Impact Factor ISSN: 1449-1907
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(12):1365-1372. doi:10.7150/ijms.26186 This issue Cite
Research Paper
Acid-electrolyzed functional water induces extracellular matrix metalloproteinase inducer, a possible novel alarmin, secretion from oral squamous cell carcinoma cell lines
Masafumi Kusunoki1, Eri Sata2,3, Kensuke Nishio4, Takayoshi Tanaka5,6, Tetsuya Nishida1,7, Naoyuki Sugano1,7, Shuichi Sato1,7, Masatake Asano8,9, ![]()
1. Department of Periodontology, Nihon University School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan
2. Department of Orthodontics, Nihon University School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan
3. Oral Structural and Functional Biology, Nihon University Graduate School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan
4. Department of Complete Denture Prosthodontics, Nihon University School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan
5. Maxillofacial Prosthetic Clinic, Nihon University School of Dentistry, Tokyo, Japan
6. Department of Oral and Maxillofacial Surgery, Nihon University School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan
7. Division of Advanced Dental Treatment, Dental Research Center, Nihon University School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan
8. Department of Pathology, Nihon University School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan
9. Division of Immunology and Pathobiology, Nihon University School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan
Received 2018-3-20; Accepted 2018-6-30; Published 2018-9-7
Abstract
Extracellular matrix metalloproteinase inducer (EMMPRIN) secretion was induced in the oral squamous cell carcinoma cell line HSC3 cell by acid-electrolyzed functional water (FW) stimulation. Augmented EMMPRIN secretion was not under transcriptional control; rather, it was derived from the intracellular storages. EMMPRIN secretion was also induced under oxidative stress and accompanied by the release of lactate dehydrogenase (LDH). The molecules released from cells undergoing necrosis are called as alarmins, and the secretion of IL-1α, a typical alarmin, was induced by FW stimulation and oxidative stress. Intracellular localization was examined by cell fractionation. A significant amount of EMMPRIN was localized in the triton X-100 and DNase sensitive fractions; the levels were drastically reduced following FW treatment. The function of the released EMMPRIN was examined using the monocytic cell line THP1. Culture supernatant derived from FW-treated HSC3 cells induced the expression of matrix metalloproteinases (MMPs) 1, 2, 8, 9, 13, and 14, platelet-derived growth factor, and interleukin-8. In contrast, vascular endothelial growth factor expression was reduced. Induction of these factors was abolished following eliminating of EMMPRIN by immunoprecipitation. These results indicate that EMMPRIN might be considered as a type of alarmin that transduces danger signals to the surrounding cells.
Keywords: Acid-electrolyzed functional water, Extracellular matrix metalloproteinase inducer, Oral squamous cell carcinoma, Matrix metalloproteinase
Introduction
Cancer cells degrade the surrounding tissue by producing enzymes such as matrix metalloproteinases (MMPs), thus enabling tumor invasion [1]. Initially, MMPs were thought to be produced mainly by the tumor cells; however, histological examinations of the pattern of MMP expression in cancer specimens revealed that most of them are produced by stromal fibroblasts [2]. Tumor cells can stimulate the surrounding fibroblasts to produce higher levels of MMPs within the tumor microenvironment via the action of the extracellular matrix metalloproteinase inducer (EMMPRIN), which belongs to the immunoglobulin superfamily [3]. EMMPRIN (renamed as CD147) is a transmembrane 28 kDa glycoprotein composed of an extracellular domain of 187 amino acid residues (aa), a transmembrane domain (24 aa) and a cytoplasmic domains (40 aa) [4-6]. The extracellular region contains two immunoglobulin domains and three N-glycosylation sites [7]. Depending on the degree of glycosylation, it migrates to various positions on the SDS-PAGE [6]. The glycosylation status of this protein is important for its biological functions [8]. The charged amino acid residue (glutamic acid) localized in the transmembrane region allows for the association of EMMPRIN with other transmembrane proteins [7]. Besides its role in cancer progression, EMMPRIN plays pleiotropic roles in development, reproduction, and wound healing [3].
