Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(13):1449-1457. doi:10.7150/ijms.27350 This issue Cite
Research Paper
Plasmodium falciparum Treated with Artemisinin-based Combined Therapy Exhibits Enhanced Mutation, Heightened Cortisol and TNF-α Induction
Abel Olusola Idowu1,2, Sanjib Bhattacharyya4, Steve Gradus4, Wellington Oyibo3, Zenas George5, Carolyn Black5, Joseph Igietseme5, Anthony Ajayi Azenabor1 ![]()
1. Department of Biomedical Sciences, University of Wisconsin, Milwaukee WI 53211 USA
2. Department of Pharmaceutics and Pharmaceutical Technology, University of Lagos, Nigeria
3. Department of Medical Microbiology and Parasitology, University of Lagos, Nigeria
4. City of Milwaukee Health Department Laboratories, Milwaukee, WI 53202 USA
5. Molecular Pathogenesis laboratory, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia, USA
Received 2018-5-18; Accepted 2018-8-1; Published 2018-9-7
Abstract
The artemisinin-based combined therapy (ACT) post-treatment illness in Plasmodium falciparum-endemic areas is characterized by vague malaria-like symptoms. The roles of treatment modality, persistence of parasites and host proinflammatory response in disease course are unknown. We investigated the hypothesis that ACT post-treatment syndrome is driven by parasite genetic polymorphisms and proinflammatory response to persisting mutant parasites. Patients were categorized as treated, untreated and malaria-negative. Malaria positive samples were analyzed for Pfcrt, Pfmdr1, K13 kelch gene polymorphisms, while all samples were evaluated for cytokines (TNF-α, IL-12p70, IL-10, TGF-β, IFN-γ) and corticosteroids (cortisol and dexamethasone) levels. The treated patients exhibited higher levels of parasitemia, TNF-α, and cortisol, increased incidence of parasite genetic mutations, and greater number of mutant alleles per patient. In addition, corticosteroid levels declined with increasing number of mutant alleles. TGF-β levels were negatively correlated with parasitemia, while IL-10 and TGF-β were negatively correlated with increasing number of mutant alleles. However, IL-12 displayed slight positive correlation and TNF-α exhibited moderate positive correlation with increasing number of mutant alleles. Since post-treatment management ultimately results in patient recovery, the high parasite gene polymorphism may act in concert with induced cortisol and TNF-α to account for ACT post-treatment syndrome.
Keywords: Proinflammation, corticosteroids, cortisol, dexamethasone, gene polymorphism, persistent malaria, ACT.
Introduction
Plasmodium falciparum, transmitted by the Anopheles mosquito, is the most prevalent causative agent of malaria in sub-Sahara Africa. The course of clinical malaria in endemic areas is characterized by vague symptoms that are indistinguishable from other diseases preponderant in areas of malaria endemicity, resulting in difficulty in malaria definition bringing about over diagnosis coupled with overtreatment [1-2]. The clinical features of malaria include febrile illness and chills that can be induced by endogenous agents such as inflammatory cytokines and stress hormones. Malaria fever onset is parasite threshold dependent. The acceptable pyrogenic threshold is a minimum of 600 to 1500 parasite per ml of blood for P. falciparum [3]. This concept of the relatedness of commencement of clinical malaria fever to parasitemia pyrogenic threshold is very important. However, the perpetual high prevalence of asymptomatic parasitemia in endemic areas makes parasite threshold alone insufficient for disease definition. Thus, it is compelling to determine the role of other central players in those manifestations associated with persistent complaints of unwellness usually designated as malaria. In this respect, although there exists an emerging field of investigations regarding the altered physiological steady state evoked by possible elaborations of stress hormones and cytokines [4-7], this is not factored into therapeutic judgement due to the emphasis on the emergence of drug resistant genes.
Resistance, defined as “the ability of a parasite to survive and/or multiply in the presence of a drug” [8], is of growing concern. Since the global adoption of Artemisinin Combination Therapy (ACT) as the first-line treatment for uncomplicated malaria in 2006, there are reports of decline in incidence of malaria and malaria-related mortalities between 2010 and 2015 [9], however, artemisinin resistance has been reported in Western Cambodia [10] and Southeast Asia [11] suggesting a possibility of occurrence in other parts of the world [12-14]. The selection of resistant genotypes has been associated with inadequate drug exposure either due to improper dosing, incorrect choice of therapeutic modality such as lack of commitment to use of the recommended ACT [15-16], poor pharmacokinetic properties, fake drugs, or other infections acquired during the drug elimination phase [8,17]. Several genes have been attributed to antimalarial drug resistance, for instance, chloroquine resistance in falciparum malaria is associated with point mutations in pfcrt gene [18-20], Pfmdr1 mutations have been associated with resistance to chloroquine, quinine, monodesethylamodiaquine, mefloquine, lumefantrine, and dihydroartemisinin [21-22]. If genetic polymorphism translates into effective and functional phenotypes, then, the emerging polymorphisms in the kelch 13 (K13) propeller gene associated with artemisinin resistance in Southeast Asia [23-24] will arguably have devastating consequence on global plasmodium treatment. Importantly, there have not been any consistent reports of this mutation being present in Africa with one exception [25].
