Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(13):1564-1572. doi:10.7150/ijms.27424 This issue Cite
Research Paper
A SHOX2 loss-of-function mutation underlying familial atrial fibrillation
Ning Li1*, Zhang-Sheng Wang2*, Xin-Hua Wang3*, Ying-Jia Xu2, Qi Qiao2, Xiu-Mei Li2, Ruo-Min Di2, Xiao-Juan Guo2,4, Ruo-Gu Li1, Min Zhang1, Xing-Biao Qiu1 ![]() , Yi-Qing Yang2,4,5
, Yi-Qing Yang2,4,5 ![]()
1. Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China
2. Department of Cardiology, The Fifth People′s Hospital of Shanghai, Fudan University, 801 Heqing Road, Shanghai 200240, China
3. Department of Cardiology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 1630 Dongfang Road, Shanghai 200127, China
4. Department of Cardiovascular Research Laboratory, The Fifth People′s Hospital of Shanghai, Fudan University, 801 Heqing Road, Shanghai 200240, China
5. Department of Central Laboratory, The Fifth People′s Hospital of Shanghai, Fudan University, 801 Heqing Road, Shanghai 200240, China
* These two authors contributed equally to this work.
Received 2018-5-23; Accepted 2018-8-29; Published 2018-10-20
Abstract
Atrial fibrillation (AF), as the most common sustained cardiac arrhythmia, is associated with substantially increased morbidity and mortality. Aggregating evidence demonstrates that genetic defects play a crucial role in the pathogenesis of AF, especially in familial AF. Nevertheless, AF is of pronounced genetic heterogeneity, and in an overwhelming majority of cases the genetic determinants underlying AF remain elusive. In the current study, 162 unrelated patients with familial AF and 238 unrelated healthy individuals served as controls were recruited. The coding exons and splicing junction sites of the SHOX2 gene, which encodes a homeobox-containing transcription factor essential for proper development and function of the cardiac conduction system, were sequenced in all study participants. The functional effect of the mutant SHOX2 protein was characterized with a dual-luciferase reporter assay system. As a result, a novel heterozygous SHOX2 mutation, c.580C>T or p.R194X, was identified in an index patient, which was absent from the 476 control chromosomes. Genetic analysis of the proband's pedigree revealed that the nonsense mutation co-segregated with AF in the family with complete penetrance. Functional assays demonstrated that the mutant SHOX2 protein had no transcriptional activity compared with its wild-type counterpart. In conclusion, this is the first report on the association of SHOX2 loss-of-function mutation with enhanced susceptibility to familial AF, which provides novel insight into the molecular mechanism underpinning AF, suggesting potential implications for genetic counseling and individualized management of AF patients.
Keywords: Atrial fibrillation, Molecular genetics, Transcription factor, SHOX2, Reporter gene assay
Introduction
Atrial fibrillation (AF), characterized by rapid chaotic oscillations of atria, is the most common sustained cardiac arrhythmia globally, and it is associated with a substantially increased risk of cerebral stroke, congestive heart failure, and demise [1-4]. The prevalence of AF is estimated to be approximately 1% in the general population, and it markedly increases as the population ages, occurring in nearly 10% of individuals over 80 years of age [1,5]. There are more than 33 million people affected with AF worldwide in 2010, and this number is anticipated to increase steadily over the next several decades due to advancing ages [6,7]. AF confers a five-fold increased risk for the incidence of stroke and a two-fold increased risk for heart failure, hence is rapidly becoming a heavy societal and monetary burden in the world [1]. Despite significant clinical importance, the defined etiology and pathogenesis of AF in an overwhelming majority of patients remain incompletely understood.
Previous studies have substantiated that AF is associated with both environmental and genetic risk factors [7-10]. The environmental risk factors are numerous, including advanced age, obesity, hypertension, concomitant cardiac diseases and even chronic inflammation and tumor [7,10-12]. However, aggregating compelling evidence has demonstrated that genetic defects play a crucial role in the pathogenesis of AF, and mutations in a wide range of genes have been causally linked to AF, encompassing those coding for ion channels and their accessory subunits, gap junction channels, signaling molecules, and transcription factors among others [7-10,12-28]. Nevertheless, given that the above-mentioned genetic mutations occur in low prevalence in patients with AF, it is justifiable to make a hypothesis that additional disease-causing genes are still to be identified.
As another member of the homeodomain-containing transcription factors, SHOX2 has been shown to be pivotal for normal cardiac development, especially for the development of the sinoatrial node, the primary cardiac pacemaker [29-32]. In mice, homozygous deletion of Shox2 led to embryonic lethality between embryonic day 11.5 and embryonic day 13.5, due to severe cardiovascular defects, including bradycardia and hypoplastic sinoatrial node and sinus valves, and the aberrant expression of Cx40 and Cx43 as well as Nkx2-5, which were previously associated with AF [33-35], was verified in vivo specifically within the sinoatrial nodal region [30]. Additionally, in zebrafish embryos, knockdown of Shox2 also caused severe bradycardia [30]. Moreover, a common Shox2-dependent genetic program has been demonstrated to prime the pacemaker cells in the pulmonary vein myocardium, creating a vulnerable substrate for AF [36]. Furthermore, clinical studies and animal experiments have established a pathogenic link between sinus node dysfunction and AF [37]. These findings make it warranted to screen SHOX2 as a prime candidate gene for human AF.
Materials and Methods
Study participants
Between January 2015 and May 2017, a cohort of 162 unrelated patients with familial AF was recruited from the Chinese Han population. The available family members of the index patient harboring an identified SHOX2 mutation were also included in this study. A total of 238 unrelated healthy individuals without AF, who were matched for ethnicity, age and gender, were enlisted as controls. The study participants experienced a comprehensive clinical investigation, including individual and familial history, detailed physical examination, standard 12-lead electrocardiogram and cardiac echocardiography as well as review of medical records. Diagnosis and classification of AF was made as previously described [14]. Subjects with hypertension, coronary heart disease, valvular heart disease, congenital heart disease, congestive heart failure, metabolic disorders, or any other recognized risk factor of AF were excluded from the study. The investigation was conducted in conformity with the ethical standards of the Declaration of Helsinki set forth in 1975. The study protocol was approved by the local Institutional Ethical Committee. Peripheral venous blood samples were drawn from the study participants after they gave informed consents. By using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), genomic deoxyribonucleic acid (DNA) was isolated from blood leukocytes.
