International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907
- Current Issue
- Volume 22; 2025
- Volume 21; 2024
- Volume 20; 2023
- Volume 19; 2022
- Volume 18; 2021
- Archive
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(11):1118-1128. doi:10.7150/ijms.24460 This issue Cite
Research Paper
Drug repurposing: Ibrutinib exhibits immunosuppressive potential in organ transplantation
Qing Zhang1,#, Jicheng Chen1,#, Hanchao Gao2,1,#, Song Zhang3, Chengjiang Zhao1,2, Cuibing Zhou1, Chengjun Wang1, Yang Li4, Zhiming Cai1, Lisha Mou1, ![]()
1. Shenzhen Xenotransplantation Medical Engineering Research and Development Center, Institute of Translational Medicine, Shenzhen Second People's Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen University School of Medicine, Shenzhen University Health Science Center, Shenzhen, China
2. Shenzhen Longhua District Central Hospital, Shenzhen, China
3. The Department of Anesthesiology, Weifang Medical University, Weifang, China
4. School of Information Science and Engineering, Shandong Agricultural University, Tai'an, China
# These authors contributed equally to the work.
Received 2017-12-20; Accepted 2018-4-12; Published 2018-7-13
Citation:
Zhang Q, Chen J, Gao H, Zhang S, Zhao C, Zhou C, Wang C, Li Y, Cai Z, Mou L. Drug repurposing: Ibrutinib exhibits immunosuppressive potential in organ transplantation. Int J Med Sci 2018; 15(11):1118-1128. doi:10.7150/ijms.24460. https://www.medsci.org/v15p1118.htm
Other stylesAbstract
Long-term administration of classic immunosuppressants can induce severe adverse effects. The development of novel immunosuppressants confronts great challenges and opportunities. Ibrutinib, an approved drug for B-cell lineages and chronic graft versus host disease (cGVHD), exhibits immunosuppressive efficacy in autoimmune diseases. Ibrutinib's potential as an immunosuppressant in organ transplantation has not been investigated to date. In a xeno-artery patch model ex vivo, ibrutinib inhibited the proliferation of PBMCs (POD 14-42), mainly CD3+CD4+ and CD3+CD8+ T cells ex vivo. The secretion of cytokines (IL-6, IL-2 and IFN-γ) was suppressed in response to ibrutinib. In allo-skin transplantation models, ibrutinib delayed the rejection of grafted skins. Ibrutinib decreased the amount of T/B cells and lymphocyte infiltration. Altogether, ibrutinib exhibited immunosuppressive potential through cytokine regulation and T cell inhibition ex vivo and in vitro. Repositioning of ibrutinib as an immunosuppressant will greatly facilitate novel immunosuppressant development.
Keywords: Ibrutinib, Immunosuppressant, Immune rejection, Allo-transplantation, Xeno-transplantation
Introduction
Immune rejection after organ transplantation usually results from the innate immune system[1] (the complement system[2]) and T-cell mediated immune rejection[3]. Except for surgical manipulation, long-term administration of immunosuppressants is necessary to alleviate immune rejection (especially T-cell mediated immune rejection)[4]. However, numerous adverse effects (such as nephrotoxicity, malignancies, and autoimmune imbalance) are not negligible[5-7]. It is urgent to develop novel immunosuppressive therapies with high efficacy and favorable safety profiles. Small molecule immunosuppressants targeting key biological molecules (such as sphingosin-1-phosphate receptor (S1P)[8-10], mTORs[11, 12], kinases[13-19], and HDACs[20]) have drawn much attention in drug discovery.
Kinases are promising drug targets in the treatment of malignancies[21, 22]. However, the study of kinase inhibitors in autoimmune disease and other immune disorders remains rare. To date, several kinase inhibitors (Supplementary Table 1) have attracted attention in the field of organ transplantation[14, 16, 17]. These kinase inhibitors have exhibited potent immunosuppressive effects in organ transplantation. Tec kinases ITK and RLK are important regulators of the development of CD4+ and CD8+ T cells, which were demonstrated in ITK- and RLK/ITK-deficient mice[23].
Ibrutinib is an irreversible inhibitor of Bruton's tyrosine kinase (Btk)[24, 25] and IL-2 inducible T cell kinase (Itk)[26, 27]. The FDA has approved Ibrutinib for the treatment of several B-cell lineages and cGVHD. The potency of ibrutinib in immune disorders, such as autoimmune diseases[24] and graft-versus-host disease[28], is now drawing much attention. However, the immune-regulating potential of ibrutinib in organ transplantation has not been reported. As an approved clinical drug, the safety profile and pharmacokinetic effects of ibrutinib have been confirmed. Moreover, in previous study[29], ibrutinib allowed for recovery of humoral immune function in patients with chronic lymphocytic leukemia (CLL). The innocuity to normal immune system gave it superiority over traditional immunosuppressants. Repositioning ibrutinib as an immunosuppressant will be of great value to drug discovery by saving costs and time[30].
In this article, we present a study of ibrutinib as a potential immunosuppressant in allo- and xeno- transplantation. A xeno-artery patch model has been employed to evaluate the anti-immune response effects of ibrutinib ex vivo. An allo-skin transplantation model from C57BL/6 to BALB/c mice has been established to study the potential of ibrutinib in vivo. In this study, we evaluated the immunosuppressive effects of ibrutinib by T/B cell count, cytokine detection, histological analysis and other tests. We found that ibrutinib exhibited potent inhibitory effects on T cell proliferation and cytokine secretion in the xeno-artery patch model ex vivo. In allo-skin transplantation model, ibrutinib delayed and abated the graft rejection via inhibiting T cells and B cells.
Materials and Methods
Reagents and cell culture
Ficoll-Paque PLUS (Cat. No. 17-1440-03) was purchased from GE Healthcare. Cell Counting Kit-8 (CCK8) was purchased from Dojindo Laboratories (Kumamoto, Japan). CellTrace™ CFSE Cell Proliferation Kit (Cat. No. C34554) was purchased from ThermoFischer Scientific. Phytohemagglutinin-M (PHA-M) (Cat. No. 11082132001) was purchased from Roche. BD™ Cytometric Bead Array NHP Th1/Th2 Cytokine Kit (Cat. No. 557800) was purchased from BD pharmingen. PE/Cy7 anti-mouse CD4 (Cat. No. 100421), PE anti-mouse CD3 (Cat. No. 100205), FITC anti-mouse CD20 (Cat. No. 150407), APC anti-mouse CD8α (Cat. No. 100711) were purchased from Biolegend. PE-Cy7 anti-human CD4 (Cat. No. 557852), PE anti-human CD8 (Cat. No. 555367), FITC anti-human CD3ε (Cat. No. 556611), APC anti-human CD20 (Cat. No. 560853) were purchased from BD pharmingen. FITC-conjugated goat-derived anti-human IgM (μ chain-specific) (Cat. No. 62-7511) and IgG (γ chain-specific) polyclonal antibody (Cat. No. 62-8411), fetal bovine serum (Cat. No. 10099141), Penicillin-Streptomycin-Glutamine (100×) (P/S, Cat. No. 10378016), and RPMI-1640 medium (Cat. No.11875119) were purchased from ThermoFischer Scientific. Optimal Cutting Temperature (OCT) compound was purchased from Agar Scientific (Cat. No. AGR1180).
