Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(12):1320-1328. doi:10.7150/ijms.26774 This issue Cite
Short Research Communication
Estrogen Receptor 1 (ESR1) Enhances Slc2a4/GLUT4 Expression by a SP1 Cooperative Mechanism
João Nilton Barreto-Andrade, Luciana Alves de Fátima, Raquel Saldanha Campello, José Augusto Cipriano Guedes, Helayne Soares de Freitas, Maristela Mitiko Okamoto Ubiratan Fabres Machado ![]()
Departament of Physiology and Biophysics, Institute of Biomedical Sciences, Universidade de São Paulo, São Paulo, Brasil.
Received 2018-4-19; Accepted 2018-7-26; Published 2018-8-10
Abstract
Background: Estrogens are involved in glycemic regulation, playing an important role in the development and/or progression of insulin resistance. For that, estrogens regulate the expression of the glucose transporter protein GLUT4 (codified by the solute carrier family 2 member 4 gene, Slc2a4), thus modulating adipose and muscle glucose disposal. This regulation is a balance between ESR1-mediated enhancer effect and ESR2-mediated repressor effect on Slc2a4 gene. However, molecular mechanisms involved in these effects are poorly understood. Since the specificity protein 1 (SP1) participates in several ESR-mediated genomic regulations, the aim of the present study is to investigate the participation of SP1 in the ESR1/2-mediated regulation of Slc2a4 gene.
Methods: Differentiated 3T3-L1 adipocytes were 24-hour challenged with 10 nM estradiol (E2) and 10 nM ESR1 agonist (PPT) or 100 nM ESR2 agonist (DPN), added or not with E2. Slc2a4 and Sp1 mRNAs (RT-qPCR), total GLUT4 and nuclear ESR1, ESR2 and SP1 proteins (Western blotting), SP1 binding activity into Slc2a4 promoter (EMSA), and nuclear complexation of SP1/ESR1 (immunoprecipitation) were analyzed.
Results: E2 and PPT increased (25-50%) whereas DPN reduced (20-45%) Slc2a4 and GLUT4 expression. Nuclear content of ESR1 and ESR2 remained unchanged. Nuclear content of SP1 increased (50 to 90%) by PPT and DPN added or not with E2; the highest effect observed with PPT alone. PPT also increased the nuclear content of SP1/ESR1 complex and the SP1 binding into the Slc2a4 promoter.
Conclusions: ESR1 activation in adipocytes increased the nuclear content of SP1 protein, the SP1/ESR1 interaction and SP1 binding into the Slc2a4 gene promoter, culminating with increased Slc2a4/GLUT4 expression. No involvement of SP1 seems to occur in ESR2-mediated repressor effect on Slc2a4. We expect that this ESR1/SP1 cooperative effect can contribute to the development of new approaches for prevention or treatment of insulin resistance and diabetes mellitus.
Keywords: Estradiol, PPT, DPN, ESR2, 3T3-L1 adipocytes
Background
Estrogens have been known for their role in the female physiology of reproduction; however, their participation in the physiology of the male reproductive system is a topic of growing importance [1, 2]. Besides, estrogens have been reported to participate in other physiological systems, in both female and male, as the regulation of glycemic homeostasis [3], thus becoming a potential player in the pathophysiology of diabetes mellitus (DM). In females, insulin resistance has been related to alterations in 17β-estradiol (E2) concentrations both in physiological states, such as menstrual cycle phase, pregnancy and menopause [4 -8], and in pathological conditions, such as gestational diabetes mellitus, polycystic ovary syndrome and Turner syndrome [4, 5, 7, 9-11]. Besides, insulin resistance has also been detected in estrogen-deficient men [12, 13]. Interestingly, these conditions of impaired glycemic homeostasis include both hyper and hypoestrogenic states, advancing that estrogen modulation of insulin sensitivity must be a very complex phenomenon.
Insulin resistance is a key mechanism in the physiopathology of type 2 diabetes mellitus (T2DM), and is characterized, among other features, by a reduced capacity of adipose and muscle tissue uptake glucose in response to the hormone [14, 15]). In this process, the insulin-regulatable glucose transporter GLUT4 (solute carrier family 2, facilitated glucose transporter member 4), codified by the Slc2a4 gene (solute carrier family 2 member 4 gene), plays an important role [14, 15], and regulation of Slc2a4/GLUT4 expression has become a target for prevention or treatment of insulin resistance [16]. Considering the above, we hypothesized that estrogen-related regulation of glycemic homeostasis would involve regulation of Slc2a4/GLUT4 expression.
The most important biological effects of estrogens are mediated by action of estrogen receptors ESR1 and ESR2, formers ER-alpha and ER-beta, respectively. ESRs can act by: 1) direct binding to palindromic sequences, the estrogen-responsive element (ERE), in the DNA of target genes [17-19]; 2) interaction with other transcriptional factors, which bind and regulate transcription of target genes [17-20]; and 3) extra-nuclear activation of several cellular signaling events, such as calcium mobilization, nitric oxide production and MAPK, SRC or AKT pathways [21-23].