Electrolytically-generated acid functional water (FW), obtained by electrolyzing low concentrations of saline, is widely used as a disinfectant, in clinical practice [9]. In a previous report, FW was demonstrated to augment basic fibroblast growth factor (bFGF) secretion in HeLa cells [10]. As FW-treatment significantly induced the secretion of lactate dehydrogenase (LDH), a marker of cell damage, it was expected to cause some amount of damage to the cells. The molecules released from the damaged cells are referred to as alarmins [11]. In the present study, we attempted to examine the effect of FW on oral squamous cell carcinoma-derived cell lines. FW-treatment was found to induce the secretion of EMMPRIN; therefore, the aim of the study was to present EMMPRIN as a novel alarmin.
Materials and Methods
Reagents
The FW (actual chloride concentration (ACC) 30 ppm, pH 2.7, oxidation-reduction potential (ORP) of more than 1100 mV) was kindly provided by Miura Denshi (Akita, Japan). Anti-human CD 147 antibody (Ab) (anti-EMMPRIN Ab) was purchased from BioLegend (San Diego, USA).
Cell culture and FW stimulation
Human oral squamous cell carcinoma cell lines HSC3 and Ca9-22, and the human monocytic cell line THP1 were obtained from the Health Science Research Resources Bank (Osaka, Japan). The cells were maintained in RPMI1640 medium supplemented with 10% FCS, 50 mg/ml streptomycin, and 50 U/ml penicillin (10% FCS-RPMI). The HSC3 cells were plated on a 24-well plate (IWAKI, Tokyo, Japan) at a density of 1 × 105 /well. The cells were washed with PBS and treated with FW for 30 s. The stimulation was stopped by adding 10% FCS-RPMI and further cultured for the indicated time periods.
Cytokine Array Experiment
HSC3 cells were plated on a 10 cm cell culture dish (Greiner, Tokyo, Japan) at a density of 1 × 106 /dish on the day before the experiment. Following stimulation with FW for 30 s, the cells were further cultured for 18 h. The culture supernatants were collected and subjected to cytokine array experiments using the Human XL Cytokine Array kit (R&D systems) according to the manufacture's protocol. Images were taken using ChemiDoc XRS (BioRad, Tokyo, Japan).
Enzyme-Linked Immunosorbent Assay (ELISA)
The cells were cultured for 1, 3, 6 and 12 h and the culture supernatants were harvested to measure the concentration of EMMPRIN. The cells were washed with PBS and lysed with cell lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.5 % TritonX-100). EMMPRIN concentrations were measured using the DuoSet ELISA Development System (R&D Systems).
Real-time PCR
Total RNA was purified using the RNeasy mini kit (QIAGEN, Tokyo, Japan). cDNA was synthesized using Superscript III reverse transcriptase (Invitrogen, San Diego, CA, USA) and subjected to real-time PCR, as described previously [12]. Real-time PCR was performed using the CFX96-Real-Time-System (BioRad, Tokyo, Japan) with SYBR green (TaKaRa, Tokyo, Japan). The primers used in this study are listed in Table 1.
Cell stimulation and LDH measurement
Lactate dehydrogenase (LDH) was measured by treating the cells with either FW or 10 mM H2O2, or culturing them at 42°C. After treatment, the cells were further cultured for 3 h. The culture supernatants were harvested and subjected to LDH measurement using an LDH activity assay kit (Sigma-Aldrich, Tokyo, Japan). The samples were also subjected to EMMPRIN and IL-1α concentration measurements using the DuoSet ELISA Development System (R&D Systems).
Primers used in this study

Cell fractionation experiment
HSC3 cells were plated on a 15 cm dish (1 × 107/dish). The cells were washed twice with PBS, and stimulated with or without FW for 30 s and further cultured for 1 h. The cells were collected, washed with PBS and subjected to cell fractionation as described previously [13]. Briefly, the cells were re-suspended with Triton X-100 buffer (0.1% Triton X-100, 10 mM HEPES, pH7.3, 100 mM NaCl, 0.1 mM MgCl2, 0.1 mM PMSF, 1.0 mM DTT and proteinase inhibitor) and incubated on ice for 10 min. The cells were pelleted by centrifugation at 1,000 × g at 4°C for 5 min. The supernatant was collected as the “1st fraction”. Next, the cell pellets were re-suspended with Triton X-100 buffer and pelleted by centrifugation at 1,000 × g for 5 min. The supernatant was collected as the “Wash fraction”. The cell pellets were re-suspended with Triton X-100 buffer containing 1000 U/ml DNase and incubated at 25°C for 1 h. Then, the cells were pelleted by centrifugation at 1,000 × g for 5 min. The supernatant was collected as the “DNase fraction”. The cell pellets were re-suspended with 2M NaCl (0.1% Triton X-100, 10 mM HEPES, pH7.3, 2M NaCl, 0.1 mM MgCl2, 0.1 mM PMSF, 1.0 mM DTT and inhibitor) and incubated on ice for 10 min. The cells were pelleted by centrifugation at 1,000 × g for 5 min and the supernatant was collected as the “NaCl fraction”. Finally, the cell pellets were re-suspended with 1% SDS (10 mM HEPEs, pH7.3, 100 mM NaCl, 0.1 mM MgCl2, 0.1 mM PMSF, 1.0 mM DTT, 1% SDS and inhibitor) and incubated at room temperature for 10 min. Then, the cells were pelleted by centrifugation at 15,000 × g at 25°C for 5 min and the supernatants were collected as “SDS fraction”. The protein concentrations of each fraction were measured using the Bio Rad protein assay kit (BioRad, Tokyo, Japan) and subjected to Western blotting.