Peripheral blood mononuclear cells (PBMCs) were harvested from cynomolgus monkeys after artery patch. Isolated PBMCs were cultured with RPMI-1640 containing 10% (vol/vol) FBS, 1% (vol/vol) P/S at 37 °C with 5% CO2. Mouse spleen cells were harvested and cultured in RPMI-1640 containing 10% (vol/vol) FBS, 1% (vol/vol) P/S at 37 °C with 5% CO2.
Animals
C57BL/6 (8-10 weeks, male, 20-30 g) and BALB/c (6-10 weeks, female, 20-25 g) mice were purchased from Guangdong Medical Lab Animal Center. Female Bama minipigs (age 2~4 months), the donors of artery patch grafts, were purchased from BGI Ark Biotechnology (Shenzhen, China). Male cynomolgus monkeys (M15001: nine years old, 7.5 kg; M15003: nine years old, 9.0 kg; M16003: fourteen years old, 4.0 kg), the recipients of pig artery patch grafts, were purchased from Guangdong Landao Biotechnology (Guangzhou, China).
Artery patch transplantation
Pig-to-monkey artery patch xenotransplantation was performed at Guangdong Landao Biotechnology under full inhalational anesthesia as previously described[31]. Three independent pig-to-monkey artery patch xenotransplantations were performed and named as M15001, M15003, and M16003. The animal experiments were approved by the Institutional Review Board on Bioethics and Biosafety of Beijing Genomics Institute (BGI-IRB) (following IACUC-approved protocols published by the Yerkes Primate Center, Atlanta, GA, USA). All animal experiments were performed in accordance with the Ministry of Health guidelines for the care and use of laboratory animals (GB 14925-2001), and the procedures were approved by the Laboratory Animal Ethics Committee of the Sun Yat-sen University. All experiments were performed in accordance with relevant guidelines and regulations.
IgG/IgM binding
The washed PBMCs were suspended in staining buffer (PBS containing 1% bovine serum albumin (Invitrogen) and 0.1% NaN3) for IgM/IgG binding assays. Serum from cynomolgus monkeys after xeno-artery patch was collected at designated time. Binding of serum from cynomolgus monkeys after xeno-artery patch to PBMCs of wild type Bama pig was measured by flow cytometry using the relative geometric mean (rGM), as previously described[32]. Briefly, PBMCs (1×105 / tube) were incubated with 5 μl pooled monkey serum for 0.5 h at 4 °C. After incubation, cells were washed with staining buffer to remove unbinding monkey serum and were blocked with 10% goat serum (Sigma) for 20 min at 4 °C. After further washing with staining buffer, FITC-conjugated goat-derived anti-human IgM (mμ chain-specific) or IgG (γ chain-specific) polyclonal antibody (concentration 1:100 for pPBMCs; Invitrogen) was added, and the cells were incubated for 30 min at 4 °C. After washing with staining buffer, 200 μl fixation buffer was added, and the cells were allowed to sit at 4 °C for 30 min before adding 100 μl staining buffer. Flow cytometry was carried out using BD Aria II (BD, San Jose, CA).
Cell viability
Peripheral blood mononuclear cells (PBMCs) (1×106, 100 μl) stimulated with PHA (5 μg/ml) were seeded into 96-well plates (round bottom) and treated with ibrutinib (final conc. 1 μM and 5 μM) for 0, 1 and 5.5 days. 10 μl CCK8 was added to the supernatant for 2 hours. Two hours later, the absorbance values of wells were measured with OD450, which was read using a multiscan GO spectrophotometer (Thermo Scientific, Waltham, MA, USA). The cell viability was calculated by the following formula. Cell viability % = (OD450 sample- OD450 medium)/(OD450 DMSO- OD450 medium) × 100.
Proliferation
Adjust the cell density of peripheral blood mononuclear cells (PBMCs) to 2×106 / ml in 1640 medium supplemented with 10% FBS. Remove supernatant from the cell pellets. Add CellTrace™ CFSE (1:1000 dilution) staining solution and gently re-suspend the cells. Incubate at 37 °C for 20 minutes, protected from light. Add complete 1640 culture medium and mix. Incubate at 37 °C for another 5 minutes. Remove the supernatant and adjust the cell density to 1×106 / ml. Resuspend the cell pellets in fresh, pre-warmed complete 1640 culture medium and stimulate with PHA (5 μg/ml). Add DMSO and ibrutinib (final conc. 1 μM) for 5.5 days' incubation. Collect cell pellets and re-suspend in FACS buffer for flow cytometry.
T/B cell count
The washed PBMCs were suspended in staining buffer (PBS containing 1% bovine serum albumin (Invitrogen) and 0.1% NaN3) for T/B cell count. Briefly, PBMCs (1×105 / tube) were suspended in 100 μl staining buffer. PE-Cy7 anti-human CD4 (5 μl per test), PE anti-human CD8 (2.5 μl per test), FITC anti-human CD3ε (10 μl per test) and APC anti-human CD20 (10 μl per test) were added, and the cells were incubated for 30 min at 4 °C protected from light. After washing with FACS buffer (0.5% BSA in PBS), 200 μl FACS buffer was added, and the cells allowed to sit at 4°C until running flow cytometry. Flow cytometry was carried out using BD Aria II (BD, San Jose, CA).
The washed spleen cells were suspended in staining buffer (PBS containing 1% bovine serum albumin (Invitrogen) and 0.1% NaN3) for T/B cell count. Briefly, spleen cells (1×106 / tube) were suspended in 100 μl staining buffer. PE/Cy7 anti-mouse CD4 (12.5 μl per test), PE anti-mouse CD3 (12.5 μl per test), FITC anti-mouse CD20 (10 μl per test) and APC anti-mouse CD8α (12.5 μl per test) were added, and the cells were incubated for 30 min at 4 °C protected from light. After washing with FACS buffer (0.5% BSA in PBS), 200 μl FACS buffer was added, and the cells allowed to sit at 4 °C until running flow cytometry. Flow cytometry was carried out using BD Aria II (BD, San Jose, CA).