Individual pathophysiologic response to antimalarial treatment varies and may be associable with stress, culminating in transient or sustained post-treatment unwellness without a defined role for parasite genetic polymorphism. For instance, glucocorticoids, such as cortisol and dexamethasone, have been shown to suppress the proinflammatory cytokines and their receptors, tipping the scale from Th1 to Th2 development and proinflammatory responses to anti-inflammatory responses [26]. Proinflammatory cytokines such as TNF-α, IL-12, and IFN-γ have been demonstrated to be essential mediators of protective immunity against erythrocytic malaria [27-28]. There is a lack of knowledge on whether overproduction of proinflammatory cytokines enhances the ability to control malaria infection or predisposes the patient to progress to severe malaria [29], which will require analysis of clinical samples in endemic areas. The relevant question here is whether post-treatment persistence of parasite in the blood accompanied by sustained unwellness of the patient is solely attributable to an infection by drug resistant malaria. We hypothesize that malaria-like post-treatment symptoms, even when malaria parasite is present in blood film in malaria-endemic areas, could be due to a combination of factors such as ACT drug effects, such as fatigue, mild to severe fever, nausea, mild chills, and dizzy spells, proinflammatory conditions, and impairment of regulatory cytokine functions. We tested this hypothesis by analyzing malaria positive blood samples that were categorized as (i) treated (previously received ACT treatment); and (ii) untreated (had not received ACT treatment); and an additional 8 malaria negative individuals as controls. Thus, this case-controlled design (ACT treated and untreated cases attending clinic) satisfied random interference with results by use of unprescribed drugs. The malaria-positive samples were analyzed for mutations in Pfcrt, Pfmdr1, and K13 kelch genes, while all samples were analyzed for levels of cytokines and corticosteroids.
Materials and Methods
Study Population and Samples
The Ethics and Human Subjects Committee of College of Medicine, University of Lagos approved the protocol used for this study (Protocol # CMUL/HREC/05/17/205). Study participants and/or their guardians provided written informed consent and/or assent. The collaborating investigators at Centers for Disease Control and Prevention (CDC) obtained permission from the CDC Human Subjects Office to participate in the genotyping of the samples under anonymous conditions without access to any personal identifying information.
Patient population represents individuals attending outpatient clinics in Lagos, Nigeria with complaints of clinical symptoms associated with malaria. All subjects of study were required to complete professionally supervised questionnaires. Twenty six patients with malaria positive blood (confirmed by microscopy and qPCR) were categorized into (i) 12 who were attending clinics after previous treatment for malaria using ACT (treated cases) and (ii) 14 who were attending clinics for the first time with complaints associated with malaria and tested malaria-positive (untreated cases). The final serum sample size used for each category varies from 3 to 10 depending on serum availability. An additional 8 samples were collected from a control group consisting of individuals who were without symptoms of acute illness during screening, had no evidence or history of chronic illness, and had no parasitemia in their blood upon examination. The drug of choice in this study is artemisinin-based combined therapy, which is a combination of artemether and lumefantrine. In all cases, venous blood collected in EDTA was examined for malaria parasites using microscopy techniques and qPCR. In addition, whole blood spotted on Whatman® #3 filter paper was preserved for molecular studies (genetic polymorphism and mutation). Extracted plasma was used for cytokine and corticosteroid studies.
DNA Extraction
Genomic DNA was isolated from blood spots or whole blood taken at enrollment using a QIAamp DNA minikit (Qiagen, Valencia, CA) following the manufacturer`s instruction and according to the methods described by [30]. The DNA was stored at -200C before use.
Multiplex Photo-induced Electron Transfer-PCR for Plasmodium DNA Amplification
DNA was screened in duplicate using the multiplex photo-induced electron transfer (PET)-PCR assay which used the P. falciparum-specific primer set, Pfrt364, according to the primer design and methods previously described by [31], forward primer 5'-CCATTTTACTCGCAATAACGCTGCAT, reverse 5'-CTGAGTCGAATGAACTAGTCGCTA C with the following primer modification; the 5`end of the reverse primers was modified with the PET tag and labeled with FAM (for the genus) and HEX (for P. falciparum).
Quantitative Real-Time PCR for Plasmodium Detection
Detection of Plasmodium species was performed via qPCR, which targeted the 18S rDNA gene, which is detected via a TaqMan real-time PCR assay as described by [31]. A cut-off CT value of 40.0 or below was used to indicate a positive result.
