC6 deacetylase activity toward the substrate [8, 9, 11-14]. By contrast, EGF receptor can phosphorylate HDAC6 and lead to reduce the enzyme activity [10]. HDAC6 promotes cell motility by catalyzing ɑ-tubulin and cortactin, thereby modulating the microtubule dynamics and dynamic F-actin assembly, respectively [42-44]. A low acetylated ɑ-tubulin level is correlated with increased cell motility. However, Zilberman et al.[45] found that HDAC6 is responsible for organizing microtubule dynamics not only through regulating tubulin acetylation. In this study, our data revealed that phospho-mimetic modification at Ser22 in HDAC6 reduced HDAC6-mediated cell migration in H1299 cells. As reported by Chen et al., the phosphorylation of HDAC6 at Ser22 by GSK3β resulted in increased deacetylation activity against ɑ-tubulin [13]. Therefore, it seems that S22E HDAC6 is not equivalent to HDAC6 Ser22 phosphorylation; consequently, the negative charge modification is insufficient to efficiently promote cell migration. An additional regulator might be involved in regulation of cell migration after HDAC6 Ser 22 phosphorylation. Phosphorylation at Ser43 increases HDAC6-mdiated cell migration but the phospho-defective mutant acts contrary regulation in H1299 cells. It is an interesting target; however, little information is available regarding its effect.. According to bioinformatics, HDAC6 is a putative substrate of NIMA and AMPK.
Notably, phosphorylation present in the NLS domain elicited active cell migration, suggesting that other HDAC6 binding partners might participate in migratory regulation. Additionally, two phosphorylation sites (Ser146 and Ser412) in the first catalytic domain of HDAC6 were evaluated in this study to determine their role in regulation of cell migration. Modification at Ser146 had no apparent impact on cell motility. The Ser412 phospho-mimetic mutant exhibited lower migration activity than did the wild type. The S412A mutant HDAC6 promoted migratory activity, which implies that Ser412 residue is an important regulatory site for cell migration.
We found that the sequence surrounding Ser22 and Ser412 in humans is similar to that of the corresponding sequence in rodents. Both Ser22 and pSer412 are categorized as the phospho-Ser/Thr-Pro motif, which is regarded as the recognition motif of Pin1. Pin1 specifically catalyzes the conformational switch of phosphorylated Pro-directed Ser/Thr (pSer/Thr-Pro) motifs. This interconversion affects protein functions [15-18]. Penela reported that Pin1 interacts with HDAC6 [31]. In this study, we also characterized interaction of HDAC6 with Pin1. A single or double mutation on HDAC6 appeared to be insufficient to fully abrogate the interaction of HDAC6 with Pin1 (Fig. 4A). Other pSer/Thr-Pro sites might be responsible for Pin1 binding or combinatorial residues. Furthermore, Penela's study suggested that Ser1060/Ser1062/Ser1068 phosphorylation on HDAC6 by GRK2 is required for interaction with Pin1 [31]. Other functional domains (except for pSer/Thr-Pro motifs) are likely also required for Pin1 binding.
The association of Pin1 with HDAC6 increases the possibility that secondary conformational changes through isomerization may influence protein function. H1299 cells have high mobility activity and expresse high levels of HDAC6 and Pin1, compared with others NSCLC cell lines. In addition to HDAC6, we also found that changes in Pin1 level through overexpression or depletion affected cell migration in H1299 cells (data not shown). Furthermore, Pin1 depletion reduced HDAC6-induced cell migration and invasion (Fig. 4B and 4C). These data indicate that Pin1 has a significant impact on HDAC6-modulating cell motility. However, the effect of Pin1 on enzymatic activity of HDAC6 remains unclear. Moreover, whether Pin1 contributes in a downstream or parallel fashion is not yet understood.
Abbreviations
CK2, casein kinase 2; EGFR, epidermal growth factor receptor; GRK2, G protein-coupled receptor kinas 2; GSK, Glycogen synthase kinase; HDAC, histone deacetylase; NSCLC, non-small cell lung cancer; Pin1, Peptidylprolyl Cis/Trans Isomerase NIMA-Interacting 1; PKC, protein kinase C; PTM, posttranslational modification; WT, wild-type.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Dr. Kun Ping Lu for Pin1 expressing constructs. We thank Dr. Yen-Yi Zhen for technical assistance and critical reading for manuscript. We thank Sequencing Center, Genome Research Center, National Yang-Ming University for sequencing. This research was supported by Academia Sinica and Ministry of Science and Technology [MOST 106-2314-B-037-038-MY3] to Chih-Jen Yang, and in part by Kaohsiung Municipal Ta-Tung Hospital [grant number KMTTH-105-044].
Author contributions
HHC designed and performed most of the experimental works, analyzed the data and wrote the manuscript; PHW performed experimental works. YPL designed part of the experiment; MSH, MH and CJY coordinated the project, planned experiments and wrote the manuscript. All authors read and approved the final paper.
Conflict of interest
The authors declare that they have no conflict of interest.