THP1 stimulation
The conditioned medium derived from FW-stimulated or un-stimulated HSC3 cells were collected for THP1 cell stimulation. The THP1 cells were cultured with or without the conditioned medium for 24 h, harvested and total RNA was extracted. cDNA was generated and real-time PCR was performed as described earlier. For the inhibition experiments, the EMMPRIN in the conditioned medium was immunoprecipitated with anti-EMMPRIN antibody followed by protein G-sepharose (HE Healthcare, Tokyo, Japan). The samples were collected after centrifugation (10,000 × g, 3 min) and subjected to THP1 stimulation.
Statistical analysis
Student's t-test or two-way ANOVA with Tukey tests were used for the statistical analysis. Results are presented as mean ± standard deviation (SD). P values < 0.05 were considered as statistically significant.
Results
The cytokine secretion profiles were compared between the PBS- and FW-treated samples (Fig 1). The expression levels of most of the cytokines secreted in the PBS-treated samples were decreased following FW-treatment. On the other hand, the levels of EMMPRIN, IL-1α, IL-1ra and IL-32 were significantly augmented in the FW-treated samples (Fig 1, lower panel). Among them, EMMPRIN level was most prominently increased; hence, we focused on this molecule in the following experiments.
Electrolytically-generated acid functional water (FW) treatment significantly augmented EMMPRIN secretion. HSC3 cells were treated with PBS (upper panel) or FW (lower panel) for 30 s. The treatment was stopped by adding 10% FCS-RPMI and the cells were washed. After wash, the cells were further cultured for 18 h, the supernatants were harvested and subjected to a cytokine array experiment. The positions of the cytokines were spotted on the membrane. The dots in rectangles showed 1. EMMPRIN, 2. IL-1α, 3. IL-1 receptor antagonist (IL-1ra) and 4. IL-32, respectively. All these factors were augmented by FW-treatment. The representatives of two independent experiments are shown.

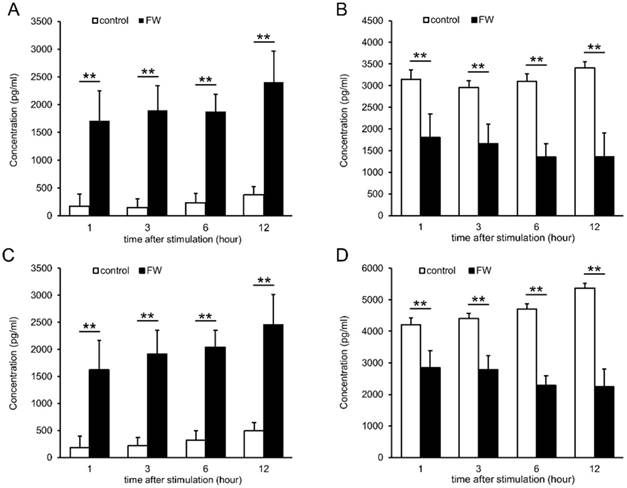
To confirm the increase in EMMPRIN levels, HSC3 and Ca9-22 cells were treated with FW, and the culture supernatants were harvested and subjected to ELISA (Fig 2). EMMPRIN levels had increased to 1700 pg/ml in the HSC3 cells and 1620 pg/ml in the Ca9-22 cells after 1 h of culture (Fig 2A and C). The levels were further increased to 2400 (HSC3) and 2450 pg/ml (Ca9-22) at 12 h (Fig 2A and C). In PBS-treated samples, EMMPRIN concentration was less than 500 pg/ml in both cell lines, whereas, in the cell lysates EMMPRIN concentrations had decreased drastically, in a culture time-dependent manner, following FW-treatment (Fig 2 B, D).