Detection of K13, pfcrt, and pfmdr1 Gene Polymorphisms
PCR was used to identify gene polymorphisms in the K13, pfcrt, and pfmdr1 genes. The K13 propeller domain was investigated using methods previously described by [32]. The forward primer 5'-CGAAATAGTATCTCGAAT and reverse primer 5'-CTGGGAACTAATAAAGAT were utilized. The pfcrt and pfmdr1 genes were investigated using methods previously described by [33]. For pfcrt codons 72 to 76, TTTTTCCCTTGTCGACCTTAAC (F) and AGGAATAAACAATAAAGAAATAATCATAC (R) primers were utilized. For pfmdr1 codons 1034, 1042, and 1246, GCATTTAGTTCAGATGATGAAATG (F) and CCATATGGTCCAACATTTGTATC (R) primers were utilized. Artemisinin-resistant laboratory control samples 3D7, 7G8, W2, HB3, and Dd2 were also included for comparison. Data analysis and sequencing of the PCR products was performed using the next generation sequencing and Sanger methods at the Centers for Disease Control and Prevention in Atlanta, GA. Data quality filtering and analysis were performed using Geneious R9 (www.geneious.com). Only paired reads that perfectly mapped to the reference Pfk13 sequence (PF3D7_1343700) were used for subsequent single nucleotide polymorphism (SNP) calls (e.g., using the Geneious map to reference and SNP/variation caller).
Cytokines Assay
The elaboration of IFN-γ, TNF-α, IL-12p70, IL-10, and TGF-β1 in patient blood samples was determined using commercial ELISA kits (R&D systems Duoset®) according to the manufacturer's instructions.
Corticosteroids Assay
Cortisol levels were determined using Thermofisher cortisol ELISA kit, a solid phase competitive Enzyme-linked Immunosorbent Assay kit that detects and quantifies the total cortisol in serum. Subsequent steps complied with the manufacturer's instructions. The assay for Dexamethasone-Induced protein (used as an index of dexamethasone levels), was performed using MyBioSource® ELISA kit according to the manufacturer's instructions.
The optical density (OD) for all assays was measured at a wavelength of 450 nm corrected at 540nm on a Microplate reader (Biotek Synergy HT Microplate reader, BioTek Instruments Inc. Vermont USA).
Statistical Analysis
Data were analyzed by student's t-test using the difference between means of two treatments and p-values of ≤ 0.05 were reported as significant. The cytokine and corticosteroid levels between the treated, untreated, and control groups were analyzed by one-way analysis of variance (ANOVA). P-value of ≤ 0.05 was adopted as significant. The correlation between parasite load and corticosteroids and cytokines and the correlation between number of mutant alleles and corticosteroids and cytokines were analyzed, correlation coefficients and R2-value were determined to demonstrate positive or negative correlations.
Results
The effect of ACT treatment on parasitemia
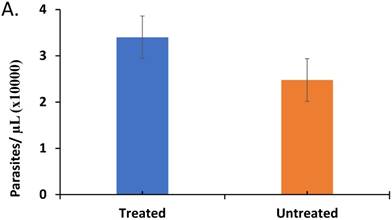
In order to determine if ACT post-treatment syndrome is driven by parasite activity in the patients, we initially assessed the status of Plasmodium falciparum in the malaria-positive samples, by measuring the parasite load in blood (parasitemia). The results from experiments comparing the average number of parasites/µL in treated to untreated patients (Figure 1A) indicates heightened parasitemia in the treated patients (p ≤0.581), which can be accounted for by disturbed immune response affecting IL-10, IL-12, and TGF-β (see discussion).
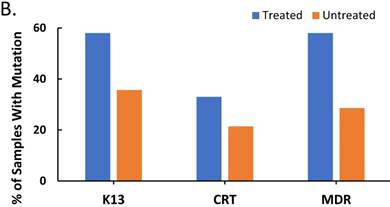
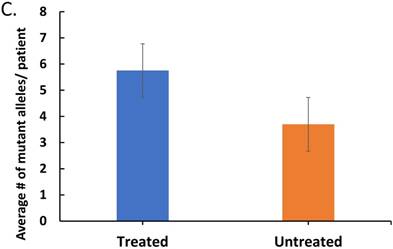
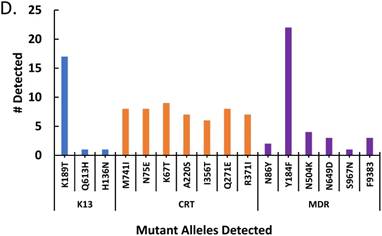
The effect of treatment on parasite load and frequency of antimalarial drug resistant gene polymorphisms in the malaria-positive sample population is represented. Average parasite load, as determined by microscopy and qPCR, is depicted in (A) between patients that were treated (n=12, treated previously with ACT) and untreated (n=14, never treated for this episode of malaria) (p ≤ 0.581). The percentage of treated and untreated patients that exhibited at least one mutant allele in the K13, Pfcrt (CRT), or Pfmdr1 (MDR) genes is depicted in (B). The average number of mutant alleles in the above genes detected per patient in mutation-positive treated and untreated patient samples is depicted in (C) (p ≤ 0.342). The prevalence of each of the observed mutant alleles across all malaria-positive patients is depicted in (D).