References
1. Verdel A, Khochbin S. Identification of a new family of higher eukaryotic histone deacetylases. Coordinate expression of differentiation-dependent chromatin modifiers. J Biol Chem. 1999;274:2440-5
2. Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A. 1999;96:4868-73
3. Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S. et al. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035-44
4. Bertos NR, Gilquin B, Chan GK, Yen TJ, Khochbin S, Yang XJ. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J Biol Chem. 2004;279:48246-54
5. Li Y, Shin D, Kwon SH. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013;280:775-93
6. Sen A, Nelson TJ, Alkon DL. ApoE4 and Abeta Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J Neurosci. 2015;35:7538-51
7. Yang CJ, Liu YP, Dai HY, Shiue YL, Tsai CJ, Huang MS. et al. Nuclear HDAC6 inhibits invasion by suppressing NF-kappaB/MMP2 and is inversely correlated with metastasis of non-small cell lung cancer. Oncotarget. 2015;6:30263-76
8. Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351-63
9. Watabe M, Nakaki T. Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J Cell Sci. 2011;124:1519-32
10. Deribe YL, Wild P, Chandrashaker A, Curak J, Schmidt MH, Kalaidzidis Y. et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal. 2009;2:ra84
11. Williams KA, Zhang M, Xiang S, Hu C, Wu JY, Zhang S. et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J Biol Chem. 2013;288:33156-70
12. Lafarga V, Aymerich I, Tapia O, Mayor F Jr, Penela P. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 2012;31:856-69
13. Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010;5:e10848
14. Zhu J, Coyne CB, Sarkar SN. PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and beta-catenin. EMBO J. 2011;30:4838-49
15. Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544-7
16. Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU. et al. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278:1957-60
17. Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875-86
18. Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904-16
19. Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V. et al. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001;20:3459-72
20. Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol. 2001;3:793-801
21. Liou YC, Ryo A, Huang HK, Lu PJ, Bronson R, Fujimori F. et al. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc Natl Acad Sci U S A. 2002;99:1335-40
22. Zhou XZ, Lu KP. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat Rev Cancer. 2016;16:463-78
23. Rustighi A, Zannini A, Campaner E, Ciani Y, Piazza S, Del Sal G. PIN1 in breast development and cancer: a clinical perspective. Cell Death Differ. 2017;24:200-11
24. Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727-37
25. Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J. et al. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244-51
26. Tan X, Zhou F, Wan J, Hang J, Chen Z, Li B. et al. Pin1 expression contributes to lung cancer: Prognosis and carcinogenesis. Cancer Biol Ther. 2010;9:111-9
27. Angelucci F, Hort J. Prolyl isomerase Pin1 and neurotrophins: a loop that may determine the fate of cells in cancer and neurodegeneration. Ther Adv Med Oncol. 2017;9:59-62
28. Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784-8
29. Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283:12686-90
30. Gradilone SA, Habringer S, Masyuk TV, Howard BN, Masyuk AI, Larusso NF. HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis. Am J Pathol. 2014;184:600-8
31. Nogues L, Reglero C, Rivas V, Salcedo A, Lafarga V, Neves M. et al. G Protein-coupled Receptor Kinase 2 (GRK2) Promotes Breast Tumorigenesis Through a HDAC6-Pin1 Axis. EBioMedicine. 2016;13:132-45
32. Ben Ke YC, Wei Tu, Ting Ye, Xiangdong Fang, Liping Yang. Inhibition of HDAC6 activity in kidney diseases: a new perspective. Molecular Medicine. 2018;24:33-8
33. Simoes-Pires C, Zwick V, Nurisso A, Schenker E, Carrupt PA, Cuendet M. HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs? Mol Neurodegener. 2013;8:7
34. Rui Y, Zheng JQ. Amyloid beta oligomers elicit mitochondrial transport defects and fragmentation in a time-dependent and pathway-specific manner. Mol Brain. 2016;9:79
35. Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol. 2011;2011:875824
36. Safa AR. Role of histone deacetylases 6 (HDAC6) in cancers. Journal of Pharmacology and Therapeutic Research. 2017;1:3-7
37. Yang W, Liu Y, Gao R, Yu H, Sun T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018;415:164-76
38. Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T. et al. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008;68:7561-9
39. Wang Z, Hu P, Tang F, Lian H, Chen X, Zhang Y. et al. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016;379:134-42
40. Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci U S A. 2010;107:20003-8
41. Marampon F, Megiorni F, Camero S, Crescioli C, McDowell HP, Sferra R. et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. 2017;397:1-11
42. Palazzo A, Ackerman B, Gundersen GG. Cell biology: Tubulin acetylation and cell motility. Nature. 2003;421:230
43. Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A. et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455-8
44. Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J. et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197-213
45. Zilberman Y, Ballestrem C, Carramusa L, Mazitschek R, Khochbin S, Bershadsky A. Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J Cell Sci. 2009;122:3531-41
Author contact
![]() Corresponding author: Dr. Chih-Jen Yang: Tel: + 886-7-3121101 ext.5651; Fax: +886-7-3161210; E-mail: chjeyakmu.edu.tw
Corresponding author: Dr. Chih-Jen Yang: Tel: + 886-7-3121101 ext.5651; Fax: +886-7-3161210; E-mail: chjeyakmu.edu.tw