Skin transplantation
Animals were maintained under specific pathogen-free conditions. Skin from C57BL/6 mice was transplanted to BALB/c recipients as previously described [33]. Ibrutinib (30 mg/kg·d) was administrated orally daily starting from two days before skin transplantation (no dosage at the day of operation). For skin transplantation, 80% of necrosis, ulceration, progressive shrinkage and desquamation were considered to be rejected. Skin status was evaluated daily according to the standard of rejection. Photographs, skin grafts, and recipient mouse spleens were harvested at day 1, 3, 6, 10, 15, 21, 28 after operation. At least three mice were recorded per group (three for ibrutinib, three for control) at designated time (day 1, 3, 6, 10, 15, 21, 28).
All animal experiments were performed in accordance with the Ministry of Health guidelines for the care and use of laboratory animals (GB 14925-2001), and all the procedures were approved by the Laboratory Animal Ethics Committee of the Sun Yat-sen University.
Graft survival
Graft viability was analyzed by observation and photo documentation of the transplanted skin at designated time. During the rejection process, the blood supplied into the graft was progressively restricted, which could be observed as an increasing area of necrosis within the graft. Transplants were classified as vivid if the necrotic part was less than 80%.
Histological analyses
The skin grafts were removed at designated time (POD 1, 3, 6, 10, 15, 21, 28), rinsed in cold saline, placed in Optimal Cutting Temperature (OCT) compound and immediately frozen in liquid nitrogen for histological analysis. The fixed skin grafts were embedded with paraffin, and sectioned. Hematoxylin and eosin (H&E) staining was performed as described previously[33].
Figure 1
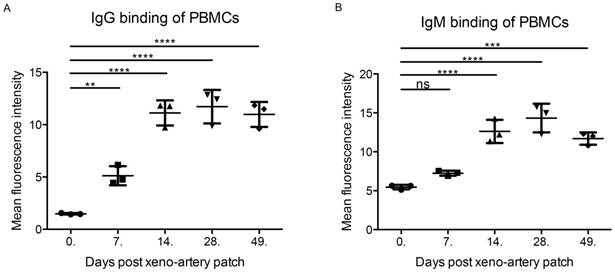
IgG and IgM binding of PBMCs. (A) IgG binding of PBMCs after xeno-artery patch. (B) IgG binding of PBMCs after xeno-artery patch. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.001, ns: not significant; n=3).
Statistical analysis
The data collected were analyzed using Graphpad Prism for the independent Student's t-tests. Experimental data were presented as the mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, ns = not significant vs without ibrutinib. All experiments were replicated at least three times. At least three mice for ibrutinib-treated group and control group separately at seven time points.
Results
Ibrutinib inhibited proliferation of PBMCs (POD14) with slight cell cytotoxicity
The recipient cynomolgus monkey exhibited a strong immune response after an artery patch during POD14 to 42, which was validated by IgG and IgM binding (Figure 1). PBMCs (POD14) were employed as the evaluation system ex vivo. After incubating PBMCs (POD14) with ibrutinib (final concentration: 1 μM and 5 μM) for 24 hours and 5.5 days separately, the cell viability was detected by CCK8. A 24 hours' incubation with ibrutinib was intended to evaluate the cytotoxicity of ibrutinib on PBMCs. After 24 hours' incubation with ibrutinib, the cell viability decreased slightly at the final concentration of both 1 μM and 5 μM (Figure 2A). In the following experiments, a final concentration of 1 μM was employed as the treating concentration. When PBMCs (POD 14) were exposed to ibrutinib for 5.5 days, which was a general evaluating assay for proliferation of PBMCs, the cell density was greatly decreased (Figure 2B). The cell viability assay showed that ibrutinib significantly decreased the cell viability of PBMCs (POD 14) after 5.5 days' incubation (Figure 2C), showing that ibrutinib inhibited the proliferation of PBMCs (POD 14). CFSE-labeled PBMCs (POD 14) were further analyzed by FACS to evaluate effects on proliferation of PBMCs. FACS analysis showed that the proliferation of CFSE-labeled PBMCs (POD 14) was almost halted after incubation with ibrutinib (1 μM) for 5.5 days (Figure 2D). IgG/IgM binding of PBMCs gradually decreased to base level after POD 49, implying the immune response gradually eliminated. We chose PBMCs at POD 0, 75 and 360 to evaluate the effects of ibrutinib on PBMCs with normal immune response. After incubation with ibrutinib (final conc. 1 μM) for 5.5 days, the cell viability of PBMCs was slightly influenced (Figure 2E). The different effects implied that ibrutinib (final conc. 1 μM) inhibited proliferation of PBMCs with strong immune response, but slightly influenced PBMCs with weak immune response. This difference made ibrutinib a priority over traditional immunosuppressants in minimizing adverse effects when long-term administrated. Adjusting therapeutic agents dosing could artificially control therapeutic effects and adverse effects.
Ibrutinib mainly inhibited the proliferation of T/B cells
In the previous assays, we found that ibrutinib efficiently inhibited the proliferation of PBMCs (POD 14), while exhibiting minor cytotoxicity and influence on PBMCs (> POD 49). In this part, a T/B cell count assay was employed to investigate the influence of ibrutinib (final conc. 1 μM) on subpopulations of PBMCs (POD 14). The results showed that ibrutinib significantly decreased the CD3+CD8+ and CD3+CD4+ T cell populations, but slightly increased the CD3-CD20+ B cell population (Figure 3). The influence of ibrutinib on T cell subpopulations was much more significant than B cells, implying that ibrutinib mainly interfered with T-cell-mediated rejection.