Sequence analysis of SHOX2
In this investigation, the longest transcript (transcript 1) of SHOX2 including a primate specific exon 2 (NM_003030.4), which encodes the longest isoform (b), was analyzed. Direct polymerase chain reaction (PCR)-sequencing of the coding exons and flanking introns as well as partial 5'- and 3'-untranslated regions of SHOX2 was performed in all study participants, as described previously [38-41]. The primer pairs used for amplification of SHOX2 are listed in Table 1. The primers were designed using genomic DNA sequences of SHOX2 from GenBank (https://www.ncbi.nlm.nih.gov/nuccore/NG_047079.1?from=5040&to=15192&report=genbank), with an accession number of NG_047079.1. For an identified sequence variation, a repeated PCR-sequencing was performed to verify it. In addition, the identified sequence variation was queried in the single nucleotide polymorphism (SNP) database (https://ncbi.nlm.nih.gov/projects/SNP), the Human Gene Mutation (HGM) database (http://www.hgmd.cf.ac.uk/ac/index.php), the 1000 Genome Project (1000GP) database (http://www.internationalgenome.org), the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/), and the Exome Variant Server (EVS) database (http://evs.gs.washington.edu/EVS) to confirm its novelty.
Primers to amplify the coding exons and flanking introns of the SHOX2 gene.
| Coding exon | Forward primer (5´ to 3´) | Reverse primer (5´ to 3´) | Amplicon (bp) |
|---|---|---|---|
| 1 2 | TCTGCTGGCAGAGGTTGAGCG CCTCTAGCGCAGAGTTTGCC | GACCGAGCATACCACCGGAC CAGGCACCAAGTGCCAAATCAA | 567 446 |
| 3 4, 5 6 | GCGGTGAGTCGAGGTACGTT TGCTGTATCTCCCAATTCTTGTCT GTCGGAACAAGATGCACAGCC | CACCACCTCCCGAGTGTGTC TGGGCTCAGAGACAGGTGATGTT TGCCTCGTGAGATCCCTGGT | 577 661 608 |
Plasmid constructs and site-targeted mutagenesis
The expression plasmid SHOX2-pcDNA3, which contains full-length cDNA of human SHOX2 (NM_003030.4) as well as the reporter plasmids of bone morphogenetic protein 4-luciferase (BMP4-luc) and islet 1-luciferase (ISL1-luc) was constructed as described previously [42,43]. The reporter plasmids of BMP4-luc and ISL1-luc both express Firefly luciferase. The mutation discovered in AF patients was introduced into the wild-type SHOX2 by PCR with a complementary pair of primers using the QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, California, USA). The mutant-type SHOX2-pcDNA3 plasmid was selected by DpnI (TaKaRa, Dalian, Liaoning, China) digestion and validated by direct sequencing.
Cellular transfection and luciferase assays
Human embryonic kidney (HEK)-293 cells were grown at 37℃ in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum as well as 100 μg/ml streptomycin and 100 U/ml penicillin. Cells were cultured in 12-well plates 24 h before transient transfection. HEK-293 cells were transfected with 1.0 μg of wild-type or mutant SHOX2-pcDNA3, 1.0 μg of BMP4-luc and 0.04 μg of pGL4.75 (Promega, Madison, WI, USA) by using LipofectamineTM 3000 transfection reagent (Invitrogen, Carlsbad, California, USA). The plasmid pGL4.75, which expresses Renilla luciferase, was used as an internal control to normalize transfection efficiency. For co-transfection experiments, 0.5 μg of wild-type SHOX2-pcDNA3 together with 0.5 μg of mutant SHOX2-pcDNA3 or 0.5 μg of empty pcDNA3 plasmid was used in the presence of 1.0 μg of BMP4-luc and 0.04 μg of pGL4.75. For the analysis of the transcriptional activation of the ISL1 promoter by SHOX2, the reporter plasmid ISL1-luc was used instead of BMP4-luc. The luciferase activity was measured 36 h after transfection with the Dual-Luciferase® Reporter Assay System (Promega), according to the manufacturer's protocol. The activity of the BMP4 or ISL1 promoter was expressed as fold activation of Firefly luciferase relative to Renilla luciferase. Experiments were done independently at least three times in triplicate for either wild-type or mutant SHOX2 with consistent results.
Statistics
The baseline clinical characteristics of study participants were expressed as means ± standard deviations (SD) for continuous variables or counts and percentages for categorical variables. Participant's characteristics were compared between two groups with unpaired Student's t test or Mann-Whitney U statistic for continuous variables or chi-square or Fisher's exact test for categorical variables as appropriate. For luciferase assays, data are presented as means ± SD from three independent experiments in triplicate. Comparisons between experimental groups were made with unpaired Student's t test or one-way ANOVA, with a P value < 0.05 indicated statistical difference. All statistics were performed using SAS (version 9.2; SAS Institute Inc., Cary, NC, USA).
Results
Baseline clinical characteristics of the study population
In the current study, 162 unrelated patients affected with AF were clinically evaluated in contrast to 238 unrelated individuals without AF. The study participants had no recognized environmental risk factors predisposing to AF, such as essential hypertension, coronary artery disease, valvular heart disease, previous cardiac surgery, chronic obstructive pulmonary disease and hyperthyroidism [1]. There is higher incidence of familial AF, ischemic stroke and implanted pacemaker, and a larger left atrial diameter in the patient group than in the control group. No significant difference in gender, age, or race existed between the two groups. The baseline clinical features of the 162 unrelated cases with AF are summarized in Table 2.