The effect of ACT treatment on the onset and frequency of parasite genetic polymorphisms
Since post-treatment persistence of the Plasmodium parasite in the blood is typically attributed to antimalarial drug resistant gene polymorphisms [11, 18], we also examined the frequency of polymorphisms in the K13, Pfcrt (CRT), and Pfmdr1 (MDR) genes, which are associated with antimalarial drug resistance, in the Plasmodium positive samples from our study population. In addition, we examined the pattern of expression of these polymorphisms with treatment. Plasmodium falciparum mutants, defined in this study as containing a minimum of one single nucleotide polymorphism (SNP) in the above genes, were compared to wildtype Plasmodium falciparum, defined in this study as the absence of mutation in the above genes. Figure 1B represents the percentage of treated and untreated patient samples, which contained mutant P. falciparum. There is a higher percentage of mutant P. falciparum amongst the treated patients compared to the untreated patients, which supported the hypothesis of drug pressure on mutation. Worthy of note is the fact that our studies revealed the heightened percentage of K13 and MDR mutations in treated patients (ACT treated), consistent with reports that artemisinin and lumefantrine are associated with K13 and MDR-mediated resistance, respectively [10, 22]. In total, 57.7% of our malaria positive patient samples (both treated and untreated) exhibited at least one SNP in the genes in this study, however, several patients exhibited multiple SNPs.
In order to determine the effect of drug pressure on the number of SNPs, the treated and untreated patients with mutant P. falciparum were analyzed for average number of SNPs. Figure 1C depicts a higher number of SNPs per patient in the treated patients compared to untreated patients indicating that treatment is not only associated with increased prevalence of mutant Plasmodium in the population, but also increased number of mutations. The prevalence of the specific mutant alleles across all malaria-positive patients is depicted in Figure 1D, with the most prevalent alleles being the K13- K189T allele and the MDR- Y184F allele. Our findings revealed that there is a high preponderance of drug resistant gene polymorphisms, which is not reflective of the known final recovery from clinical malaria.
The relationship between P. falciparum load and secretion of cytokines and corticosteroids during infection
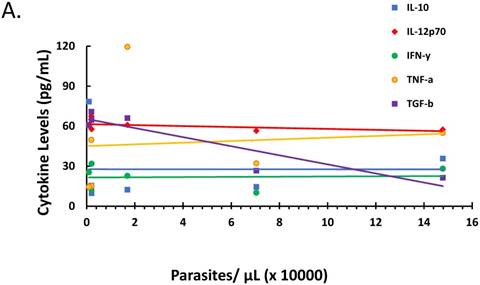
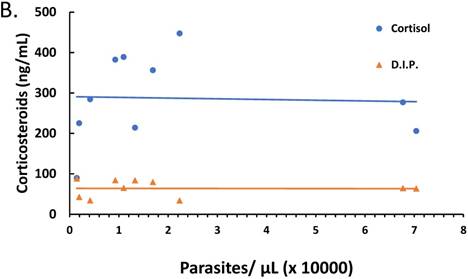
The ratio of proinflammatory cytokines to anti-inflammatory cytokines can be a predictor of the course of clinical infection and may affect the status of the patient during the infection [26-28]. Also, fever onset, the predominant clinical symptom of malaria infection, is known to be parasite threshold-dependent with cytokines and corticosteroids being endogenously expressed stimulators of fever development. We investigated the role of parasitemia on cytokine and corticosteroid secretion that may drive ACT post-treatment syndrome. There was marked negative correlation between parasitemia and TGF-β (ρ ≤ -0.923), yet parasitemia had minimal effect on IL-10, IL-12, IFN-γ, and TNF-α as depicted in Figure 2A. However, changes in parasitemia did not result in corresponding change in corticosteroids (Figure 2B) suggesting that parasitemia has minimal effect on corticosteroid elaboration.
The relationship between parasitemia and cytokines and corticosteroid elaboration in the malaria-positive sample population is depicted. Parasitemia was determined by microscopy and qPCR. Levels of IL-10, IL-12, IFN-γ, TNF-α, TGF-β were determined by ELISA and expressed as pg/mL (true TGF-β values are in pg/mL x 10). The correlation between parasitemia and cytokines is represented in (A) with a significant correlation noted for TGF-β (ρ ≤ -0.923). Levels of cortisol and dexamethasone-induced protein (D.I.P.) were determined by ELISA and expressed as ng/mL. The correlation between parasitemia and cortisol and D.I.P. is represented in (B). Correlation coefficients: cortisol (ρ ≤ -0.042) and D.I.P. (ρ ≤ -0.014).