Figure 2
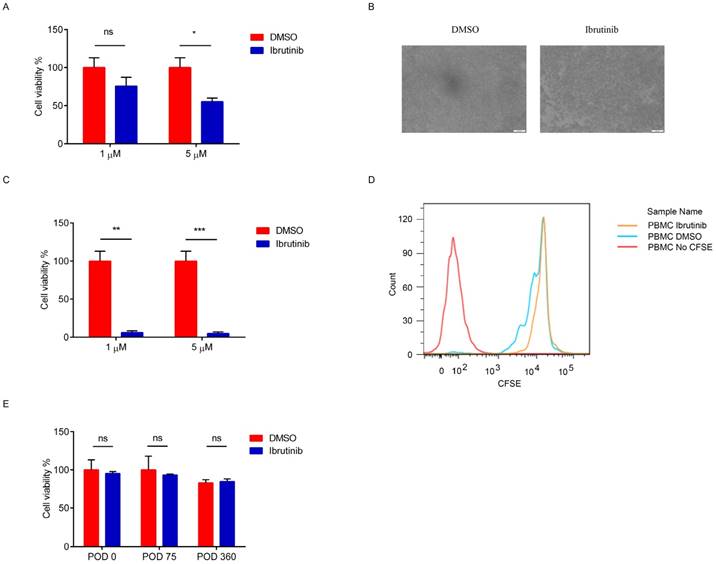
Effects of ibrutinib on PBMCs. (A) Cell viability of PBMCs (POD 14) after treatment with ibrutinib (final conc. 1 μM) for 24 hours. (B) Image of PBMCs (POD14) after treatment with ibrutinib (final conc. 1 μM). (C) Cell viability of PBMCs (POD14) after treatment with ibrutinib (final conc. 1 μM) for 5.5 days. (D) Inhibition of PBMCs (POD14) proliferation after treatment with ibrutinib (final conc. 1 μM) for 5.5 days. (E) Cell viability of PBMCs (POD 0, 75, 360) after treatment with ibrutinib (final conc. 1 μM) for 5.5 days. (*p<0.05, **p<0.01, ***p<0.001, ns = not significant vs without ibrutinib; n=3).
Figure 3
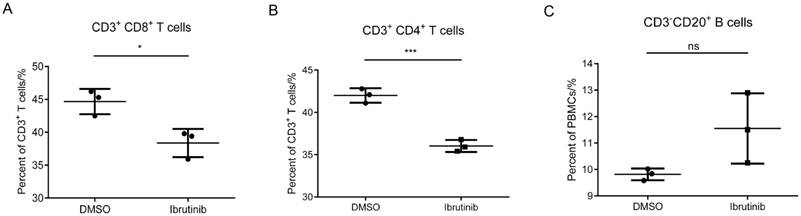
Effect of ibrutinib on T/B cells. Statistics of three independent T/B cell counts. (*p<0.05, **p<0.01, ***p<0.001, ns = not significant vs without ibrutinib; n=3).
Ibrutinib inhibited the secretion of immune rejection related cytokines
Cytokines are the key mediators of immune balance, the expression and secretion of which are under strict spatial and temporal control. Cytokines storm may be responsible to the immune rejection after organ transplantation. A cytometric bead array was used to detect cytokines secreted in the supernatant of PBMCs (POD 14) after treated with ibrutinib (final conc. 1 μM). The results revealed that cytokines (IFN-γ, IL-2 and IL-6) were sensitive to ibrutinib treatment (Figure 4). However, TNF-α, IL-4, and IL-5 displayed no obvious change in both groups (Supplementary Figure 1). Influence of ibrutinib on PBMCs (POD 14) was divergent depending on the cytokine types. Ibrutinib may exhibit immunosuppressive potential via modulating the secretion of cytokines (IFN-γ, IL-2 and IL-6).
Figure 4
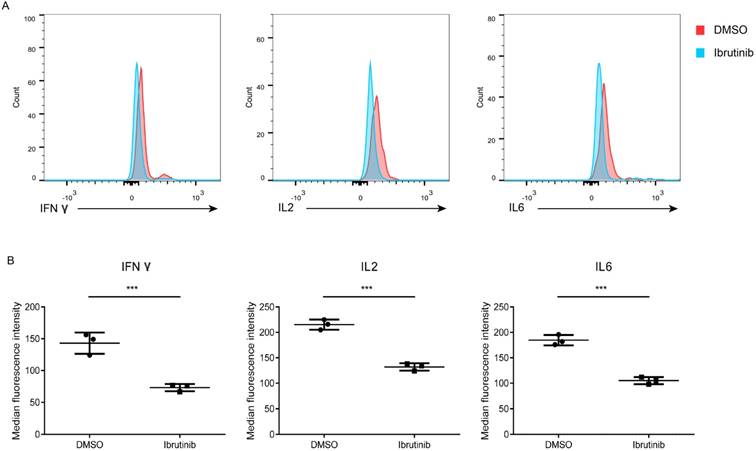
Influence of ibrutinib on cytokines secretion. (A) Representative result of cytokines (IFN-γ, IL-2 and IL-6) secreted from PBMCs (POD 14) after treatment with ibrutinib (final conc. 1 μM). (B) Statistics of MFI of cytokines (IFN-γ, IL-2 and IL-6) secreted from PBMCs (POD 14) after treatment with ibrutinib (final conc. 1 μM). Datas are representative of at least three independent experiments (mean±SEM). (*p < 0.05, **p < 0.01, ***p < 0.001 by Student's t test.)
Figure 5
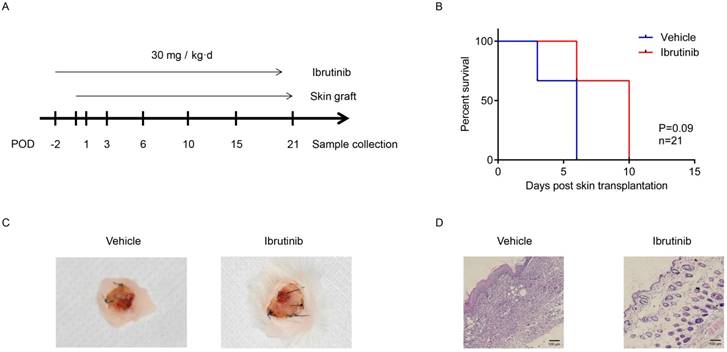
Effects of ibrutinib on allo-skin transplantation. (A) Main sketch of allo-skin transplantation. (B) Graft survival of the grafted skin between the ibrutinib-treated group and the control. (C) The appearance of grafted skins. (D) H&E staining of the grafted skin at POD 10. At least three mice for ibrutinib-treated group and control group separately at seven time points. (*p < 0.05, **p < 0.01, ***p < 0.001 by Student's t test.)