Baseline demographics and clinical features of the study population.
| Parameter | Patient group (n =162) | Control group (n =238) | p-value |
|---|---|---|---|
| Age (years) | 55.7 ± 9.3 | 56.4 ± 10.1 | 0.4828 |
| Gender (male/female) | 85/77 | 125/113 | 0.9919 |
| Family history of atrial fibrillation (%) | 38 (23) | 0 (0) | <0.0001* |
| History of ischemic stroke (%) | 7 (4) | 0 (0) | 0.0011* |
| History of implanted pacemaker (%) | 5 (3) | 0 (0) | 0.0094* |
| Body mass index (kg/m2) | 23.5 ± 2.1 | 23.8 ± 2.4 | 0.1978 |
| Systolic blood pressure (mmHg) | 128.4 ± 10.6 | 129.7 ± 12.3 | 0.2736 |
| Diastolic blood pressure (mmHg) | 83.1 ± 7.2 | 82.7 ± 6.8 | 0.5732 |
| Fasting blood glucose (mmol/L) | 4.6 ± 0.7 | 4.5 ± 0.6 | 0.1272 |
| Total cholesterol (mmol/L) | 3.7 ± 0.8 | 3.6 ± 0.6 | 0.1543 |
| Triglyceride (mmol/L) | 1.5 ± 0.5 | 1.5 ± 0.4 | 1.0000 |
| Left atrial diameter (mm) | 38.5 ± 8.1 | 36.2 ± 7.0 | 0.0026* |
| Left ventricular ejection fraction (%) | 63.0 ± 8.5 | 63.7 ± 7.6 | 0.3894 |
| Resting heart rate (beats/min) | 75.2 ± 18.5 | 74.8 ± 12.3 | 0.7952 |
| History of alcohol consumption (%) | 15 (9) | 23 (10) | 0.8922 |
| History of smoking (%) | 12 (7) | 20 (8) | 0.7185 |
Data are expressed as mean with standard deviation for a continuous variable or number with percentage for a categorical variable.
* indicates significant difference between the two groups.
Identification of a novel SHOX2 mutation
By sequencing all coding exons and flanking introns as well as parts of the 5'- and 3'-untranslated regions of the SHOX2 gene, a heterozygous mutation was detected in an index patient suffered from AF. Specifically, a substitution of thymine for cytosine at nucleotide 580 (c.580C>T), predicting to generate a truncated protein with only amino-terminal 193 amino acids (p.R194X), was identified in the proband from family 1. The DNA sequence electropherograms showing the detected heterozygous SHOX2 mutation and its wild-type control sequence are displayed in Figure 1A. The schematic diagrams of SHOX2 proteins showing the homeobox domain and location of the mutation identified in this study are presented in Figure 1B. The nonsense mutation was neither discovered in 476 control chromosomes nor found in the SNP, HGM, 1000GP, ExAC and EVS databases, which were consulted again on April 29, 2018, indicating that it is a novel mutation.
A novel SHOX2 mutation associated with familial atrial fibrillation. (A) Sequence chromatograms displaying the heterozygous SHOX2 mutation and its homozygous wild-type control. The arrow points to the homozygous nucleotides of C/C in a healthy subject (wild type) or the heterozygous nucleotides of C/T in the proband (mutant). The rectangle denotes the nucleotides constituting a codon of SHOX2. (B) Schematic diagrams of human SHOX2 proteins. The truncated protein associated with atrial fibrillation was shown with half structural domain of homeobox. NH2, amino-terminus; COOH, carboxyl-terminus. (C) Pedigree structure of the family with atrial fibrillation. The family was arbitrarily designated as family 1. Family members are identified by generations and numbers. Squares indicate male family member; circles, female members; a symbol with a slash, the deceased member; closed symbols, affected members; open symbols, unaffected members; the arrow, the proband; “+”, carriers of the heterozygous mutation; “-”, non-carriers.
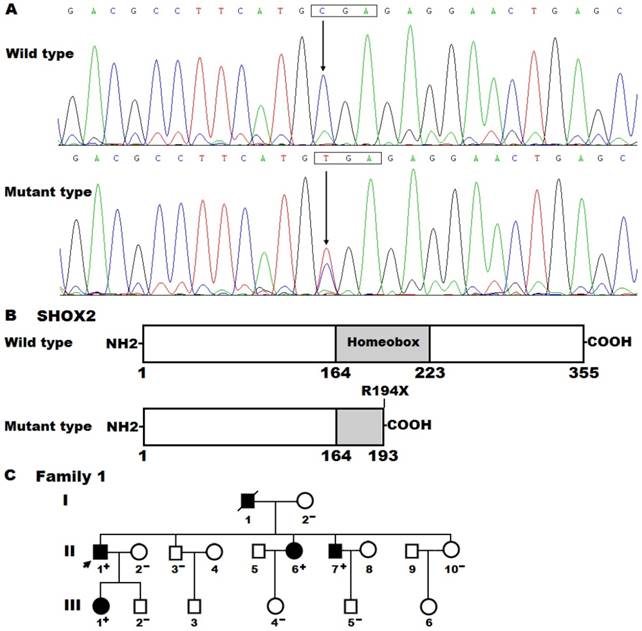
A genetic screen of the proband′s family members available revealed that the same mutation was present in all affected family members, but absent in unaffected family members. Analysis of the pedigree showed that in the family AF was transmitted in an autosomal dominant pattern with complete penetrance. The pedigree structure of the family with AF is shown in Figure 1C. The phenotypic features and mutational status for SHOX2 of the affected pedigree members are given in Table 3.