The effect of P. falciparum antimalarial drug resistant gene mutations on cytokines and corticosteroids secretion
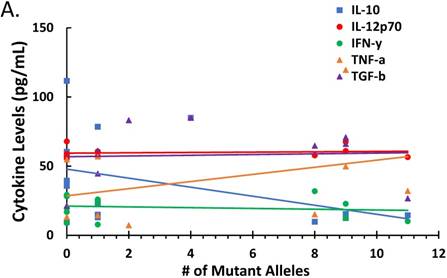
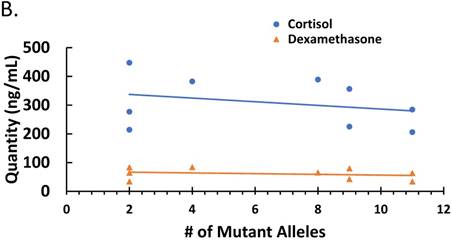
Since parasitemia had a negative correlation with some cytokines (Figure 2), we determined whether there was a correlation between treatment-induced parasite genetic polymorphism and cytokine profiles or corticosteroid levels. The correlation between numbers of mutant alleles detected in the antimalarial drug resistant genes investigated in this study with cytokine levels is depicted in Figure 3A. There was a negative correlation between number of mutant alleles and IL-10 and TGF-β, indicating that when a patient sample has a higher number of mutant alleles, less IL-10/ TGF-β is produced (ρ ≤ -0.373 and ≤ -0.307, respectively). For TNF-α there was a positive correlation indicating that its production increases with increasing number of mutant alleles (ρ ≤ 0.350); although it was uncertain, whether the mutant parasites were better TNF-α inducers. However, there was a negative correlation between corticosteroids, cortisol and dexamethasone-induced protein, and number of mutant alleles (ρ ≤ -0.2849 and ≤ -0.2485, respectively) (Figure 3B). This indicates that corticosteroid elaboration decreases with increasing mutation.
The effect of mutations in the malaria-positive patient samples on levels of cytokines and corticosteroids is represented. Total levels of IL-10, IL-12p70, IFN-γ, TNF-α and TGF-β cytokines detected by ELISA and expressed in pg/mL (true TGF-β values are in pg/mL x 10). The correlation between the number of mutant alleles and cytokine levels is depicted in Figure 3A. Significant correlation coefficients include IL-10 (ρ ≤ -0.404) and TNF-α (ρ ≤ 0.332). The correlation between cortisol and dexamethasone-induced protein (D.I.P.) levels detected by ELISA and expressed in ng/mL in patients with mutant versus wildtype P. falciparum is depicted in Figure 3C. Correlation coefficients: cortisol (ρ ≤ -0.2849) and D.I.P. (ρ ≤ -0.2485).
The effect of ACT drug treatment on cytokines and corticosteroids secretion
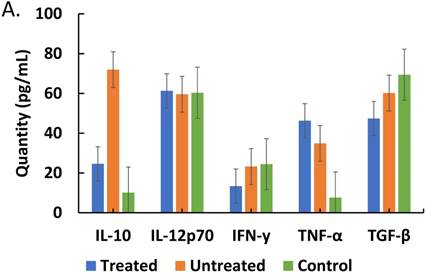
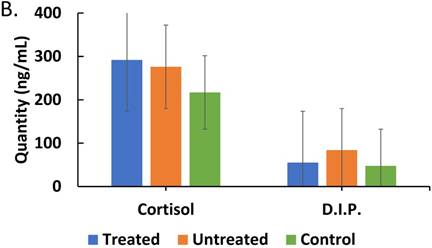
In order to investigate how treatment affects interplay between corticosteroids and cytokines during the course of malaria infection, we assessed the impact of treatment on cytokine profiles, we determined if our findings could represents a pattern of correlation between treatment and lack of it on parasite moderation of cytokine patterns. We directly assessed the effect of ACT treatment on cytokines and corticosteroid secretion. It was found that there was a correlation pattern showing that in treated patients, with persistent parasites, IL-10, IFN-γ and TGF-β levels were markedly lower than in untreated patients (Figure 4A). Therefore, arguably, treatment is impactful on effect of parasitemia on the regulatory cytokines (IL-10 and TGF-β) profile in malaria patients. These findings are consistent with an impaired role for IL-10 and TGF-β in the course malaria infection. On the effect of ACT treatment on corticosteroids secretion, Figure 4B reveals an increased level of cortisol but a decreased level of dexamethasone-induced protein in treated compared with untreated patients.
Comparisons of cytokine and corticosteroid levels across three categories, treated (malaria-positive and previously treated with ACT but returned with complaints of persistent unwellness), untreated (malaria-positive and visiting the health facility for the first time with a new infection) and control (malaria-negative samples) is represented. Total levels of IL-10, IL-12p70, IFN-γ, TNF-α and TGF-β cytokines detected by ELISA and expressed in pg/mL (true values of TGF-β in pg/ml x 10) in treated, untreated, and control patients is depicted in Figure 4A. ANOVA was performed to compare the three categories: IL-10 (p ≤ 0.199), IL-12 (p ≤ 0.758), IFN-γ (p ≤ 0.001), TNF-α (p ≤ 0.0910), and TGF-β (p ≤ 0.287). Total cortisol and dexamethasone-induced protein levels detected by ELISA and expressed in ng/mL in treated, untreated, and control patient samples is depicted in Figure 4B. ANOVA was performed to compare the three categories: cortisol (p ≤ 0.400) and D.I.P. (p ≤ 0.0102).