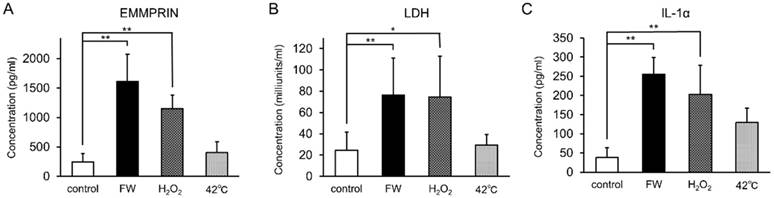
To examine whether augmented EMMPRIN secretion was regulated at the transcriptional level, total RNA was purified after 0.5, 1, 3, 6, and 12 h of stimulation and subjected to real-time PCR. EMMPRIN mRNA levels were not drastically altered during the indicated time points in either of the cell lines (Fig S1). FW-treatment was expected to exert a certain amount of stress on the cells, and the induction of EMMPRIN might have been the result of this stress-related response. To examine this possibility, HSC3 cells were cultured in the presence of 10 mM H2O2 or at 42°C for 3 h and the concentrations of secreted EMMPRIN were measured (Fig 3). Significant EMMPRIN secretion was observed with FW (1,610 pg/ml) and 1% H2O2 (1,150 pg/ml), whereas culturing at 42°C induced a slight increase in EMMPRIN secretion (400 pg/ml), statistical significance notwithstanding (Fig 3A). The extent of cell damage was monitored by measuring LDH concentrations (Fig 3B). The most prominent LDH release was observed after FW-treatment (127 nkat/ml) followed by the 1% H2O2 (123 nkat/ml) treatment, and to a lesser extent after the 42°C culture (49 nkat/ml; Fig. 3B). The molecules released from the stressed cells are called as alarmins. To examine the candidature of EMMPRIN as an alarmin, the release of the representative alarmin cytokine IL-1α was measured (Fig 3C). IL-1α secretion was significantly augmented after FW and 1% H2O2 treatment, but only slightly after culturing at 42°C (Fig 3C).
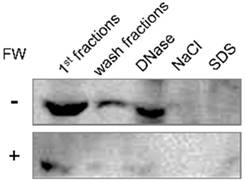
Intracellular localization of EMMPRIN in HSC3 cells was examined by the cell fractionation assay. The FW-treated or un-treated HSC3 cells were sequentially processed with Triton X-100 based buffer containing various reagents. Each fraction was subjected to Western blotting. The main 66 kDa band was detected in the 1st, wash and DNase fractions (Fig 4, upper panel). The same fractionation experiment was performed after FW treatment and compared with the control. The 66 kDa band was detected in the same fractions as in the control; however, the intensities were significantly lower in FW-treated sample (Fig 4, lower panel). These findings indicated that FW treatment drastically reduced EMMPRIN levels in the cells.
FW-treatment significantly augmented the secretion of EMMPRIN. HSC3 (A, B) and Ca9-22 (C, D) cells were treated with or without FW for 30 sec. After wash, the cells were further cultured for 1, 3, 6 and 12 h. The culture supernatants and the cell lysates were harvested and subjected to ELISA. In both cell lines, EMMPRIN in the culture supernatants augmented drastically. In contrast, EMMPRIN in cell lysates dropped significantly. The mean ± standard deviation (SD) of three independent experiments are shown. (** p<0.01)
EMMPRIN secretion paralleled LDH release and IL-1α secretion. HSC3 cells were cultured under stress conditions (FW-stimulated, 10 mM H2O2 treatment or cultured at 42°C) for 3 h. The culture supernatants were harvested and EMMPRIN (A), LDH (B) and IL-1α (C) concentrations were measured. FW- and 10 mM H2O2 -treatment induced both EMMPRIN, LDH and IL-1α secretion to the culture supernatnats. IL-1α secretion was only slightly augmented at 42°C. The mean ± SD of three independent experiments are shown. (*p<0.05, ** p<0.01)
The intracellular localization of EMMPRIN before and after FW-treatment. HSC3 cells were treated with or without FW for 30 s, and further cultured for 1 h. The cells were collected and processed sequentially with Trion X-100 buffer twice (1st and wash fractions), DNase buffer (DNase), NaCl buffer (NaCl) and SDS buffer (SDS). The extracted samples were loaded on SDS-PAGE and subjected to Western blotting. EMMPRIN in the 1st fraction disappeared rapidly after FW-treatment. The representative of four independent experiments are shown.