Remarkable studies in transgenic mice have demonstrated that ESR1 is an enhancer whereas ESR2 is an repressor of Slc2a4/GLUT4 expression in muscle and adipose tissues [24-27]. Furthermore, some studies have demonstrated that ESR1 plays a dominant effect on adipocytes, whereas ESR2 plays a dominant effect on skeletal muscle [26, 28-30]. Consequently, E2 increases and decreases Slc2a4/GLUT4 expression in adipocytes and muscle cells, respectively [28, 29, 31].
However, despite some studies reporting E2-induced and ESRs-mediated regulation of Slc2a4 gene expression, the molecular mechanisms through which this regulation occurs are still obscure. Importantly, the Slc2a4 promoter contains neither the consensus palindromic estrogen responsive element (ERE) sequence AGGTCANNNTGACCT, nor a highly homologous domain. This suggests that ESRs regulation of Slc2a4 expression should involve non-classical genomic pathway, such as the ESR/SP1 (specificity protein 1) cooperative mechanism, in which ESR activation triggers the SP1 binding and regulation of target genes [20]. SP1 is a transcription factor member of the Krüppel-like family, which has been described to bind into a consensus sequence 5′-(G/T)GGGCGG(G/A)(G/A)(C/T)-3′ in target genes [20]. Regarding that, a SP1 binding site was identified in the Slc2a4 promoter, through which the SP1 triggers an enhancing effect on Slc2a4 transcription [32]. Thus, considering that direct ESR activation of Slc2a4 gene is unlikely, we hypothesized that E2-induced regulations could be indirectly mediated by the ESR/SP1 interaction. In this paper, we demonstrated in adipocytes 3T3L1 that activation of ESR1 increases nuclear SP1 content, the SP1/ESR1 interaction and SP1 binding into the Slc2a4 promoter. Collectively, the results reveal an indirect mechanism through which E2 enhances the Slc2a4/GLUT4 expression, contributing to the improvement of glycemic homeostasis. We expect that this regulation will contribute to determining new targets for the regulation of Slc2a4 expression, which could contribute to the prevention or treatment of insulin resistance and diabetes mellitus.
Methods
3T3-L1 Cell Culture
Mouse 3T3-L1 fibroblasts [American Type Culture Collection, Rio de Janeiro Cell Bank, Rio de Janeiro, RJ, Brazil (ATCC® Number: CL-173TM)] were propagated and differentiated as previously described [28-30]. Differentiated cells were starved in DMEM, without phenol red (Sigma-Aldrich, St Louis, MO, USA) and with dextran carbon-treated 10% FBS (Sigma-Aldrich, St Louis, MO, USA) for 24 hours prior to treatment. Cells were subjected to differentiation at passages 8 to 10.
Cell treatments
Differentiated adipocytes were cultivated for 24 hours in the absence of any stimulus (control, C) or in the presence of 1) 10 nM of water-soluble 17β-estradiol (E4389; Sigma-Aldrich, St Louis, MO, USA) (10E2); 2) 10 nM of e ESR1 agonist [4,4',4''-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol] - PPT (H6036, Sigma-Aldrich, St. Louis, MO, USA); or 3) 100 nM of ESR2 agonist- DPN (1494, Tocris, St. Louis, MO, USA) [28, 29]. PPT and DPN were additionally used in the presence of 10 nM E2 in some experiments, as indicated. Besides, PPT was solubilized in dimethyl sulfoxide (DMSO), and thus, DMSO was added to other media that contained no PPT.
mRNA analysis by RT-qPCR
RNA was reverse-transcribed (RT) using the ImProm-II® Reverse Transcription System (Promega Corporation, Madison, WI, USA). Gene expression was analyzed by real-time polymerase chain reaction (qPCR), carried out in a StepOne Plus Instrument (Applied Biosystems Inc., Foster City, CA, USA). Slc2a4 and Gapdh expression was determined using TaqMan® PCR master mix kit (Applied Biosystems Inc., Foster City, CA, USA), assay identifiers ID: Mm01245502_m1 and Mm99999915_g1. Sp1 gene expression was determined by RT-qPCR using Platinum® SYBR® Green qPCR SuperMix UDG (TermoFisher Scientific) and using the following primers: Sp1 forward: GGCTACCCCTACCTCAAAGGA and reverse: TTGGAAGACTCGCTGCCATT; Gapdh forward: GAAGGTCGGTGTGAACGGAT reverse: AAGACACCAGTAGACTCCACGA.
Protein analysis by Western blotting
Total cellular GLUT4 content was measured in cells homogenate, using an anti-GLUT4 antibody (07-1404, EMD Millipore, Billerica, MA, USA), as previously described [28, 29].
Nuclear protein extract was obtained from treated cells as previously described [32, 34], and used for quantification of SP1, ESR1 and ESR2 proteins as previously described (29, 30, 34]. Primary anti-SP1 (# 5931, Cell Signaling), anti-ESR1 (#06-935, Millipore) and anti-ESR2 (#sc-6821, Santa Cruz Biotechnology Inc.) antibodies were used, followed by the appropriate secondary antibody and final chemiluminescence analysis. Blots were quantified by optical densitometry (ImageScanner III, GE Healthcare, Uppsala, Sweden). Protein-loaded normalization was undertaken by analyzing the Ponceau-stained membrane, and results were further normalized considering the mean of control values as 1.0.