Ibrutinib delayed and alleviated immune rejection in allo-skin transplantation
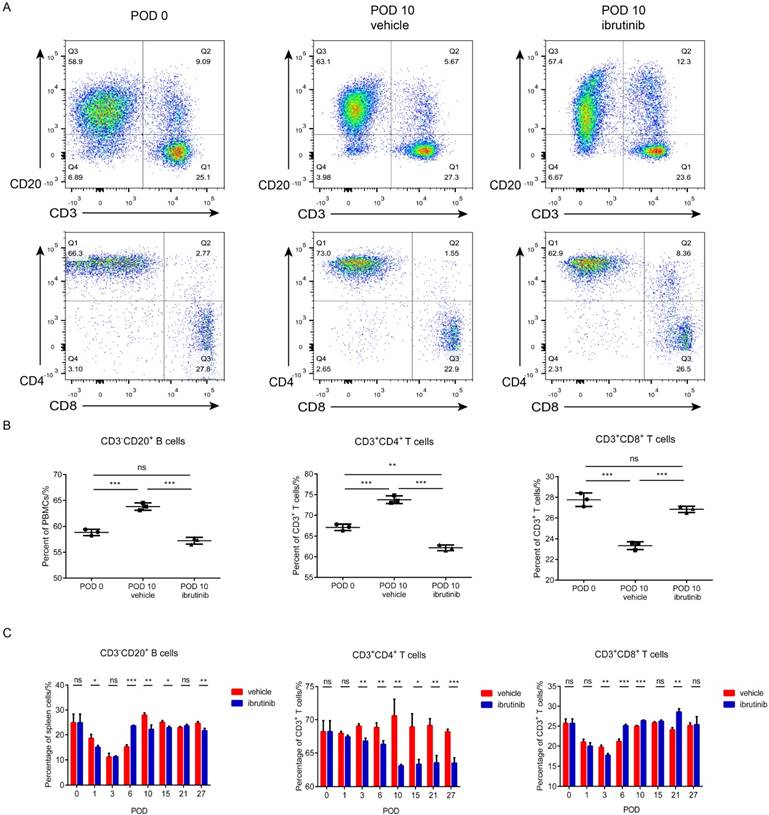
Murine skin transplantation is a routine model for evaluating immunosuppressant candidates in vivo. In this study, a C57BL/6 to BALB/c full-thick skin transplantation model was established to evaluate the potential of ibrutinib as an immunosuppressant. A dosage of 30 mg/kg·d of ibrutinib and vehicle were orally administered to recipient BALB/c mice daily starting from two days before the operation. The status of the grafted skin and recipient mice were monitored and recorded daily. Recipient mouse spleens and grafted skin were harvested at designated time (Figure 5A). The graft survival indicated that ibrutinib improved the status of grafted skin and postponed the rejection compared with the vehicle group, which was summarized in Figure 5B. Approximate 15% of the grafted skin was rejected in the ibrutinib-treated group, while 50% was rejected in the vehicle group at POD 10 (Figure 5C). T/B cells of the recipient spleens harvested at the designated time were detected (Figure 6). The T/B cell count demonstrated that the immune response was intense during the interval of POD 7 to 21 (Figure 6C) in coincidence with the appearance. At POD 10, CD3-CD20+ B cells and CD3+CD4+ T cells were decreased in the ibrutinib-treated group compared with vehicle (Figure 6A and 6B). H&E staining of the grafted skin at POD 10 revealed that the histology of the skin grafts in the ibrutinib-treated group exhibited more intact tissue alignment and less lymphocytic inflammatory infiltrate (Figure 5D). In conclusion, ibrutinib delayed and alleviated the immune rejection in allo-skin transplantation via interefering with CD3-CD20+ B cells and CD3+CD4+ T cells.
Figure 6
Effect of ibrutinib on T/B cells in allo-skin transplantation. (A) Representive figure of T/B cells on POD 0 and POD 10 (vehicle and ibrutinib). (B) Statistics of T/B cells on POD 0 and POD 10 (vehicle and ibrutinib). (C) Statistics of T/B cells on POD 0, 1, 3, 6, 10, 15, 21, 27 (vehicle and ibrutinib). At least three mice for ibrutinib-treated group and control group separately at seven time points. (*p < 0.05, **p < 0.01, ***p < 0.001 by Student's t test.)
Discussion
In organ transplantation, after the acute immune rejection, immunosuppressants are necessary for maintenance therapy to alleviate immune rejection and increase long-term survival. Chemical immunosuppressants have the advantages of being convenient, inexpensive, and easily optimized. However, traditional chemical immunosuppressants (such as anti-proliferative agents, steroids and calcineurin inhibitors) cause serious issues either poor immunosuppressive effects or severe adverse effects (such as high risk of infection, malignancies, nephrotoxicity, hepatotoxicity, and other sequelae). The development of novel immunosuppressants with high efficacy and a favorable safety profile is urgent and challenging. Ibrutinib, an approved drug for B-cell lymphomas and cGVHD, has been recently reported to be an irreversible inhibitor of ITK and exhibited potential therapeutic effects in autoimmune diseases and graft-versus-host disease. In the present study, we evaluated the potential of ibrutinib as an immunosuppressant in allo- and xeno- transplantation. The repositioning of ibrutinib as an immunosuppressant would be of great significance to drug development.
The artery patch model of wild type or genetically modified pigs to cynomolgus monkeys is a convenient and reliable xenotransplantation model. The physiological status of the recipient monkey is good enough for further evaluation without any immunosuppressants. Besides, the grafts can activate the immune system and induce anti-pig antibodies and cell-mediated immune rejection. David Cooper has firstly monitored xeno-immune rejection in xeno-artery patch model [34]. In the artery patch model of Bama wild-type pig to cynomolgus monkey, IgG/IgM binding of recipient PBMCs demonstrated that the immune response was relatively strong for 14-42 days after the artery patch. Comparing the effects of ibrutinib on PBMCs with the levels of immune response, ibrutinib inhibited PBMCs with a strong immune response, but showed minor effects on normal PBMCs. This finding may reflect the specialty of ibrutinib over traditional immunosuppressants. T-cell mediated rejection is the major barrier to graft long-term survival [35, 36] and participates in antibody-mediated rejection (ABMR) [37]. T-cell mediated rejection is treatable under the control of effective immunosuppressants, such as T-cell costimulatory blockades [38] and T cell inhibitors [39]. The potential biological targets of ibrutinib in PBMCs might be ITK and BTK, which are the key mediators of T/B cells. The T/B cell count assay indicated that ibrutinib induced a decrease in CD3+CD4+ and CD3+CD8+ T cells ex vivo, but slightly increased CD3-CD20+ B cells. These results indicate that ibrutinib may have a predominant effect on the T-cell mediated immune response, implying the potential of ibrutinib as a maintenance therapy agent.