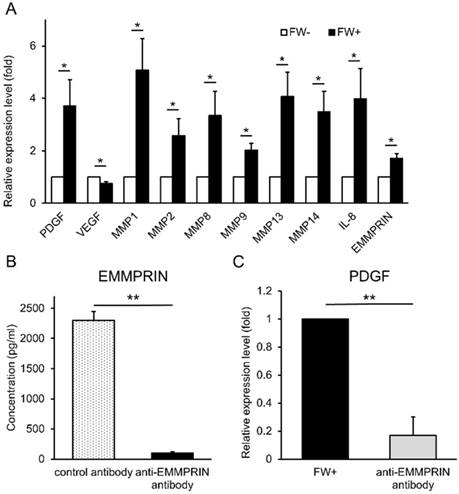
Next, we examined the functionality of the released EMMPRIN. HSC3 cells were treated with or without FW and cultured for 3 h. At the end of the culture, the supernatants (conditioned media) were collected. The human monocytic cell line, THP1 was cultured with these conditioned media and the expression of various EMMPRIN target genes were examined by real-time PCR. The FW-treated conditioned medium (FW (+)) significantly induced the expression of PDGF, MMP1, MMP2, MMP8, MMP9, MMP13, MMP14 and IL-8 (Fig 5A); in contrast, only VEGF expression was reduced (Fig 5A). To further confirm EMMPRIN-mediated induction of its target genes, the EMMPRIN in the conditioned medium was eliminated by immunoprecipitation. EMMPRIN concentration in Ab-treated FW (+) was significantly reduced to less than 100 pg/ml when compared with Ab-treated conditioned medium in the control (2,300 pg/ml; Fig 5B). Using these conditioned media, the THP1 cells were stimulated and PDGF mRNA expression was examined. The treatment of THP1 cells with Ab-treated FW (+) resulted in reduced PDGF levels (Fig 5C) indicating an EMMPRIN-mediated induction of PDGF in the THP1 cells.
FW-induced EMMPRIN augmented the expression of its target genes in THP1 cells. (A) HSC3 cells were stimulated with or without FW for 30 s and further cultured for 3 h. The culture supernatants from FW-treated (FW+) or -untreated (FW-) were harvested. THP1 cells were stimulated with the FW+ or FW- for 3 h. At the end of culture, the total RNA was purified and subjected to real-time PCR. The basal expression levels of each target gene was set as 1 and relative expression level was shown as fold-induction. Most of the target genes, but VEGF, were augmented by FW+. (B) The anti-EMMPRIN antibody or control antibody was added to FW+ and rotated for 18 h followed by protein G-sepharose to eliminate EMMPRIN from FW+. After immunoprecipitation, EMMPRIN concentration was measured by ELISA. Addition of anti-EMMPRIN antibody totally eliminated EMMPRIN from FW+. (C) THP1 cells were stimulated with the immunoprecipitated (anti-EMMPRIN antibody) or control antibody-treated conditioned medium (FW+). Total RNA was purified and subjected to real-time PCR to examine PDGF expression. FW+ augmented the expression of PDGF and this value was set as 1. In contrast, anti-EMMPRIN antibody-treated sample failed to augment PDGF expression in THP-1 cells. The mean ± SD of at least three independent experiments are shown. (*p<0.05, ** p<0.01)
Discussion
FW and H2O2 stimulation resulted in the augmented secretion of EMMPRIN in the culture medium. We examined the two-different cell lines, HSC3 and Ca9-22, to demonstrate that the FW-induced EMMPRIN secretion is a general phenomenon for oral squamous cell carcinoma cells. This change was independent of the transcriptional regulation. Moreover, intracellular EMMPRIN levels were concomitantly reduced. These results clearly indicated that EMMPRIN was released from the intracellular storage in response to the stimuli. In order to examine the intracellular localization of EMMPRIN, cell fractionation experiments were performed. EMMPRIN is a transmembrane glycoprotein belonging to the immunoglobulin superfamily [4-6], and was thus expected to be localized exclusively on the cell surface. The 1st and wash fractions contained cytoplasmically localized proteins, and most of the EMMPRIN was localized in these fractions. The triton-X100-based buffer is not strong enough to extract the transmembrane proteins; therefore, a part of the EMMPRIN might be attached to the cell membrane or localized in the cytoplasm by unknown mechanisms. A typical alarmin IL-1α may demonstrate membrane attachment; the precursor form of IL-1α bound to the plasma membrane via a lectin-like interaction and this interaction is dissociated by D-mannose treatment [14]. Interestingly, EMMPRIN can be detected in DNase-sensitive fractions. DNase destroys the basic genomic DNA structure and this fraction is expected to contain the proteins interacting with DNA. If this is the case, EMMPRIN might be localized in the nucleus. Although the implication of this finding remains elusive, the intracellular localization of EMMPRIN needs to be examined further. FW-treatment drastically reduced EMMPRIN in the all fractions in this study suggesting that the EMMPRIN secreted by FW stimulation is derived from cytoplasmic or nuclear storages.