SP1/ESR1 nuclear complex analysis
Nuclear interaction of SP1/ESR1 was analyzed by SP1 immunoprecipitation followed by ESR1 immunodetection in samples of cells treated with ESR1 agonist PPT. Briefly, 400 μg of nuclear protein (prepared as described above) was incubated with 5 μL of anti-SP1 antibody (# 5931, Cell Signaling), at 4 oC, for 24 hours; thus, protein A-Sepharose ® (P3391, SIGMa, Merk KGaA, Darmstadt, Germany) was added, following an additional 24-hour incubation at 4 oC. After that, the antibody/protein complexes were precipitated by centrifugation (1,000 x g, 2 min) and washed 4 times. The pellet was used for measurement of ESR1 protein by Western blotting as described above. Results were normalized considering the mean of control values as 1.0.
SP1/Slc2a4 DNA binding activity
SP1 protein binding into the Slc2a4 gene promoter was analyzed by electrophoretic mobility shift assay (EMSA), as previously described [28]. Nuclear proteins were extracted from treated cells as described above. The oligonucleotide used reproduces the -149/-125 segment of mouse Slc2a4 promoter sequence, highlighting that this segment contains the SP1-binding-site previously characterized in rat and human genes [32]. Position of the segment is in accordance with the transcriptional start site of the mouse Slc2a4 promoter sequence described in www.ensembl.org.
Double-stranded oligonucleotide was end-labeled with T4 polynucleotide kinase (Invitrogen Life Technologies, Carlsbad, CA, USA) and γ-32P-ATP (PerkinElmer Life, Waltham, MA, USA) in a final binding buffer [60 mM HEPES, pH 7.6, 150 mM KCl, 10% glycerol, 0.6 mM EDTA, 1.93 mg/mL, bovine serum albumin, 2.3 mM dithiothreitol, and 0.25 μg polydeoxyinosinicdeoxycytidylic acid (polydI-dC)] (GE Health Care Life Sciences), for 20 min at room temperature. Competition binding assays were conducted with the addition of 100-fold molar of unlabeled oligonucleotide or 5 μL anti-SP1 antibody (# 5931, Cell Signaling). The DNA-protein complexes were electrophoresed on 4% non-denaturing polyacrylamide gel. The gel was dried, subjected to autoradiography and their blots were quantified by optical densitometry (ImageScanner III, GE Healthcare, Uppsala, Sweden). Results were expressed as arbitrary units, considering the mean of control values as 1.0.
Data analysis
Results were expressed as mean ± standard error of mean (SEM). Means were compared by one-way ANOVA (Tukey's post-test) or by unpaired two tailed Student t-test, as appropriate, after confirming the normality of the data distribution (Shapiro-Wilk test). P values ≤ 0.05 were considered statistically significant.
Results
ESR1 stimulates E2-induced expression of Slc2a4/GLUT4
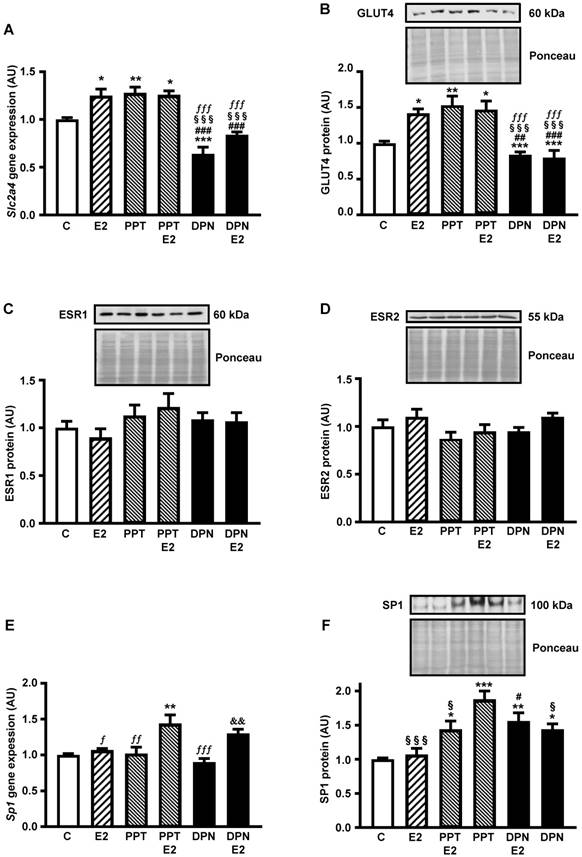
Figures 1A and 1B show that the expression of both Slc2a4 mRNA and GLUT4 protein increases in response to E2 (by 30% and 40%, respectively; P<0.05 vs. control). Besides, the results show that PPT (ESR1 agonist) increases (similarly to E2; P<0.01 vs. control), whereas DNP (ESR2 agonist) decreases the Slc2a4/GLUT4 expression (by 35% and 20%, respectively; P<0.001 vs. control), confirming a preponderant effect of ESR1 in adipocytes. Furthermore, addition of 10 nM E2 to PPT or DPN did not change the isolated effect of the agonists, indicating that their effects were maximal.