Phenotypic features and status of SHOX2 mutation of the affected living pedigree members.
| Subject information | Phenotype | Electrocardiogram | Echocardiogram | Genotype | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Identity | Gender | Age at time of study (years) | Age at diagnosis of AF (years) | AF (Classification) | Heart rate (beats/min) | QRS interval (ms) | QTc | LAD (mm) | LVEF (%) | SHOX2 mutation |
| Family 1 | R194X | |||||||||
| II-1 | M | 53 | 41 | Permanent | 87 | 100 | 447 | 35 | 62 | +/- |
| II-6 | F | 47 | 43 | Permanent | 62 | 116 | 528 | 38 | 60 | +/- |
| II-7 | M | 45 | 39 | Persistent | 71 | 102 | 450 | 33 | 63 | +/- |
| III-1 | F | 20 | 20 | Paroxysmal | 75 | 96 | 435 | 30 | 65 | +/- |
AF: atrial fibrillation, F: female, M: male, QTc: corrected QT interval, LAD: left atrial diameter, LVEF: left ventricular ejection fraction, +/-: heterozygote.
Additionally, all the mutation carriers in this family had severely reduced heart rates (sinus bradycardia) and significantly prolonged PR interval as well as RR and QRS lengths prior to AF occurrence. There is no significant difference in corrected QT intervals between the affected individuals and the unaffected individuals. Besides, the affected individuals had shorter stature when compared to their healthy parents, but no obvious craniofacial or brain anomalies were observed.
No transcriptional activation of the BMP4 promoter by R194X-mutant SHOX2
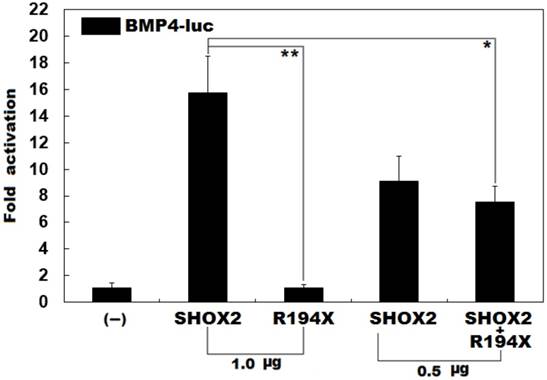
Previous studies have validated that SHOX2 transactivates the expression of BMP4 [42]. To ascertain whether the identified mutation exerts an impact on the transactivational activity of SHOX2, luciferase reporter assays were performed. Consistent with its role as a transcription factor, wild-type SHOX2 activated the promoter of its target gene BMP4, while the R194X-mutant SHOX2 failed to activate its target molecule. As shown in Figure 2, the wild-type and R194X-mutant SHOX2 proteins activated the BMP4 promoter by ~16 folds and ~1 fold, respectively. When the wild-type and R194X-mutant SHOX2 proteins were co-expressed, the induced activation of the BMP4 promoter was ~7-fold, indicating that the R194X-mutant SHOX2 protein fails to transactivate the BMP4 promoter compared with its wild-type counterpart.
Failure to transcriptionally activate the BMP promoter by R194X-mutant SHOX2. Activation of the BMP4-driven luciferase in HEK-293 cells by wild-type or R194X-mutant SHOX2, alone or in combination, showed that the R194X-mutant SHOX2 protein failed to transcriptionally activate the BMP4 promoter. Three independent experiments were carried out in triplicates. The data are expressed as means and standard deviations. ** indicates t = 11.3237, p = 0.00035; * indicates t = 5.90352, p = 0.00412, when compared with wild-type SHOX2.
Failure to transactivate the ISL1 promoter by R194X-mutant SHOX2
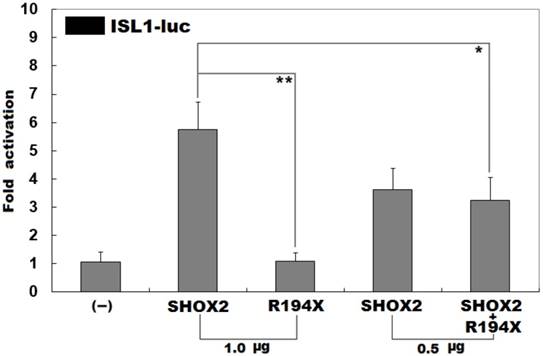
Previous investigations have substantiated that ISL1 is a direct transcriptional target of SHOX2 [43]. To evaluate the functional consequence of the found nonsense mutation, luciferase reporter analyses of wild-type SHOX2 and R194X-mutant SHOX2 were performed. As expected, wild-type SHOX2 transcriptionally activated the promoter of its target gene ISL1, while the R194X-mutant SHOX2 was not able to activate its target. As shown in Figure 3, the wild-type and R194X-mutant SHOX2 proteins activated the ISL1 promoter by ~6 folds and ~1 fold, respectively. When the wild-type and R194X-mutant SHOX2 proteins were co-expressed, the induced activation of the ISL1 promoter was ~3-fold, indicating that the R194X-mutant SHOX2 protein has no transcriptional activation of the ISL1 promoter.
Inability to transcriptionally activate the ISL1 promoter by R194X-mutant SHOX2. Activation of the ISL1-driven luciferase in HEK-293 cells by wild-type or R194X-mutant SHOX2, alone or in combination, showed that the R194X-mutant SHOX2 protein was unable to transcriptionally activate the ISL1 promoter. Three independent experiments were carried out in triplicates. The data are given as means and standard deviations. ** indicates t = 7.94861, p = 0.00136; * indicates t = 3.42684, p = 0.02661, when compared with wild-type SHOX2.
Discussion
In the present study, a novel heterozygous nonsense mutation in SHOX2, p.R194X, was identified in a family with AF. The mutation co-segregated with AF in the family and was absent from the 476 reference chromosomes. The nonsense mutation was predicted to create a truncated protein with only half homeobox left. The homeobox domain is responsible for target DNA sequence recognition and binding to the consensus motif, hence is a functionally pivotal structural domain [44]. Functional analyses demonstrated that the mutant SHOX2 protein had no transcriptional activity. Therefore it is very likely that SHOX2 loss-of-function mutation contributes to AF in mutation carriers.