EMMPRIN secretion paralleled LDH release in the cells, which were thought to present with a certain degree of damage. Various alarmins such as high mobility group box-1 protein (HMGB1) and IL-1α have been described to date [11]. The common features of these alarmins include the following: intranuclear localization, lack of signal peptides, ER-Golgi-independent secretion pathway, and other immunogenic functions.
Consistent with other alarmins rapid EMMPRIN secretion was noted following FW-treatment. Cell fractionation analysis suggested an intranuclear localization of the protein. The localization of other alarmins such as IL-1α and HMGB-1 is generally observed in the nucleus due to nuclear localizing signals (NSL) [15, 16]. Moreover, IL-1α has been reported to contribute to the transcriptional expression of its target genes [17]. On the other hand, EMMPRIN does not have NLS. The intranuclear localizing mechanisms and functions of EMMPRIN are unclear, and merit further studies.
The most important function of EMMPRIN is to induce the production of MMPs [4]. THP1 cells were stimulated with conditioned medium derived from the FW-treated or -untreated HSC3 cells in order to examine whether the EMMPRIN released after FW stimulation was functional. The conditioned medium successfully induced the expression of various MMPs, PDGF and IL-8. Although both EMMPRIN and PDGF contribute to the wound healing, no reports correlating these two factors have been observed. For these reasons, we concentrated on the EMMPRIN-induced PDGF up-regulation. Moreover, the enhancing effect was abolished when the EMMPRIN in the conditioned medium was removed by immunoprecipitation. These results indicated that EMMPRIN exerted its effects on the monocytic cells. The monocyte is one of the most important immune cells in the body. EMMPRIN was able to activate the THP1 cells in the present study, thus fulfilling the fourth criteria of alarmins.
Two different kinds of EMMPRIN receptors have been described, the receptor for advanced glycation end product (RAGE) and toll-like receptor 4 (TLR4) [3]. The expressions of both receptors in THP1 cells were confirmed by reverse transcriptase-PCR (data not shown), and EMMPRIN signal should be transduced by these receptors. Among the genes analyzed, only VEGF was down regulated by EMMPRIN. This observation was inconsistent with previous reports showing EMMPRIN-mediated VEGF induction [18, 19]. Another recent study further demonstrated EMMPRIN-mediated bone-marrow-derived cell recruitment to the cancer region [20]. Although the reasons for the discrepancy between the findings are unknown, it may be noteworthy that the previous studies used lung cancer and endothelial cells. EMMPRIN might exert various effects in a cell type-specific manner. Thus, the downstream signaling pathways leading to MMPs induction or VEGF reduction might be different between cells, and will need to be clarified in more detail in future.
The expression levels of EMMPRIN are strongly associated with the prognosis of cancer [21]. In the present study, FW was shown to induce the secretion of EMMPRIN from the cellular storages of the oral cancer cells. As FW is useful for disinfection and the acceleration of wound healing, it is indispensable to know whether FW-treatment can augment EMMPRIN secretion from normal cells such as fibroblasts or epithelial cells. The results obtained in this study suggested that its clinical use in the field of cancer warrants careful attention.
Supplementary Material
Supplementary figure.