ESR1 activity increases Slc2a4/GLUT4 expression and nuclear content of SP1 in adipocytes Adipocytes (3T3-L1) were treated (24 hours) with no stimulus (C), with estradiol (E2), ESR1 agonist alone (PPT) or with E2 (PPT+E2), and ESR2 agonist alone (DPN) or with E2 (DPN+E2). Slc2a4 mRNA (A), total cellular GLUT4 protein (B), nuclear ESR1 (C), nuclear ESR2 (D), Sp1 mRNA (E) and nuclear SP1 (F) contents were measured. For each protein analyzed, a representative immunoblot and respective Ponceau stained membrane are shown; lanes are in the same sequence of the graph bars. Data are means ± SEM of 5 different samples, compared by one-way-ANOVA, followed by Tukey's post-test, after to confirm the normality of the data distribution by the Shapiro-Wilk test. *P<0.05, **P<0.01 and ***P<0.001 vs C; #P<0.05, ##P<0.01 and ###P<0.001 vs E2; §P<0.05 and §§§P<0.001 vs PPT; fP<0.05. ffP<0.01 and fffP<0.001 vs PPT+E2; &&P<0.01 vs DPN.
E2 does not alter nuclear content of ESR1 or ESR2
Once we determined the participation of ESR1 and ESR2 on E2-induced regulation of the Slc2a4/GLUT4 expression, we examined whether this participation was a consequence of changes in nuclear content of ESRs. The results revealed that neither the nuclear content of ESR1 (Figure 1 C) nor the nuclear content of ESR2 (Figure 1D) was altered after treatment with E2 and or the selective agonists.
ESR1 and ESR2 modulate Sp1 gene expression and nuclear content of SP1 protein
Figure 1E shows that PPT+E2 enhanced Sp1 mRNA (by 45%; P<0.01 vs. control); however, curiously, neither PPT nor E2 alone was capable of altering Sp1 expression. Besides, comparison between DPN and DPN+E2 showed a statistical difference (45%; P<0.01), although these groups were not different from either C or E2 groups. Differently, PPT and DPN, added or not with E2, increased the nuclear content of SP1 (from 44% to 88%; P<0.05 to P<0.001 vs. control); however, E2 alone did not alter the SP1. Interestingly, PPT alone induced the largest increase, significantly different from the increase of PPT+E2 (30% higher; P<0.05).
Slc2a4 gene promoter has a SP1-binding site close to putative ESR-binding sites
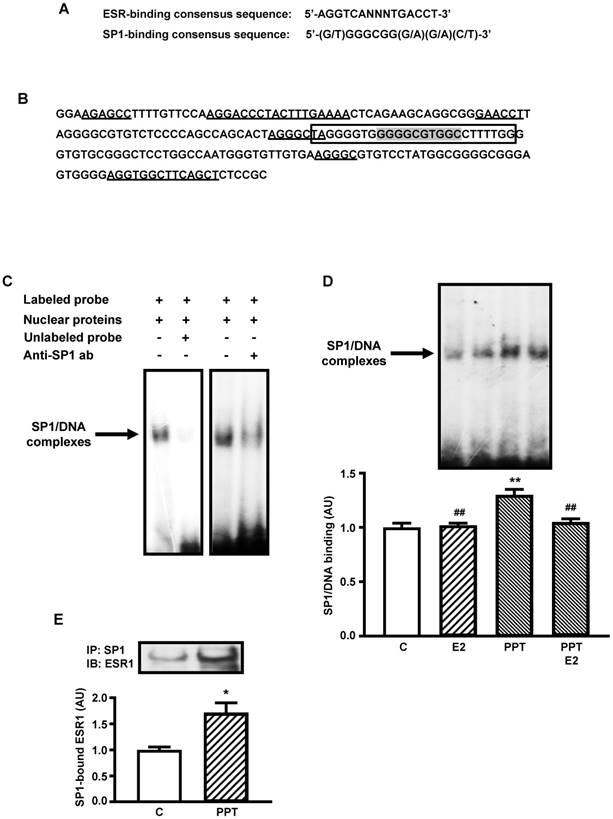
Figure 2A shows the consensus sequence of SP1- and ESR-binding sites [20, 32, 34] used for analyses in the present study. Figure 2B shows a 200 nucleotides segment of mouse Slc2a4 promoter, which contains the SP1 responsive element [32] analyzed in the present study. Several sequences are indicated in the segment, such as: the oligonucleotide constructed for EMSA analysis containing the core of the SP1-binding site, 2 sequences homologous to the complete (palindromic) ESR-binding site, 3 sequences homologous to the first half-site of the ESR-binding site and 1 sequence homologous to the second half-site of the ESR-binding site. These data reveal how this SP1-binding site is surrounded by several putative ESRs-binding sites.