Discussion
In an effort to determine the impact of ACT, as a drug of choice, during malaria episode, we decided to assess the recovery of P. falciparum infected patients from malaria and the pathophysiologic consequences of drug on treated compared with untreated patients. Our investigations show a compelling evidence of drug-induced consequences manifesting as (i) parasite genetic mutation, (ii) perpetuation of stressor factors and proinflammatory conditions, and (iii) impairment of regulatory cytokine functions. This combination of factors may arguably account for the post-treatment malaria-like-illness commonly designated as drug-resistant malaria instead of ACT-syndrome. We investigated the frequency of P. falciparum genes associated with resistance to antimalarial drugs in malaria patients and our findings (Figure 1) show that there is high preponderance of P. falciparum genetic mutation associated with resistance to antimalarial drugs. If such genetic mutation effectively translates into functional phenotypes, then this would not be compatible with known facts of the final recovery from the disease in an endemic area, since further post-treatment management of patients usually results in recovery. The adoption of combination therapies in several countries is accompanied by the expression, in parasites, of genetic mutations supposedly associated with drug resistance [22, 34]. It is difficult to ascertain that the resistance is mechanistically an outright drug resistant process involving efficient and effectively expressed alternative form of genes as genetic phenotypes. In this study, we have high gene polymorphism levels, which, if functionally effective, will mean a disastrous situation in the whole of sub-Sahara Africa, which largely the situation in Lagos, Nigeria will reflect.
Although the likelihood is that parasites carrying mutant genes are less likely to be as fit compared with the wild type [35], there is the possibility that they can alter the physiologic steady state of the host. This important feature was investigated to determine if mutant plasmodium could influence the stress hormones and the inflammatory components of the immune system in a manner that translates into clinical treatment failure. In our studies (Figures 1A, 1B, 1C, and 1D), we have shown that there is heightened parasitemia and parasite mutations despite treatment as previously reported [10, 36] but this alone may not be entirely consistent with an argument for patient unwellness. This is because our data (Figures 2A, 2B, 3A, and 3B) also showed that parasite load and type (wildtype and mutants) can result in sustained stressor factors (cortisol and TNF-α) elaboration despite treatment. In addition, therapeutic intervention with ACT, in particular, is shown (Figures 4A and 4B) to increase stressor factors. Taken together these interacting factors may in part, account for patient unwellness despite treatment. This condition could result from ACT-induced altered internal environmental factors, which could create a picture of (i) enhanced proliferation of resistant genes, (ii) sustained proinflammation and (iii) heightened stressor conditions meted by stress hormones culminating in persistent post-treatment unwellness. In addition, our findings (Figure 4A) reveal that the pattern of TNF-α is such that drives malaria infection course in the direction of proinflammation, which is sustained after treatment and may account for the post-treatment complaints, even in the case of IL-12, although not significantly increased, treatment did not attenuate its level. Other than the proinflammatory cytokine profile pattern, the elevated values of post-treatment proinflammatory cytokines compared with control values in healthy individuals reveals a probable opportunity for heightened effects which could mete out those unwellness consequences associated with persistent malaria.
Noteworthy is the unique implications of IL-10 decreased levels in treated patients (Figure 4A); this is a cytokine principally derived from Th2 cells and it is an inhibitory factor which, under normal circumstances, would impair the sustained build-up of proinflammatory cytokines at the recovery phase of clinical malaria. In addition, it has ability to turn off macrophage elaboration of IL-12, thus, a compromise of its elaboration as exhibited here, implies, a room for protracted and sustained proinflammatory situation. Closely connected with this is the post-treatment impairment of the elaboration of TGF-β whose intrinsic role includes the induction of the development and propagation of regulatory T-cells (T-reg cells). Since T-reg cells have the capacity to induce CD4+-T-cell development from naïve T-cells [37], it is clear why impairment of CD4+ T-cells will result in compromised elaboration of IL-10. Thus, causing a situation of persistence of proinflammatory cytokine expression. On basis of those variations that exist in steady state, the expression of impaired IL-10 will be variable and this could account for the various cases of “usually sick” malaria cases that exhibit protracted symptoms of malaria illness in endemic areas. Although uncertain, it would appear that the observed negative correlation between parasitemia and IL-10 elaboration (Figure 2A) is a revelation of parasite direct effect while Figure 4A shows the attenuating effect of drug used during treatment and pathophysiologic change on IL-10 elaboration. Also, the decreased level of IFN- γ reported in Figure 4A is of importance because of its pivotal role in the activation of macrophages, which is needed for plasmodium clearance. Taken together, these events could account for chronic progressive malaria disease, which can be mistakenly attributed to drug resistance.
Although the role of chronic stress, as typified by our reported heightened cortisol levels, in relation to the steady state of the immune system is unclear, previous studies have reported both favorable and unfavorable events associated with the immune response in related conditions [38-39]. Our findings show an evidence of slight negative correlation between post- and pre-treatment parasitemia and corticosteroid levels without a statistically significant difference with treatment, thus, revealing the impactful nature of corticosteroids. Corticosteroids are readily bound to their receptors on cells in a biphasic, concentration-dependent manner [40-41]. The implication of this phasic binding potential is likely to mete differential protracted action on patients and this could explain the differences in post-treatment unwellness exhibited by malaria patients. Expectedly, corticosteroids should downregulate inflammatory cytokine (TNF-α) elaboration but in the case of malaria infection, patients' sera show the reverse (Figure 4A), which cannot be ignored considering the post-treatment complaints of illness. Also, corticosteroids may have a role for delayed immune clearance of malaria on one hand and an impaired elaboration of IL-10 on the other hand, resulting in unwanted sustained inflammatory condition. This is understandable if one considers the role of IL-10 in proinflammatory cytokine impairment. This perspective may provide explanation for carefully controlled use if not total discontinuity of corticosteroids in cerebral malaria therapy. In conclusion, the ACT-meted-syndrome consists of post-treatment malaria-like-illness, enhanced genetic polymorphism in parasite that may not be effective phenotypes, and proinflammatory conditions accompanied by regulatory cytokine impairment.