Acknowledgements
This study was supported in part by research grants from Sato Fund from the Nihon University School of Dentistry and a grant from the Dental Research Center, Nihon University School of Dentistry, a Nihon University multidisciplinary research grant and Individual Research Grant and MEXT-Supported Program for the Strategic Research Foundation at Private Universities 2013-2017. Grant-in-Aid for Scientific Research (C) (MEXT).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
2. Basset P, Wolf C, Chambon P. Expression of the stromelysin-3 gene in fibroblastic cells of invasive carcinomas of the breast and other human tissues: a review. Breast Cancer Res Treat. 1993;24:185-193
3. Iacono KT, Brown AL, Greene MI. et al. CD147 immunoglobulin superfamily receptor function and role in pathology. Exp Mol Pathol. 2007;83:283-295
4. Fossum S, Mallett S, Barclay AN. The MRC OX-47 antigen is a member of the immunoglobulin superfamily with an unusual transmembrane sequence. Eur J Immunol. 1991;21:671-679
5. Miyauchi T, Masuzawa Y, Muramatsu T. The basigin group of the immunoglobulin superfamily: complete conservation of a segment in and around transmembrane domains of human and mouse basigin and chicken HT7 antigen. J Biochem. 1991;110:770-774
6. Biswas C, Zhang Y, DeCastro R. et al. The human tumor cell-derived collagenase stimulatory factor (renamed EMMPRIN) is a member of the immunoglobulin superfamily. Cancer Res. 1995;55:434-439
7. Muramatsu T, Miyauchi T. Basigin (CD147): a multifunctional transmembrane protein involved in reproduction, neural function, inflammation and tumor invasion. Histol Histopathol. 2003;18:981-987
8. Sun J, Hemler ME. Regulation of MMP-1 and MMP-2 production through CD147/extracellular matrix metalloproteinase inducer interactions. Cancer Res. 2001;61:2276-2281
9. Kubota A, Goda T, Tsuru T. et al. Efficacy and safety of strong acid electrolyzed water for peritoneal lavage to prevent surgical site infection in patients with perforated appendicitis. Surg Today. 2015;45:876-879
10. Saiki A, Motoyoshi M, Motozawa K. et al. EMMPRIN inhibits bFGF-induced IL-6 secretion in an osteoblastic cell line, MC3T3-E1. Int J Med Sci. 2017;14:1173-1180
11. Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359-365
12. Omagari D, Mikami Y, Suguro H. et al. Poly I: C-induced expression of intercellular adhesion molecule-1 in intestinal epithelial cells. Clin Exp Immunol. 2009;156:294-302
13. Furuta M, Kose S, Koike M. et al. Heat-shock induced nuclear retention and recycling inhibition of importin α. Genes Cells. 2004;9:429-41
14. Brody DT, Durum SK. Membrane IL-1: IL-1 alpha precursor binds to the plasma membrane via a lectin-like interaction. J Immunol. 1989;143:1183-1187
15. Wesndorf JH, Garfinkel S, Zhan X. et al. Identification of a nuclear localization sequence within the structure of the human interleukin-1 alpha precursor. J Biol Chem. 1993;268:22100-22104
16. Maier JA, Statuto M, Ragnotti G. Endogenous interleukin 1 alpha must be transported to the nucleus to exert its activity in human endothelial cells. Mol Cell Biol. 1994;14:1845-1851
17. Werman A, Werman-Venkert R, White R. et al. The precursor form of IL-1α is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci U S A. 2004;101:2434-2439
18. Tang Y, Nakada MT, Rafferty P. et al. Regulation of vascular endothelial growth factor expression by EMMPRIN via the PI3K-Akt signaling pathway. Mol Cancer Res. 2006;4:371-377
19. Bougatef F, Quemener C, Kellouche S. et al. EMMPRIN promotes angiogenesis through hypoxia-inducible factor-2alpha-mediated regulation of soluble VEGF isoforms and their receptor VEGFR-2. Blood. 2009;114:5547-5556
20. Chen Y, Gou X, Kong DK. et al. EMMPRIN regulates tumor growth and metastasis by recruiting bone marrow-derived cells through paracrine signaling of SDF-1 and VEGF. Oncotarget. 2015;6:32575-32585
21. Weidle UH, Scheuer W, Eggle D. et al. Cancer-related issues of CD147. Cancer Genomics Proteomics. 2010;7:157-169
Author contact
![]() Corresponding author: Department of Pathology, Nihon University, School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan. +81-3-3219-8114; asano.masatakeac.jp
Corresponding author: Department of Pathology, Nihon University, School of Dentistry, 1-8-13 Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan. +81-3-3219-8114; asano.masatakeac.jp