ESR1-mediated stimulation of Slc2a4 gene transcription involves a SP1's cooperative mechanism. A: ESR-binding and SP1-binding consensus sequences [20, 35]. B: -239/-40 segment of Slc2a4 promoter depicting: the -149/-125 sequence used for EMSA analysis (in the box), containing the SP1-binding site (shaded); and 2 large sequences homologous to the complete (palindromic) ESR-binding site, 3 short sequences homologous to the first half-site of the ESR-binding site and 1 short sequence homologous to the second half-site of the ESR-binding site (underlined). C: EMSA analysis of SP1 binding into the -149/-125 segment of Slc2a4 gene promoter. D: SP1 binding activity into Slc2a4 promoter measured in 3T3-L1 cells 24-hour treated in culture medium alone (C) or supplemented with estradiol (E2), ESR1 selective agonist (PPT) or both (PPT+E2). At the top, representative experiment shows blots of the SP1/DNA complexes, in the same sequence of the graph bars. Data are means ± SEM of 5 different samples, compared by one-way-ANOVA, followed by Tukey's post-test, after to confirm the normality of the data distribution by the Shapiro-Wilk test. **P<0.01 vs C and ##P<0.01 vs E2. E: ESR1 protein measured by Western blotting in SP1-immunoprecipitated nuclear proteins from 3T3-L1 cells after 24-hour treatment in culture medium alone (C) or supplemented with ESR1 selective agonist (PPT). At the top representative images corresponding to the graph bars. Data are means ± SEM of 4 control and 6 PPT samples, compared by unpaired two-tailed Student t-test. *P<0.05 vs C.
ESR1 stimulates SP1 binding activity into Slc2a4 gene promoter
Figure 2C shows typical image of nuclear proteins binding into the SP1 binding site of the Slc2a4 gene promoter (EMSA), using the oligonucleotide indicated in Figure 2B. Competition with 100-fold molar unlabeled probe depicts the specificity of the protein/DNA complex. Besides, reduction in the binding activity observed with addition of anti-SP1 antibody confirmed the presence of SP1 protein in the complex.
Considering that only the ESR1-mediated regulation of Slc2a4 expression could involve the participation of SP1, EMSA analysis was performed in cells treated with E2, PPT or both. Figure 2D shows that only PPT treatment induced a significant increase in the SP1 binding activity (by 30%; P<0.01 vs control, E2 and PPT+E2), although E2 and PPT+E2 have shown a slight tendency to increase as well.
Finally, to confirm whether this PPT-induced increase in SP1 binding activity could be related to a molecular interaction between ESR1/SP1, SP1 protein was immunoprecipitated from nuclear protein extract, and the immunoprecipitated samples were subjected to ESR1 quantification by Western blotting. Figure 2E shows that PPT treatment significantly increased (72%; P=0.0388) the amount of ESR1 arrested by SP1 immunoprecipitation.
Discussion
Estrogen has been proposed as a modulator of the glycemic homeostasis, and thus involved in the pathophysiology of obesity, insulin resistance and diabetes [3]. That is related to the estrogen capacity to modulate muscle and adipose tissues glucose disposal, an effect in which the GLUT4 plays an important role. In fact, estrogen-induced regulation of GLUT4 protein expression has been reported as a balance between ESR1-mediated increase and ESR2-mediated decrease of Slc2a4/GLUT4 expression [25, 27]. However, the mechanisms by which estrogen receptors trigger those inverse effects are still unknown. In this study, we determined the participation of SP1 protein in ESR1/2-mediated regulation of Slc2a4 gene expression in adipocytes. We demonstrated that ESR1 activation increases the nuclear content of SP1 protein, the SP1/ESR1 interaction and the SP1 binding into the Slc2a4 gene promoter. No involvement of SP1 seems to occur in ESR2-mediated repressor effect on Slc2a4 gene.
Firstly, we investigated the expression of Slc2a4/GLUT4. PPT (ESR1 agonist) increased whereas DPN (ESR2 agonist) decreased Slc2a4/GLUT4 expression; besides, E2 also increased the expression, revealing the preponderant effect of ESR1 in adipocytes. Predominance of ESR1 in nucleus of adipose cells has already been described (Dieudonne et al., 2004, Barros et al., 2009), explaining that the effect of E2 follows the ESR1 effect. Besides, those regulations of the GLUT4 have already been confirmed to be accompanied by parallel regulations of cellular glucose uptake [28], thus evincing a mechanism by which E2 can modulate glycemic homeostasis.
ESRs-induced effects may be determined by changes in the amount of receptors. Although 24-hour E2 treatment was shown to increase Esr1 mRNA [30], the present results depict that nuclear content of ESR1 and ESR2 was unchanged in response to E2 and/or to their selective agonists. Recently, it was demonstrated by immunocytochemistry that 24-hour E2 can induce a clear translocation of ESR1 from nucleus to the plasma membrane [29]. Whether this different result is or not a consequence of distinct methodological approaches must be determined. Importantly, the present study aimed to demonstrate the potential ESR/SP1 mediated regulation of Slc2a4 expression; thus, we must focus on nuclear content of ESR proteins.