Acknowledgements
This work was supported by funds from College of Health Sciences, University of Wisconsin-Milwaukee and the University of Lagos, Nigeria. In addition, the Fulbright foreign student program provided funds for the economic upkeep of Mr. Abel Idowu during the entire period of research. The Milwaukee Health Department Laboratories in Milwaukee WI and the Division of Parasitic Diseases and Malaria, Centers for Disease Control and Prevention, Atlanta, Georgia funded the aspects of studies on malaria gene polymorphism. We thank Eldin Talundzic, Naomi Lucci, and Venkatachalam Udhayakumar of the Centers for Disease Control and Prevention, Atlanta, Georgia for their enormous technical support in the studies of malaria gene polymorphism.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Berkley JA, Maitland K, Mwangi I. et al. Use of clinical syndromes to target antibiotic prescribing in seriously ill children in malaria endemic area: observational study. Brit Med J. 2005;330:995-1001
2. Jakka S, Veena S, Atmakuri R, Eisenhut M. Characteristic abnormalities in cerebrospinal fluid biochemistry in children with cerebral malaria compared to viral encephalitis. Cerebrospin Flu Res. 2006;3:1-7
3. Rogier C, Trape J, Commenges D. Evidence for an Age-Dependent Pyrogenic Threshold of Plasmodium falciparum Parasitemia in Highly Endemic Populations. Am J Trop Med Hyg. 1996;54:613-9
4. Boström S, Giusti P, Arama C. et al. Changes in the levels of cytokines, chemokines and malaria-specific antibodies in response to Plasmodium falciparum infection in children living in sympatry in Mali. Malar J. 2012;11:109-15
5. Gonçalves RM, Scopel KG, Bastos MS, Ferreira MU. Cytokine Balance in Human Malaria: Does Plasmodium vivax Elicit More Inflammatory Responses than Plasmodium falciparum? PLoS ONE. 2012;7:1-10
6. Gomes PS, Bhardwaj J, Rivera-Correa J, Freire-De-Lima CG, Morrot A. Immune Escape Strategies of Malaria Parasites. Front Micro. 2016;7:1617-23
7. Tran TM, Jones MB, Ongoiba A. et al. Transcriptomic evidence for modulation of host inflammatory responses during febrile Plasmodium falciparum malaria. Sci Rep. 2016;6:312-22
8. Costa GL, Amaral LC, Fontes CJ, Carvalho LH, Brito CF, Sousa TN. Assessment of copy number variation in genes related to drug resistance in Plasmodium vivax and Plasmodium falciparum isolates from the Brazilian Amazon and a systematic review of the literature. Malar J. 2017;16:1-11
9. World Health Organization. Malaria. World Health Organization, Geneva, Switzerland. 2017
10. Dondorp AM, Nosten F, Yi P. et al. Artemisinin Resistance in Plasmodium falciparum Malaria. N Eng J Med. 2009;361:455-67
11. Ashley EA, Dhorda M, Fairhurst RM. et al. Spread of Artemisinin Resistance in Plasmodium falciparum Malaria. N Eng J Med. 2014;371:411-23
12. Roper C. Intercontinental Spread of Pyrimethamine-Resistant Malaria. Sci. 2004;305:1124-9
13. Summers RL, Martin RE. Functional characteristics of the malaria parasite's “chloroquine resistance transporter”: Implications for chemotherapy. Virul. 2010;1:304-8
14. Mita T, Venkatesan M, Ohashi J. et al. Limited Geographical Origin and Global Spread of Sulfadoxine-Resistant dhps Alleles in Plasmodium falciparum Populations. J Infect Dis. 2011;204:1980-8
15. Mangham LJ, Cundill B, Ezeoke O, Nwala E, Uzochukwu BS, Wiseman V, Onwujekwe O. Treatment of uncomplicated malaria at public health facilities and medicine retailers in south-eastern Nigeria. Malar J. 2011;10:155-62
16. Ezeoke OP, Ezumah NN, Chandler CC, Mangham-Jefferies LJ, Onwujekwe OE, Wiseman V, Uzochukwu BS. Exploring health providers' and community perceptions and experiences with malaria tests in South-East Nigeria: a critical step towards appropriate treatment. Malar J. 2012;11:368-75
17. Blasco B, Leroy D, Fidock DA. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nature Med. 2017;23:917-28
18. Babiker HA, Pringle SJ, Abdel-Muhsin A, Mackinnon M, Hunt P, Walliker D. High-Level Chloroquine Resistance in Sudanese Isolates of Plasmodium falciparum Is Associated with Mutations in the Chloroquine Resistance Transporter Gene pfcrt and the Multidrug Resistance Gene pfmdr1. J Infect Dis. 2001;183:1535-8
19. Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine Resistance in Plasmodium falciparum Malaria Parasites Conferred by pfcrt Mutations. Sci. 2002;298:210-3
20. Bray PG, Martin RE, Tilley L, Ward SA, Kirk K, Fidock DA. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol Micro. 2005;56:323-33
21. Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Krishna S. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364:438-47
22. Wurtz N, Fall B, Pascual A. Role of Pfmdr1 in In Vitro Plasmodium falciparum Susceptibility to Chloroquine, Quinine, Monodesethylamodiaquine, Mefloquine, Lumefantrine, and Dihydroartemisinin. Antimicrob Agent Chemother. 2014;58:7032-40
23. Ariey F, Witkowski B, Amaratunga C. et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50-5
24. Kamau E, Campino S, Amenga-Etego L, Drury E, Ishengoma D, Johnson K, Djimde A. K13-propeller polymorphisms in Plasmodium falciparum parasites from sub-Saharan Africa. J Infect Dis. 2014;608:1352-5
25. Lu F, Culleton R, Zhang M. et al. Emergence of Indigenous Artemisinin-Resistant Plasmodium falciparum in Africa. N Eng J Med. 2017;376:991-3
26. Elenkov I, Chrousos G. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Neuroendo Immun Basis Rheum Dis. 2002;966:290-303
27. Dodoo D, Omer FM, Todd J, Akanmori BD, Koram KA, Riley EM. Absolute Levels and Ratios of Proinflammatory and Anti-inflammatory Cytokine Production In Vitro Predict Clinical Immunity to Plasmodium falciparum Malaria. J Infect Dis. 2002;185:971-9
28. Dunst J, Kamena F, Matuschewski K. Cytokines and Chemokines in Cerebral Malaria Pathogenesis. Front Cell & Infect Micro. 2017;7:1-16
29. Artavanis-Tsakonas K, Riley EM. Innate Immune Response to Malaria: Rapid Induction of IFN- from Human NK Cells by Live Plasmodium falciparum-Infected Erythrocytes. J Immunol. 2002;169:2956-63
30. Oyola SO, Ariani CV, Hamilton WL. et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar J. 2016;15:597-609
31. Demas A, Oberstaller J, DeBarry J. et al. Applied Genomics: Data Mining Reveals Species-Specific Malaria Diagnostic Targets More Sensitive than 18S rRNA. J Clin Micro. 2011;49:2411-8
32. Chenet S, Akinyi Okoth S, Huber C. et al. Independent Emergence of the Plasmodium falciparum Kelch Propeller Domain Mutant Allele C580Y in Guyana. J Infect Dis. 2016;213:1472-5
33. Griffing S, Syphard L, Sridaran S. et al. Pfmdr1 Amplification and Fixation of pfcrt Chloroquine Resistance Alleles in Plasmodium falciparum in Venezuela. Antimicrob Agent & Chemother. 2010;54:1572-9
34. Berzosa P, Esteban-Cantos A, García L. et al. Profile of molecular mutations in pfdhfr, pfdhps, pfmdr1, and pfcrt genes of Plasmodium falciparum related to resistance to different anti-malarial drugs in the Bata District (Equatorial Guinea). Malar J. 2017;16:1-10
35. Petersen I, Eastman R, Lanzer M. Drug-resistant malaria: Molecular mechanisms and implications for public health. FEBS Let. 2011;585:1551-62
36. White NJ. Antimalarials drug resistance. J Clin Investig. 2004;113:1084-92
37. Abel S, Lückheide N, Westendorf A. et al. Strong impact of CD4 Foxp3 regulatory T cells and limited effect of T cell-derived IL-10 on pathogen clearance during Plasmodium yoelii infection. J Immunol. 2012;188:5467-77
38. Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol & Cell Endocrin. 2011;335:2-13
39. Padgett DA, Glaser R. How stress influences the immune response. Trends Immunol. 2003;24:444-8
40. Derijk R, Schaaf M, Kloet ED. Glucocorticoid receptor variants: clinical implications. J Steroid Biochem & Mol Bio. 2002;81:103-22
41. Müller MB, Holsboer F, Keck ME. Genetic modification of corticosteroid receptor signalling: Novel insights into pathophysiology and treatment strategies of human affective disorders. Neuropep. 2002;36:117-31
Author contact
![]() Corresponding author: Dr. Anthony A. Azenabor, Room 459, Enderis Hall, University of Wisconsin, 2400 E. Hartford Avenue, Milwaukee, WI 53211 USA. Phone: (414) 446-7694; Fax: (414) 229-2619; Email: aazenaboedu
Corresponding author: Dr. Anthony A. Azenabor, Room 459, Enderis Hall, University of Wisconsin, 2400 E. Hartford Avenue, Milwaukee, WI 53211 USA. Phone: (414) 446-7694; Fax: (414) 229-2619; Email: aazenaboedu