In humans, SHOX2 maps on chromosome 3q25.32, coding for three isoforms of proteins, encompassing isoform a (with 331 amino acids), isoform b (with 355 amino acids) and isoform c (with 319 amino acids). Previous studies have substantiated that SHOX2 transcriptionally regulates expression of many genes in the heart, including HCN4, NKX2-5, CX40, CX43 and NPPA [44]. Furthermore, mutations in several target genes mediated by SHOX2 have been causally linked to AF [24,33-35,45-49]. In the present study, the functional characteristics of the novel SHOX2 mutation identified in patients with familial AF were analyzed by dual-luciferase assays and the results indicated that the mutant SHOX2 protein failed to transcriptionally activate the BMP4 or ISL1 promoter. These observational results imply that SHOX2 haploinsufficiency resulted from a mutation is potentially an alternative molecular mechanism of AF.
Association of genetically compromised SHOX2 with increased susceptibility to AF may be partially attributed to abnormal development of the heart, especially the sinoatrial node. During embryonic cardiac development, SHOX2 is highly expressed in the sinus venosus region, including the sinoatrial nodes and the sinus valves [30,31], and similar expression profile of SHOX2 was also observed in the human embryonic heart [29,50]. In mice, homozygous deletion of Shox2 resulted in embryonic lethality between embryonic days 11.5 and 13.5 due to cardiac defects, including severe hypoplasia of sinoatrial nodes and bradycardia associated with down-regulation of the sinoatrial node-specific marker genes, Tbx3 and Hcn4, and ectopic expression of Nkx2-5, Cx40, Cx43, and Nppa within the sinoatrial node region [30,31]. In mice with Shox2 replaced by the closely related but hypomorphic Shox allele, embryonic lethality was rescued, but arrhythmias remained [50]. In zebrafish, targeted knockdown of Shox2 using morpholino-modified antisense oligonucleotides led to pericardial edema and pericardial blood congestion because of severely reduced heart rates in zebrafish embryos, and the bradycardia in Shox2 morphants could be rescued by cardiomyocyte-specific expression of wild-type SHOX2 [43]. Furthermore, gene expression analysis demonstrated that SHOX2 and NKX2-5 were expressed in a mutually exclusive manner from the earliest formation stages of the venous poles and sinoatrial nodes until at least embryonic day15.5 [30,31], and overexpression of NKX2-5 caused the same sinoatrial node and sinus valve phenotypes as that observed in Shox2-mull hearts, indicating that NKX2-5 activity is detrimental to sinoatrial nodal development and that SHOX2 plays a key role in sinoatrial nodal development through repression of Nkx2-5 [51]. Taken collectively, these findings suggest that a haploinsufficient mutation in SHOX2 predisposes to arrhythmias, including AF.
Notably, SHOX2 mutations were previously identified in patients with early-onset AF. Hoffmann and colleagues [52] made a sequence analysis of SHOX2 in 378 unrelated patients with early-onset AF, and identified two heterozygous mutations (p.G81E and p.H283Q) in two patients, respectively. Transactivation studies of the identified two missense mutations with two known SHOX2 target genes BMP4 and ISL1 unveiled that the p.G81E mutation had no effect on the transcriptional activity of SHOX2, while the p.H283Q mutation failed to activate both BMP4 and ISL1 target genes [52]. Further functional analysis in the zebrafish displayed that the p.H283Q mutation severely impaired cardiac pacemaker function. The same authors also demonstrated significantly reduced expression levels of SHOX2 in right atrial appendages of AF patients compared to patients with sinus rhythm [52]. These results together with the current study suggest that mutated SHOX2 may be a rare cause for AF.
In conclusion, the present research firstly associates a SHOX2 loss-of-function mutation with enhanced susceptibility to familial AF in humans, which provides novel insight into the molecular pathogenesis underpinning AF, suggesting potential implications for genetic counseling and personalized treatment of the patients with AF.
Acknowledgements
The authors are really thankful to the study participants for their devotion to the study. This work was financially supported by the National Natural Science Foundation of China (81470372, 81400244 and 81370400), the Natural Science Foundation of Shanghai, China (18ZR1423400), the Three-Year Action Plan for Clinical Innovation of Shanghai, China (16CR3005A), the Science and Technology Support Project of Medical Guidance (Chinese and Western Medicine), Shanghai, China (17411971000), the Grant from the Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Program, Shanghai, China (20172028), and the Experimental Animal Research Project, Shanghai, China (17140902402).
Conflict of Interest
The authors have declared no conflict of interest.