The SP1 gene and protein expression were also analyzed. Although only PPT+E2 increased the Sp1 mRNA expression, the nuclear content of SP1 protein was increased by both PPT and DPN, added or not with E2. Similar discrepancies between Sp1 mRNA expression and SP1 protein content were already described in HepG2 cell and related to a PI3K/AKT-mediated stabilization of SP1 protein [36], highlighting that activation of this pathway has already been observed in 3T3-L1 adipocytes in response to ESR1 activation [29]. That could explain the PPT effect here observed in nuclear content of SP1; however, the absence of E2 effect and the positive effect of DPN lack appropriate explanation at present. Importantly, considering that DPN is a repressor of Slc2a4 and it increased the nuclear content of SP1 (classic Slc2a4 enhancer), it seems clear that the ESR2-mediated effect must not be related to the SP1 cooperative effect, as ESR1 seems to be.
Both ESR1 and ESR2 may regulate expression of target genes by binding to promoter regions containing palindromic consensus of estrogen responsive element (ERE) AGGTCANNNTGACCT, imperfect ERE sequences, or preserved or not ERE half sites [20, 35, 37]. The ERE half sites became more and more important, since the demonstration that they can participate in the novel genomic mechanism of ERE-dependent transactivation, including cooperative ESR1/SP1 interaction [20]. In this process, SP1/ESR1 interact and bind each one to their responsive element, which includes similar ERE half-sites [20]. In several E2-responsive genes, this interaction was characterized as involving SP1 and ESR1 motifs close to each other, separated by 3 to 68 nucleotides [20]. In the Slc2a4 gene promoter, a well-documented SP1 binding site [32] is located at -141/-132 segment, and nearby this SP1 domain there are two whole EREs, three first half sites of ERE and one last half site of ERE (Figure 2B). All these domains display 67% similarity to the consensus sequence; and half-sites similarities can increase when compared to other half-sites already functionally reported [20]. Furthermore, these domains are located 8 to 88 nucleotides far from the SP1RE (Figure 2B), and thus the proximity between the SP1RE and the EREs make the SP1/ESR1 cooperativity highly probable in the Slc2a4 gene regulation.
Considering that only the ESR1 enhancer effect upon Slc2a4 gene expression might be involving the SP1 cooperativity, we continued the study focusing on the ESR1 activity. EMSA analysis reveals that PPT alone increased the SP1 binding into Slc2a4 promoter, and that must participate in the ESR1 enhancer effect on Slc2a4 expression. However, regarding SP1/DNA binding, unexpected responses were also observed such as: 1) E2 did not alter the SP1 binding, despite the isolated effect of PPT; and 2) the PPT effect disappeared when E2 was added.
Concerning the first unexpected effect, divergent responses to E2 and PPT, in cells that primarily express the ESR1, have already been reported in vivo, in cerebral areas related to feeding behavior [38], and in vitro, in HepG2 cell proliferation regulation by leptin [39]. Although the discrepancy was not discussed in the HepG2 cell study, in the feeding behavior study it was related to time after E2 or PPT stimulation (6 or 24 hours), and some mechanisms were hypothesized: 1) accessibility to nuclear compartment, 2) distinct capacity of E2 or PPT to trigger signals related to plasma membrane associated to ESR1 or to GPER1 (G protein coupled estrogen receptor 1), and 3) activation of intracellular pathways that lead to genomic effects [38]. Besides, distinct capacity of E2 or PPT to induce ESR1 nuclear exclusion should also be considered. Additionally, we must consider that E2 effects are currently described as dose-dependent [31, 38-40], and the doses of E2 and PPT used [41] may not trigger the exact same grade of ESR1 activation in the 3T3-L1 adipocytes. Thus, both modulators of biological effect (time and dose) may be involved in the present response, and we can hypothesize that handling cell treatment based on dose and/or time, the effects of E2 and PPT might be equal.
The second unexpected response regards the disappearance of the increased PPT-induced SP1 binding with the addition of E2, highlighting that E2 alone has no effect. At first sight, we might have supposed that E2 introduced an ESR2-mediated opposite effect, as described for the NFKB (nuclear factor NF-kappa-B) binding into the Slc2a4 promoter [29]. However, the data concerning ESR2 effect upon SP1 regulation do not support that supposition: firstly, because nuclear content of SP1 also increased in response to DPN (ESR2-agonist); and, secondly, because preliminary analysis suggests that DPN treatment did not alter SP1 binding into the Slc2a4 (data not shown). Thus, ESR2-mediated effect on nuclear regulation of SP1 may be important to other E2-target genes, but not to Slc2a4.
In summary, the EMSA results reveal that PPT-induced ESR1 activity stimulates SP1 binding activity, although this effect, as discussed above, could not be observed in response to E2 based on dose and time studied here. In fact, in breast cancer cells transfected with SP1 and ESR1, both E2 and the antiestrogens tamoxifen and fulvestrant are able to induce reporter gene activity of E2-responsive genes [20]. Those observations indicate that the ESR1/SP1 interactions can really promote unexpected and even contrary regulations.
Once the EMSA analysis indicated that PPT simulates the SP1 binding into Slc2a4 promoter, and that can be related to cooperative effect between SP1 and ESR1, we checked whether ESR1 and SP1 were complexing in nucleus of PPT treated cells. In fact, the preceding SP1 immunoprecipitation of nuclear proteins, followed by immunodetection of ESR1, confirmed that these proteins are complexed; besides, the amount of this complex increased after 24-hour treatment with PPT. This result reinforces the potential cooperativity of SP1/ESR1 in response to activation of ESR1.