Cytokines have been identified as strong regulators and potential biomarkers of immune responses (immune rejection, tolerance and effects of immunosuppressants) after organ transplantation [40]. The systemic regulation of cytokines plays a central role in the maintenance of immune homeostasis. IFN-γ [41] and TNF-α [42, 43] are typical Th1-cytokines, which are responsible for immune rejection [44]. IL-6 reportedly stimulates the inflammatory and autoimmune processes in many immune disorder diseases and has become a potent therapeutic target [45]. Th17 cells and IL-6 are considered to contribute to the mechanisms of rejection after organ transplantation [46, 47]. In this study, ibrutinib decreased the secretion of IFN-γ and IL-6. The secretion of IL-6 and IFN-γ might be regulated under a comprehensive network, implying that except for the direct cytokines, other regulators in the by-pathway are also mediated by ibrutinib. The cytokine analysis demonstrated that IL-6, IFN-γ and IL-2 are the main effectors sensitive to ibrutinib.
Full-thickness skin transplantation is considered to be a reliable and well-established animal model to evaluate the potential of immunosuppressant candidates. In this model, T-cell mediated rejection and graft survival are easily evaluated. In the previous ex vivo study, ibrutinib was found to suppress the proliferation of T cells and secretion of cytokines. Ibrutinib delayed the immune rejection of grafted skin in vivo and prolonged graft survival by decreasing CD3+CD4+ T cells and CD3-CD20+ B cells. However, ibrutinib delayed the immune rejection but not eliminated it, implying that the immunosuppressive effects of ibrutinib were not strong enough in the allo-skin transplantation model. Compared with solid organ transplantation, the immune response of recipient mice after skin transplantation was too mild to adequately evaluate the potential of immunosuppressant candidates. Considering the different targets and potency of ibrutinib and other classic immunosuppressants, it is difficult to determine the exact agents for comparison of immunosuppressive potential in allo-skin transplantation model. The effects of ibrutinib ex vivo and in vitro demonstrated that ibrutinib has an immunosuppressive potential via interfering with T-cell mediated rejection and cytokine regulation. A more suitable solid organ transplantation model with typical and prominent immune rejection is needed to comprehensively evaluate the potential of ibrutinib as an efficient immunosuppressant.
It was obvious that ibrutinib decreased the amount of CD3+CD4+ T cells in both PBMCs after xeno-artery patch and spleen cells after skin transplantation. Cytokine analysis showed that ibrutinib inhibited the secretion of IL-2, IFN-γ and IL-6 while IL-4, IL-5 and TNF-α were basically not influenced by ibrutinib. The cytokine analysis further demonstrated the inhibitory effect of ibrutinib on helper T cells. Ibrutinib had more obvious effects on Th1-type cytokines than Th2-type cytokines, which was not coincide with the conclusion by Dubovsky [26]. Dubovsky demonstrated ibrutinib drive a Th1-selective pressure in naïve CD4+ T lymphocytes by inhibiting ITK. But in our system, ibrutinib inhibited the secretion of both Th1 and Th2 type cytokines in PBMCs after xeno-artery patch, which exhibited strong immune responses. Ibrutinib was cytokine responsive but not Th-type responsive. Besides, ibrutinib had different effects on CD8+ T cells in PBMCs after xeno-artery patch and spleen cells after skin transplantation. Berg [23] and Schwartzberg [48] have demonstrated that Tec kinases (ITK and RLK) had an important role in the development of conventional versus innate CD4+ and CD8+ T cells in Itk-/- and Itk-/-Rlk-/- mice. However, the precise role of ITK and RLK in the differentiation of T cells is still obscure. Ibrutinib could inhibit both ITK and RLK but with different inhibitory effects. The roles (amount and activity) of ITK and RLK in PBMCs after xeno-artery patch and spleen cells after skin transplantation were not experimentally proved or reported. And the responses of ITK and RLK to ibrutinib at the working dosage were hard to determine and compare in these two different experimental models. Thus, quantitative fluorescent probes like PCI33380 [24], probe 1 [49], and Ibrutinib-SiR-COOH [50] may provide useful information about target occupation and metabolism which will help explain the influence of ibrutinib by ITK and RLK inhibition and the possible drug dosage and interval. The effects of ITK and RLK on the development of CD8+ T cell lineages, the subtype of CD8+ T cells in these two different experimental models, the interaction of CD4+ T cells, CD8+ T cells and even other T/B cells, and the different immune responses made it quite difficult to explain the effects of ibrutinib on CD8+ T cells. The separation and enrichment of subtypes of T/B cells or differentiation in vitro may be good choices to look into the precise mechanism of ibrutinib in T/B cell related immune responses.
Resembled to many drug discovery stories, therapeutic effects in vitro do no equal or even represent the effects in vivo. Whether ibrutinib is a potential immunosuppressant in organ transplantation still need comprehensive evaluation in vivo. Thus an ideal experimental model, reference immunosuppressants, administration dosage, combined therapies with known immunosuppressants (such as cyclosporine, CNIs, mTORs), and evaluation platform are necessities for novel immunosuppressants development. Kinase inhibitors for the treatment of cancers have been development successfully. Kinase inhibitors targeting T cells or B cells are potential lead compounds for immunosuppressants development. Repurposing of known kinase inhibitors with therapeutic potential to immunosuppressants would be of great value to facilitate the development of novel immunosuppressants by reducing risks and costs.
In this study, ibrutinib exhibited potential immunosuppression effects ex vivo and in vivo via suppressing T-cell mediated rejection and mediating the cytokine network. The immunosuppressive potency of ibrutinib was not ideal in the allo-skin transplantation model as expected. Ibrutinib was shown to delay immune rejection by interfering with T cells, especially CD3+CD4+ T cells. However, how ibrutinib inhibited the proliferation of T cells remained unknown. Ibrutinib had a negative influence on cytokines (IFN-γ, IL-2, and IL-6), mainly the Th1-type cytokines. The regulation of cytokine expression and secretion is a complex process and difficult to interpret, especially when multiple cytokines are involved.
Conclusion
The immunosuppressants applied clinically have been faced with severe safety issues. In this study, we found that ibrutinib, an approved drug for several B-cell lymphomas, exhibited anti-immune rejection potential by T-cell and cytokine mediation. Although the immunosuppressive effect of ibrutinib in allo-skin transplantation model was not as expected, the potential in T-cell mediated rejection and improvement in graft survival implied that ibrutinib is a promising candidate for immunosuppression in xeno- and allo- transplantation. Furthermore, the alternative application of ibrutinib to other diseases is a convenient avenue to drug development. Bypassing tedious safety evaluating processes would greatly facilitate and accelerate drug development. Ibrutinib offers a promising platform for immunosuppressants development. Structural optimization of ibrutinib and the elucidation of its mechanisms would greatly facilitate the development of novel immunosuppressants. Kinase inhibitors are potent therapeutic agents for many diseases. Repurposing of known kinase inhibitors targeting T/B cells to immunosuppressants would greatly help the development of novel immunosuppressants.