References
1. January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr, Conti JB, Ellinor PT, Ezekowitz MD, Field ME, Murray KT, Sacco RL, Stevenson WG, Tchou PJ, Tracy CM, Yancy CW; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:e1-76
2. Krijthe BP, Kunst A, Benjamin EJ, Lip GY, Franco OH, Hofman A, Witteman JC, Stricker BH, Heeringa J. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur Heart J. 2013;34:2746-51
3. Maagh P, Plehn G, Christoph A, Oernek A, Meissner A. Impact of cryoballoon ablation in hypertrophic cardiomyopathy-related heart failure due to paroxysmal atrial fibrillation: A Comparative Case Series. Int J Med Sci. 2016;13:664-72
4. Meissner A, Maagh P, Christoph A, Oernek A, Plehn G. ECG-Guided surveillance technique in cryoballoon ablation for paroxysmal and persistent atrial fibrillation: a strategy to prevent from phrenic nerve palsy. Int J Med Sci. 2016;13:403-11
5. Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, Singer DE. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370-5
6. Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH Jr, Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, Murray CJ. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014;129:837-47
7. Tucker NR, Clauss S, Ellinor PT. Common variation in atrial fibrillation: navigating the path from genetic association to mechanism. Cardiovasc Res. 2016;109:493-501
8. Christophersen IE, Rienstra M, Roselli C, Yin X, Geelhoed B, Barnard J, Lin H, Arking DE, Smith AV, Albert CM, Chaffin M, Tucker NR, Li M, Klarin D, Bihlmeyer NA, Low SK, Weeke PE, Müller-Nurasyid M, Smith JG, Brody JA, Niemeijer MN, Dörr M, Trompet S, Huffman J, Gustafsson S, Schurmann C, Kleber ME, Lyytikäinen LP, Seppälä I, Malik R, Horimoto ARVR, Perez M, Sinisalo J, Aeschbacher S, Thériault S, Yao J, Radmanesh F, Weiss S, Teumer A, Choi SH, Weng LC, Clauss S, Deo R, Rader DJ, Shah SH, Sun A, Hopewell JC, Debette S, Chauhan G, Yang Q, Worrall BB, Paré G, Kamatani Y, Hagemeijer YP, Verweij N, Siland JE, Kubo M, Smith JD, Van Wagoner DR, Bis JC, Perz S, Psaty BM, Ridker PM, Magnani JW, Harris TB, Launer LJ, Shoemaker MB, Padmanabhan S, Haessler J, Bartz TM, Waldenberger M, Lichtner P, Arendt M, Krieger JE, Kähönen M, Risch L, Mansur AJ, Peters A, Smith BH, Lind L, Scott SA, Lu Y, Bottinger EB, Hernesniemi J, Lindgren CM, Wong JA, Huang J, Eskola M, Morris AP, Ford I, Reiner AP, Delgado G, Chen LY, Chen YI, Sandhu RK, Li M, Boerwinkle E, Eisele L, Lannfelt L, Rost N, Anderson CD, Taylor KD, Campbell A, Magnusson PK, Porteous D, Hocking LJ, Vlachopoulou E, Pedersen NL, Nikus K, Orho-Melander M, Hamsten A, Heeringa J, Denny JC, Kriebel J, Darbar D, Newton-Cheh C, Shaffer C, Macfarlane PW, Heilmann-Heimbach S, Almgren P, Huang PL, Sotoodehnia N, Soliman EZ, Uitterlinden AG, Hofman A, Franco OH, Völker U, Jöckel KH, Sinner MF, Lin HJ, Guo X; METASTROKE Consortium of the ISGC; Neurology Working Group of the CHARGE Consortium, Dichgans M, Ingelsson E, Kooperberg C, Melander O, Loos RJF, Laurikka J, Conen D, Rosand J, van der Harst P, Lokki ML, Kathiresan S, Pereira A, Jukema JW, Hayward C, Rotter JI, März W, Lehtimäki T, Stricker BH, Chung MK, Felix SB, Gudnason V, Alonso A, Roden DM, Kääb S, Chasman DI, Heckbert SR, Benjamin EJ, Tanaka T, Lunetta KL, Lubitz SA, Ellinor PT; AFGen Consortium. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946-52
9. Paludan-Müller C, Svendsen JH, Olesen MS. The role of common genetic variants in atrial fibrillation. J Electrocardiol. 2016;49:864-70
10. Fatkin D, Santiago CF, Huttner IG, Lubitz SA, Ellinor PT. Genetics of atrial fibrillation: state of the art in 2017. Heart Lung Circ. 2017;26:894-901
11. Doesch C, Lossnitzer D, Rudic B, Tueluemen E, Budjan J, Haubenreisser H, Henzler T, Schoenberg SO, Borggrefe M, Papavassiliu T. Right ventricular and right atrial involvement can predict atrial fibrillation in patients with hypertrophic cardiomyopathy? Int J Med Sci. 2016;13:1-7
12. Mao L, Huang W, Zou P, Dang X, Zeng X. The unrecognized role of tumor suppressor genes in atrial fibrillation. Gene. 2018;642:26-31
13. Christophersen IE, Ellinor PT. Genetics of atrial fibrillation: from families to genomes. J Hum Genet. 2016;61:61-70
14. Wang ZC, Ji WH, Ruan CW, Liu XY, Qiu XB, Yuan F, Li RG, Xu YJ, Liu X, Huang RT, Xue S, Yang YQ. Prevalence and spectrum of TBX5 mutation in patients with lone atrial fibrillation. Int J Med Sci. 2016;13:60-7
15. Orr N, Arnaout R, Gula LJ, Spears DA, Leong-Sit P, Li Q, Tarhuni W, Reischauer S, Chauhan VS, Borkovich M, Uppal S, Adler A, Coughlin SR, Stainier DY, Gollob MH. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun. 2016;7:11303
16. Seppälä I, Kleber ME, Bevan S, Lyytikäinen LP, Oksala N, Hernesniemi JA, Mäkelä KM, Rothwell PM, Sudlow C, Dichgans M, Mononen N, Vlachopoulou E, Sinisalo J, Delgado GE, Laaksonen R, Koskinen T, Scharnagl H, Kähönen M, Markus HS, März W, Lehtimäki T. Associations of functional alanine-glyoxylate aminotransferase 2 gene variants with atrial fibrillation and ischemic stroke. Sci Rep. 2016;6:23207