Conclusions
In this study, we demonstrated in 3T3L1 adipocytes that activation of ESR1 increases nuclear SP1 content, SP1/ESR1 interaction and SP1 binding into the Slc2a4 promoter. Collectively, the results reveal a novel mechanism through which E2 enhances Slc2a4/GLUT4 expression. We hope this novel ESR1-induced regulation of Slc2a4 gene expression will help determine new targets for the development of approaches, contributing to the prevention or treatment of insulin resistance and diabetes mellitus.
Abbreviations
AKT: RAC-alpha serine/threonine-protein kinase
ANOVA: analysis of variance
DM: diabetes mellitus
DMSO: dimethyl sulfoxide
DPN: [2,3-bis(4-hydroxyphenyl)-propionitrile]
E2: 17beta-estradiol
EDTA: ethylenediamine tetraacetic acid
EMSA: electrophoretic mobility shift assay
ERE: estrogen responsive element
ESR1: estrogen receptor 1 (former ER alpha)
ESR2: estrogen receptor 2 (former ER beta)
Gapdh: glyceraldehyde-3-phosphate dehydrogenase gene (rat)
GLUT4: solute carrier family 2, facilitated glucose transporter member 4
GPER1: G protein coupled estrogen receptor 1
HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
MAPK: mitogen-activated protein kinase
NFKB: nuclear factor NF-kappa-B
PI3K: PI-3 kinase
PPT: [4,4',4''-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol]
Slc2a4: solute carrier family 2 member 4 gene (rat)
Sp1: specific protein 1 gene (rat)
SP1: specific protein 1
SP1RE: SP1 responsive element
SRC: proto-oncogene tyrosine-protein kinase Src
T2DM: type 2 diabetes mellitus
Acknowledgements
The authors thank Dr. Adauri Brezolin for English revision of the manuscript.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Funding
This research was supported by grant from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) #2016/15603-0. João N. Barreto-Andrade is a recipient of FAPESP scholarship (#2015/18715-1).
Author contributions
JNBA, JACG, MMO and HSF conducted the experiments. JNBA, RSC, LAF and UFM proposed the research hypothesis, designed the experiments. JNBA, LAF and UFM discussed and analyzed and analyzed the results. UFM wrote and revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hess RA, Bunick D, Lee KH, Bahr J, Taylor JA, Korach KS, Lubahn DB. A role for oestrogens in the male reproductive system. Nature. 1997;390:509-512
2. Hess RA, Carnes K. The role of estrogen in testis and the male reproductive tract: a review and species comparison. Anim Reprod. 2004;1:5-30
3. Meyer MR, Clegg DJ, Prossnitz ER, Barton M. Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol. 2011;203:259-269
4. Buchanan TA, Metzger BE, Freinkel N, Bergman RN. Insulin sensitivity and B-cell responsiveness to glucose during late pregnancy in lean and moderately obese women with normal glucose tolerance or mild gestational diabetes. Am J Obstet Gynecol. 1990;162:1008-1014
5. Okuno S, Akazawa S, Yasuhi I, Kawasaki E, Matsumoto K, Yamasaki H, Matsuo H, Yamaguchi Y, Nagataki S. Decreased expression of the GLUT4 glucose transporter protein in adipose tissue during pregnancy. Horm Metab Res. 1995;27:231-234
6. Solomon CG, Hu FB, Dunaif A, Rich-Edwards J, Willet WC, Hunter DJ, Colditz GA, Speizer FE, Manson JE. Long or highly irregular menstrual cycles as a marker for risk of type 2 diabetes mellitus. JAMA. 2001;286:2421-2426
7. Kaaja RJ, Greer IA. Manifestations of chronic disease during pregnancy. JAMA. 2005;294:2751-2757
8. Saucedo R, Basurto L, Zarate A, Martínez C, Hernandez M, Galván R. Effect of estrogen therapy on insulin resistance and plasminogen activator inhibitor type 1 concentrations in postmenopausal women. Gynecol Obstet Invest. 2007;64:61-64
9. Case AM, Reid RL. Menstrual cycle effects on common medical conditions. Compr Ther. 2001;27:65-71
10. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38:1165-1174
11. Gravholt CH, Nyholm B, Saltin B, Schmitz O, Christiansen JS. Muscle fiber composition and capillary density in Turner syndrome: evidence of increased muscle fiber size related to insulin resistance. Diabetes Care. 2001;24:1668-1673
12. Faustini-Fustini M, Rochira V, Carani C. Oestrogen deficiency in men: where are we today? Eur J Endocrinol. 1999;140:11-129
13. Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, specker B, Williams TC, Lubahn DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056-61
14. Smith U, Kahn BB. Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J Intern Med. 2016;280:465-475
15. Karlsson HK, Zierath JR. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem Biophys. 2007;48:103-113
16. Corrêa-Giannella ML, Machado UF. SLC2A4 gene: a promising target for pharmacogenomics of insulin resistance. Pharmacogenomics. 2013 doi:14:847-850
17. Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science. 1997;277:1508-1510
18. Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S. Ligand-, cell-, and estrogen receptor subtype (alpha/beta)-dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem. 2000;275:5379-5387
19. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Anderrsson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol. Rev. 2001;81:1535-1565
20. Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008;41:263-275
21. Burns KA, Li Y, Arao Y, Petrovich RM, Korach KS. Selective mutations in estrogen receptor alpha D-domain alters nuclear translocation and non-estrogen response element gene regulatory mechanisms. J Biol Chem. 2011;286:12640-12649
22. Levin ER, Hammes SR. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol. 2016;17:783-797
23. Vrtacnik P, Ostanek B, Mencej-Bedrac S, Marc J. The many faces of estrogen signaling. Biochemia Medica. 2014;24:329-342
24. Barros RP, Machado UF, Warner M, Gustafsson JA. Muscle GLUT4 regulation by estrogen receptors ERβ and ERα. Proc Natl Acad Sci USA. 2006;103:1605-1608
25. Barros RP, Machado UF, Gustafsson JA. Estrogen receptors: new players in diabetes mellitus. Trends Mol Med. 2006;12:425-431
26. Barros RP, Gabbi C, Morani A, Warner M, Gustafsson JA. Participation of ERalpha and ERbeta in glucose homeostasis in skeletal muscle and white adipose tissue. Am J Physiol Endocrinol Metab. 2009;297:124-33
27. Barros RP, Gustafsson JA. Estrogen receptors and the metabolic network. Cell Metab. 2011;14:289-299
28. Campello RS, Alves-Wagner AB, Lucas TF, Mori RC, Furuya DT, Porto CS, Machado UF. Estrogen receptor 1 agonist PPT stimulates Slc2a4 gene expression and improves insulin-induced glucose uptake in adipocytes. Curr Top Med Chem. 2012;12:2059-2069
29. Campello RS, Fatima LA, Barreto-Andrade JN, Lucas TG, Mori RC, Porto CS, Machado UF. Estradiol-induced regulation of GLUT4 in 3T3-L1 cells: involvement of ESR1 and AKT activation. J Mol Endocrinol. 2017;59:257-268
30. Fatima LA, Campello RS, Santos RS, Freitas HS, Frank AP, Machado UF, Clegg DJ. Estrogen receptor 1 (ESR1) regulates VEGFA in adipose tissue. Scientific Reports. 2017 DOI:10.1038/s41598.017-16686-7
31. Barros RP, Morani A, Moriscot A, Machado UF. Insulin resistance of pregnancy involves estrogen-induced repression of muscle GLUT4. Mol Cell Endocrinol. 2008;295:24-31
32. Im SS, Kwon SK, Kang SY, Kim TH, Kim HI, Hur MW. Regulation of GLUT4 gene expression by SREBP-1c in adipocytes. Biochem J. 2006;399:131-139
33. Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499
34. Okamoto MM, Anhê GF, Sabino-Silva R, Marques MF, Freitas HS, Mori RC, Melo KF, Machado UF. Intensive insulin treatment induces insulin resistance in diabetic rats by impairing glucose metabolism-related mechanisms in muscle and liver. J. Endocrinol. 2011;211:55-64
35. Liu D, Deschamps A, Korach KS, Murphy E. Estrogen-enhanced gene expression of lipoprotein lipase in heart is antagonized by progesterone. Endocrinology. 2008;149:711-716
36. Chen XH, Lu LL, Ke HP, Liu ZC, Wang HF, Wei W, Qi YF, Wang HS, Cai SH, Du J. The TGF-β-induced up-regulation of NKG2DLs requires AKT/GSK-3β-mediated stabilization of SP1. J Cell Mol Med. 2017;21:860-870
37. Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor alpha to estrogen-responsive elements. J Biol Chem. 2010;285:2676-2685
38. Thammacharoen S, Geary N, Lutz TA, Ogawa S, Asarian L. Divergent effects of estradiol and the estrogen receptor-α agonist PPT on eating and activation of PVN CRH neurons in ovariectomized rats and mice. Brain Res. 2009;1268:88-96
39. Shen M, Shi H. Estradiol and Estrogen Receptor Agonists Oppose Oncogenic Actions of Leptin in HepG2 Cells. PLoS ONE. 2016;11:doi 10.1371/journal.pone.0151455
40. Nagira K, Sasaoka T, Wada T, Fukui K, Ikubo M, Hori S, Tsuneki H, Saito S, Kobayashi M. Altered subcellular distribution of estrogen receptor alpha is implicated in estradiol-induced dual regulation of insulin signaling in 3T3-L1 adipocytes. Endocrinology. 2006;147:1020-1028
41. Garrido P, Morán J, Alonso A, González S, González C. 17β-estradiol activates glucose uptake via GLUT4 translocation and PI3K/Akt signaling pathway in MCF-7 cells. Endocrinology. 2013;154:1979-1989
Author contact
![]() Corresponding author: Ubiratan Fabres Machado (ubiratanusp.br). Department of Physiology and Biophysics, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brasil. Av. Prof. Lineu Prestes 1524, 05508-900, São Paulo (SP), Brasil
Corresponding author: Ubiratan Fabres Machado (ubiratanusp.br). Department of Physiology and Biophysics, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brasil. Av. Prof. Lineu Prestes 1524, 05508-900, São Paulo (SP), Brasil