Abbreviations
Btk: Bruton's tyrosine kinase; IFN-γ: Interferon gamma; IL-2: Interleukin-2; IL-4: Interleukin-4; IL-6: Interleukin-6; ITK: IL-2 inducible T cell kinase; RLK (TXK): Resting lymphocyte kinase; JAK: Janus kinases; PBMCs: Peripheral blood mononuclear cells; PKC: Protein kinase C; POD: Post operation days; SYK: Spleen Tyrosine Kinase; Th cells: helper T cells; TNF-α: Tumor necrosis factor alpha.
Supplementary Material
Supplementary figure and table.
Acknowledgements
Authorship
Qing Zhang: designed the study and wrote the manuscript.
Jicheng Chen and Hanchao Gao: discussed the whole design and revised the manuscript.
Chengjiang Zhao: performed the IgG/IgM binding assay.
Song Zhang, Cuibing Zhou and Chengjun Wang: performed in vitro and in vivo experiments.
Yang Li: revised the manuscript.
Zhiming Cai and Lisha Mou: conceived the study.
Funding
The work was supported by grants from National Key R&D Program of China (2017YFC1103704), Sanming Project of Medicine in Shenzhen (SZSM201412020), Fund for High Level Medical Discipline Construction of Shenzhen (2016031638), Shenzhen Foundation of Science and Technology (JCYJ20170306091928754, JCJY20160229204849975, and GJHZ20170314171357556), Shenzhen Foundation of Health and Family Planning Commission (SZBC2017028 and SZXJ2017021), China Postdoctoral Science Foundation (2017M612790), National Natural Science Foundation of China (81502410 and 11747006), Natural Science Foundation of Shandong (ZR2016BB13).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Farrar CA, Kupiec-Weglinski JW, Sacks SH. The innate immune system and transplantation. Cold Spring Harbor perspectives in medicine. 2013;3:a015479
2. Sacks SH, Zhou W. The role of complement in the early immune response to transplantation. Nature reviews Immunology. 2012;12:431-42
3. Weatherly K, Braun MY. Organ transplantation: modulation of T-cell activation pathways initiated by cell surface receptors to suppress graft rejection. Methods in molecular biology. 2011;677:419-30
4. Beimler J, Morath C, Zeier M. [Modern immunosuppression after solid organ transplantation]. Der Internist. 2014;55:212-22
5. Zaza G, Tomei P, Ria P, Granata S, Boschiero L, Lupo A. Systemic and nonrenal adverse effects occurring in renal transplant patients treated with mTOR inhibitors. Clinical & developmental immunology. 2013;2013:403280
6. Bamoulid J, Staeck O, Halleck F, Khadzhynov D, Brakemeier S, Durr M. et al. The need for minimization strategies: current problems of immunosuppression. Transplant international: official journal of the European Society for Organ Transplantation. 2015;28:891-900
7. Godman B, Baumgartel C. Are generic immunosuppressants safe and effective? Bmj. 2015;350:h3248
8. Stepkowski SM, Wang M, Qu X, Yu J, Okamoto M, Tejpal N. et al. Synergistic interaction of FTY720 with cyclosporine or sirolimus to prolong heart allograft survival. Transplant Proc. 1998;30:2214-6
9. Khattar M, Deng R, Kahan BD, Schroder PM, Phan T, Rutzky LP. et al. Novel sphingosine-1-phosphate receptor modulator KRP203 combined with locally delivered regulatory T cells induces permanent acceptance of pancreatic islet allografts. Transplantation. 2013;95:919-27
10. Gholamnezhadjafari R, Falak R, Tajik N, Aflatoonian R, Ali Keshtkar A, Rezaei A. Effect of FTY720 (fingolimod) on graft survival in renal transplant recipients: a systematic review protocol. BMJ open. 2016;6:e010114
11. Moes DJ, Guchelaar HJ, de Fijter JW. Sirolimus and everolimus in kidney transplantation. Drug discovery today. 2015;20:1243-9
12. Lim WH, Eris J, Kanellis J, Pussell B, Wiid Z, Witcombe D. et al. A systematic review of conversion from calcineurin inhibitor to mammalian target of rapamycin inhibitors for maintenance immunosuppression in kidney transplant recipients. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2014;14:2106-19
13. Zhang EY, Kong KF, Altman A. The yin and yang of protein kinase C-theta (PKCtheta): a novel drug target for selective immunosuppression. Advances in pharmacology. 2013;66:267-312
14. Higashi Y. JAK inhibitors as a new generation of small-molecule immunosuppressants. Nihon yakurigaku zasshi Folia pharmacologica Japonica. 2014;144:160-6
15. Fantus D, Thomson AW. The Ups and Downs of TORKinibs in Transplantation. Transplantation. 2015;99:e117-8
16. Liu S, Yu Z, Yu X, Huang SX, Luo Y, Wu L. et al. SHP2 is a target of the immunosuppressant tautomycetin. Chemistry & biology. 2011;18:101-10
17. Deuse T, Hua X, Taylor V, Stubbendorff M, Baluom M, Chen Y. et al. Significant reduction of acute cardiac allograft rejection by selective janus kinase-1/3 inhibition using R507 and R545. Transplantation. 2012;94:695-702
18. Stepkowski SM, Kirken RA. Janus tyrosine kinases and signal transducers and activators of transcription regulate critical functions of T cells in allograft rejection and transplantation tolerance. Transplantation. 2006;82:295-303
19. Stepkowski SM, Erwin-Cohen RA, Behbod F, Wang ME, Qu X, Tejpal N. et al. Selective inhibitor of Janus tyrosine kinase 3, PNU156804, prolongs allograft survival and acts synergistically with cyclosporine but additively with rapamycin. Blood. 2002;99:680-9
20. Zhang Q, Dai Y, Cai Z, Mou L. HDAC Inhibitors: Novel Immunosuppressants for Allo- and Xeno- Transplantation. ChemistrySelect. 2018;3:176-87
21. Chen Q, Luo H, Zhang C, Chen YP. Bioinformatics in protein kinases regulatory network and drug discovery. Mathematical biosciences. 2015;262:147-56
22. Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug discovery today. 2016;21:5-10