17. Gandara E, Barboza AG, Vazquez F. Prevalence of prothrombin G20210A mutation in patients with atrial fibrillation: A systematic review and meta-analysis. Thromb Res. 2016;147:7-9
18. Wang C, Wu M, Qian J, Li B, Tu X, Xu C, Li S, Chen S, Zhao Y, Huang Y, Shi L, Cheng X, Liao Y, Chen Q, Xia Y, Yao W, Wu G, Cheng M, Wang QK. Identification of rare variants in TNNI3 with atrial fibrillation in a Chinese GeneID population. Mol Genet Genomics. 2016;291:79-92
19. Ye J, Tucker NR, Weng LC, Clauss S, Lubitz SA, Ellinor PT. A functional variant associated with atrial fibrillation regulates PITX2c expression through TFAP2a. Am J Hum Genet. 2016;99:1281-91
20. Zhao J, Yao H, Li Z, Wang L, Liu G, Wang DW, Wang DW, Liang Z. A novel nonsense mutation in LMNA gene identified by exome sequencing in an atrial fibrillation family. Eur J Med Genet. 2016;59:396-400
21. Ma JF, Yang F, Mahida SN, Zhao L, Chen X, Zhang ML, Sun Z, Yao Y, Zhang YX, Zheng GY, Dong J, Feng MJ, Zhang R, Sun J, Li S, Wang QS, Cao H, Benjamin EJ, Ellinor PT, Li YG, Tian XL. TBX5 mutations contribute to early-onset atrial fibrillation in Chinese and Caucasians. Cardiovasc Res. 2016;109:442-50
22. Gudbjartsson DF, Holm H, Sulem P, Masson G, Oddsson A, Magnusson OT, Saemundsdottir J, Helgadottir HT, Helgason H, Johannsdottir H, Gretarsdottir S, Gudjonsson SA, Njølstad I, Løchen ML, Baum L, Ma RC, Sigfusson G, Kong A, Thorgeirsson G, Sverrisson JT, Thorsteinsdottir U, Stefansson K, Arnar DO. A frameshift deletion in the sarcomere gene MYL4 causes early-onset familial atrial fibrillation. Eur Heart J. 2017;38:27-34
23. Tucker NR, Mahida S, Ye J, Abraham EJ, Mina JA, Parsons VA, McLellan MA, Shea MA, Hanley A, Benjamin EJ, Milan DJ, Lin H, Ellinor PT. Gain-of-function mutations in GATA6 lead to atrial fibrillation. Heart Rhythm. 2017;14:284-91
24. Ishikawa T, Ohno S, Murakami T, Yoshida K, Mishima H, Fukuoka T, Kimoto H, Sakamoto R, Ohkusa T, Aiba T, Nogami A, Sumitomo N, Shimizu W, Yoshiura KI, Horigome H, Horie M, Makita N. Sick sinus syndrome with HCN4 mutations shows early onset and frequent association with atrial fibrillation and left ventricular noncompaction. Heart Rhythm. 2017;14:717-24
25. Lieve KV, Verkerk AO, Podliesna S, van der Werf C, Tanck MW, Hofman N, van Bergen PF, Beekman L, Bezzina CR, Wilde AAM, Lodder EM. Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. Int J Cardiol. 2017;236:187-93
26. Low SK, Takahashi A, Ebana Y, Ozaki K, Christophersen IE, Ellinor PT; AFGen Consortium, Ogishima S, Yamamoto M, Satoh M, Sasaki M, Yamaji T, Iwasaki M, Tsugane S, Tanaka K, Naito M, Wakai K, Tanaka H, Furukawa T, Kubo M, Ito K, Kamatani Y, Tanaka T. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat Genet. 2017;49:953-8
27. Thorolfsdottir RB, Sveinbjornsson G, Sulem P, Helgadottir A, Gretarsdottir S, Benonisdottir S, Magnusdottir A, Davidsson OB, Rajamani S, Roden DM, Darbar D, Pedersen TR, Sabatine MS, Jonsdottir I, Arnar DO, Thorsteinsdottir U, Gudbjartsson DF, Holm H, Stefansson K. A missense variant in PLEC increases risk of atrial fibrillation. J Am Coll Cardiol. 2017;70:2157-68
28. Nielsen JB, Fritsche LG, Zhou W, Teslovich TM, Holmen OL, Gustafsson S, Gabrielsen ME, Schmidt EM, Beaumont R, Wolford BN, Lin M, Brummett CM, Preuss MH, Refsgaard L, Bottinger EP, Graham SE, Surakka I, Chu Y, Skogholt AH, Dalen H, Boyle AP, Oral H, Herron TJ, Kitzman J, Jalife J, Svendsen JH, Olesen MS, Njølstad I, Løchen ML, Baras A, Gottesman O, Marcketta A, O'Dushlaine C, Ritchie MD, Wilsgaard T, Loos RJF, Frayling TM, Boehnke M, Ingelsson E, Carey DJ, Dewey FE, Kang HM, Abecasis GR, Hveem K, Willer CJ. Genome-wide study of atrial fibrillation identifies seven risk loci and highlights biological pathways and regulatory elements involved in cardiac development. Am J Hum Genet. 2018;102:103-15
29. Blaschke RJ, Monaghan AP, Schiller S, Schechinger B, Rao E, Padilla-Nash H, Ried T, Rappold G. SHOT, a SHOX-related homeobox gene, is implicated in craniofacial, brain, heart, and limb development. Proc Natl Acad Sci USA. 1998;95:2406-11
30. Blaschke RJ, Hahurij ND, Kuijper S, Just S, Wisse LJ, Deissler K, Maxelon T, Anastassiadis K, Spitzer J, Hardt SE, Schöler H, Feitsma H, Rottbauer W, Blum M, Meijlink F, Rappold G, Gittenberger-de Groot AC. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation. 2007;115:1830-8
31. Espinoza-Lewis RA, Yu L, He F, Liu H, Tang R, Shi J, Sun X, Martin JF, Wang D, Yang J, Chen Y. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nk2-5. Dev Biol. 2009;327:376-85
32. Ye W, Song Y, Huang Z, Zhang Y, Chen Y. Genetic Regulation of sinoatrial node development and pacemaker program in the venous pole. J Cardiovasc Dev Dis. 2015;2:282-98
33. Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677-88
34. Thibodeau IL, Xu J, Li Q, Liu G, Lam K, Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, Nicholson BJ, Gollob MH. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation. 2010;122:236-44
35. Yu H, Xu JH, Song HM, Zhao L, Xu WJ, Wang J, Li RG, Xu L, Jiang WF, Qiu XB, Jiang JQ, Qu XK, Liu X, Fang WY, Jiang JF, Yang YQ. Mutational spectrum of the NKX2-5 gene in patients with lone atrial fibrillation. Int J Med Sci. 2014;11:554-63
36. Ye W, Wang J, Song Y, Yu D, Sun C, Liu C, Chen F, Zhang Y, Wang F, Harvey RP, Schrader L, Martin JF, Chen Y. A common Shox2-Nk2-5 antagonistic mechanism primes the pacemaker cell fate in the pulmonary vein myocardium and sinoatrial node. Development. 2015;142:2521-32
37. Chen YC, Lu YY, Cheng CC, Lin YK, Chen SA, Chen YJ. Sinoatrial node electrical activity modulates pulmonary vein arrhythmogenesis. Int J Cardiol. 2014;173:447-52
38. Zhou YM, Dai XY, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Huang RT, Xue S, Yang YQ. HAND1 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med. 2016;54:1161-7
39. Qiu XB, Qu XK, Li RG, Liu H, Xu YJ, Zhang M, Shi HY, Hou XM, Liu X, Yuan F, Sun YM, Wang J, Huang RT, Xue S, Yang YQ. CASZ1 loss-of-function mutation contributes to familial dilated cardiomyopathy. Clin Chem Lab Med. 2017;55:1417-25
40. Huang RT, Wang J, Xue S, Qiu XB, Shi HY, Li RG, Qu XK, Yang XX, Liu H, Li N, Li YJ, Xu YJ, Yang YQ. TBX20 loss-of-function mutation responsible for familial tetralogy of Fallot or sporadic persistent truncus arteriosus. Int J Med Sci. 2017;14:323-32
41. Qiao XH, Wang F, Zhang XL, Huang RT, Xue S, Wang J, Qiu XB, Liu XY, Yang YQ. MEF2C loss-of-function mutation contributes to congenital heart defects. Int J Med Sci. 2017;14:1143-53
42. Puskaric S, Schmitteckert S, Mori AD, Glaser A, Schneider KU, Bruneau BG, Blaschke RJ, Steinbeisser H, Rappold G. Shox2 mediates Tbx5 activity by regulating Bmp4 in the pacemaker region of the developing heart. Hum Mol Genet. 2010;19:4625-33
43. Hoffmann S, Berger IM, Glaser A, Bacon C, Li L, Gretz N, Steinbeisser H, Rottbauer W, Just S, Rappold G. Islet1 is a direct transcriptional target of the homeodomain transcription factor Shox2 and rescues the Shox2-mediated bradycardia. Basic Res Cardiol. 2013;108:339
44. Liu H, Chen CH, Ye W, Espinoza-Lewis RA, Hu X, Zhang Y, Chen Y. Phosphorylation of Shox2 is required for its function to control sinoatrial node formation. J Am Heart Assoc. 2014;3:e000796
45. Christophersen IE, Holmegard HN, Jabbari J, Sajadieh A, Haunsø S, Tveit A, Svendsen JH, Olesen MS. Rare variants in GJA5 are associated with early-onset lone atrial fibrillation. Can J Cardiol. 2013;29:111-6
46. Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett JC Jr, Olson TM. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158-65
47. Duhme N, Schweizer PA, Thomas D, Becker R, Schröter J, Barends TR, Schlichting I, Draguhn A, Bruehl C, Katus HA, Koenen M. Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur Heart J. 2013;34:2768-75
48. Sun Y, Yang YQ, Gong XQ, Wang XH, Li RG, Tan HW, Liu X, Fang WY, Bai D. Novel germline GJA5/connexin40 mutations associated with lone atrial fibrillation impair gap junctional intercellular communication. Hum Mutat. 2013;34:603-9
49. Macri V, Mahida SN, Zhang ML, Sinner MF, Dolmatova EV, Tucker NR, McLellan M, Shea MA, Milan DJ, Lunetta KL, Benjamin EJ, Ellinor PT. A novel trafficking-defective HCN4 mutation is associated with early-onset atrial fibrillation. Heart Rhythm. 2014;11:1055-62
50. Liu H, Chen CH, Espinoza-Lewis RA, Jiao Z, Sheu I, Hu X, Lin M, Zhang Y, Chen Y. Functional redundancy between human SHOX and mouse Shox2 genes in the regulation of sinoatrial node formation and pacemaking function. J Biol Chem. 2011;286:17029-38
51. Espinoza-Lewis RA, Liu H, Sun C, Chen C, Jiao K, Chen Y. Ectopic expression of Nkx2.5 suppresses the formation of the sinoatrial node in mice. Dev Biol. 2011;356:359-69
52. Hoffmann S, Clauss S, Berger IM, Weiß B, Montalbano A, Röth R, Bucher M, Klier I, Wakili R, Seitz H, Schulze-Bahr E, Katus HA, Flachsbart F, Nebel A, Guenther SP, Bagaev E, Rottbauer W, Kääb S, Just S, Rappold GA. Coding and non-coding variants in the SHOX2 gene in patients with early-onset atrial fibrillation. Basic Res Cardiol. 2016;111:36
Author contact
![]() Corresponding authors: Xing-Biao Qiu, Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. Tel: +86-21-62821990, Fax: +86-21-62821105, E-mail: qxingbiaocn; Yi-Qing Yang, Department of Cardiovascular Research Laboratory, The Fifth People′s Hospital of Shanghai, Fudan University, 801 Heqing Road, Shanghai 200240, China. Tel: +86-21-24289657, Fax: +86-21-24289657, E-mail: yangyiqingcom.
Corresponding authors: Xing-Biao Qiu, Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. Tel: +86-21-62821990, Fax: +86-21-62821105, E-mail: qxingbiaocn; Yi-Qing Yang, Department of Cardiovascular Research Laboratory, The Fifth People′s Hospital of Shanghai, Fudan University, 801 Heqing Road, Shanghai 200240, China. Tel: +86-21-24289657, Fax: +86-21-24289657, E-mail: yangyiqingcom.