23. Prince AL, Yin CC, Enos ME, Felices M, Berg LJ. The Tec kinases Itk and Rlk regulate conventional versus innate T-cell development. Immunological reviews. 2009;228:115-31
24. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B. et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13075-80
25. Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC. et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. 2007;2:58-61
26. Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y. et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539-49
27. Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E966-72
28. Schutt SD, Fu J, Nguyen H, Bastian D, Heinrichs J, Wu Y. et al. Inhibition of BTK and ITK with Ibrutinib Is Effective in the Prevention of Chronic Graft-versus-Host Disease in Mice. PloS one. 2015;10:e0137641
29. Sun C, Tian X, Lee YS, Gunti S, Lipsky A, Herman SE. et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015;126:2213-9
30. Kozako T, Soeda S, Yoshimitsu M, Arima N, Kuroki A, Hirata S. et al. Angiotensin II type 1 receptor blocker telmisartan induces apoptosis and autophagy in adult T-cell leukemia cells. FEBS Open Bio. 2016;6:442-60
31. Iwase H, Ekser B, Zhou H, Liu H, Satyananda V, Humar R. et al. Further evidence for sustained systemic inflammation in xenograft recipients (SIXR). Xenotransplantation. 2015;22:399-405
32. Lee W, Hara H, Ezzelarab MB, Iwase H, Bottino R, Long C. et al. Initial in vitro studies on tissues and cells from GTKO/CD46/NeuGcKO pigs. Xenotransplantation. 2016;23:137-50
33. Cheng CH, Lee CF, Fryer M, Furtmuller GJ, Oh B, Powell JD. et al. Murine Full-thickness Skin Transplantation. Journal of visualized experiments: JoVE. 2017
34. Ezzelarab MB, Ekser B, Echeverri G, Hara H, Ezzelarab C, Long C. et al. Costimulation blockade in pig artery patch xenotransplantation - a simple model to monitor the adaptive immune response in nonhuman primates. Xenotransplantation. 2012;19:221-32
35. Wood KJ, Goto R. Mechanisms of rejection: current perspectives. Transplantation. 2012;93:1-10
36. Wood KJ, Zaitsu M, Goto R. Cell mediated rejection. Methods in molecular biology. 2013;1034:71-83
37. Mengel M, Husain S, Hidalgo L, Sis B. Phenotypes of antibody-mediated rejection in organ transplants. Transplant international: official journal of the European Society for Organ Transplantation. 2012;25:611-22
38. Clarkson MR, Sayegh MH. T-cell costimulatory pathways in allograft rejection and tolerance. Transplantation. 2005;80:555-63
39. Das S, Ariizumi K, Cruz PD Jr. T-cell inhibitors: a bench-to-bedside review. Dermatitis: contact, atopic, occupational, drug. 2012;23:195-202
40. Brunet M. Cytokines as predictive biomarkers of alloreactivity. Clinica chimica acta; international journal of clinical chemistry. 2012;413:1354-8
41. Hidalgo LG, Halloran PF. Role of IFN-gamma in allograft rejection. Critical reviews in immunology. 2002;22:317-49
42. Grewal IS. Overview of TNF superfamily: a chest full of potential therapeutic targets. Advances in experimental medicine and biology. 2009;647:1-7
43. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nature reviews Immunology. 2003;3:745-56
44. Dallman MJ. Cytokines and transplantation: Th1/Th2 regulation of the immune response to solid organ transplants in the adult. Current opinion in immunology. 1995;7:632-8
45. Rossi JF, Lu ZY, Jourdan M, Klein B. Interleukin-6 as a therapeutic target. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:1248-57
46. Reinhold SW, Straub RH, Kruger B, Kaess B, Bergler T, Weingart C. et al. Elevated urinary sVCAM-1, IL6, sIL6R and TNFR1 concentrations indicate acute kidney transplant rejection in the first 2 weeks after transplantation. Cytokine. 2012;57:379-88
47. Nakagiri T, Inoue M, Minami M, Shintani Y, Okumura M. Immunology mini-review: the basics of T(H)17 and interleukin-6 in transplantation. Transplant Proc. 2012;44:1035-40
48. Broussard C, Fleischacker C, Horai R, Chetana M, Venegas AM, Sharp LL. et al. Altered development of CD8+ T cell lineages in mice deficient for the Tec kinases Itk and Rlk. Immunity. 2006;25:93-104
49. Zhang Q, Liu H, Pan Z. A general approach for the development of fluorogenic probes suitable for no-wash imaging of kinases in live cells. Chemical communications. 2014;50:15319-22
50. Kim E, Yang KS, Kohler RH, Dubach JM, Mikula H, Weissleder R. Optimized Near-IR Fluorescent Agents for in Vivo Imaging of Btk Expression. Bioconjugate chemistry. 2015;26:1513-8
Author contact
![]() Corresponding author: Dr. Lisha Mou (lishamoucom)
Corresponding author: Dr. Lisha Mou (lishamoucom)
Citation styles
APA
Zhang, Q., Chen, J., Gao, H., Zhang, S., Zhao, C., Zhou, C., Wang, C., Li, Y., Cai, Z., Mou, L. (2018). Drug repurposing: Ibrutinib exhibits immunosuppressive potential in organ transplantation. International Journal of Medical Sciences, 15(11), 1118-1128. https://doi.org/10.7150/ijms.24460.
ACS
Zhang, Q.; Chen, J.; Gao, H.; Zhang, S.; Zhao, C.; Zhou, C.; Wang, C.; Li, Y.; Cai, Z.; Mou, L. Drug repurposing: Ibrutinib exhibits immunosuppressive potential in organ transplantation. Int. J. Med. Sci. 2018, 15 (11), 1118-1128. DOI: 10.7150/ijms.24460.
NLM
Zhang Q, Chen J, Gao H, Zhang S, Zhao C, Zhou C, Wang C, Li Y, Cai Z, Mou L. Drug repurposing: Ibrutinib exhibits immunosuppressive potential in organ transplantation. Int J Med Sci 2018; 15(11):1118-1128. doi:10.7150/ijms.24460. https://www.medsci.org/v15p1118.htm
CSE
Zhang Q, Chen J, Gao H, Zhang S, Zhao C, Zhou C, Wang C, Li Y, Cai Z, Mou L. 2018. Drug repurposing: Ibrutinib exhibits immunosuppressive potential in organ transplantation. Int J Med Sci. 15(11):1118-1128.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.