International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(10):1014-1024. doi:10.7150/ijms.25656 This issue Cite
Research Paper
Trehalose inhibits H2O2-induced autophagic death in dopaminergic SH-SY5Y cells via mitigation of ROS-dependent endoplasmic reticulum stress and AMPK activation
Zhijie Gao1#, Helei Wang2#, Bo Zhang3, Xuemei Wu3, Yanfeng Zhang3, Pengfei Ge1,4, Guangfan Chi5, Jianmin Liang3,4 ![]()
1. Department of Neurosurgery, First hospital of Jilin University, Changchun 130021, China.
2. Department of Gastrointestinal Surgery, First hospital of Jilin University, Changchun 130021, China.
3. Department of Pediatric Neurology, First hospital of Jilin University, Changchun 130021, China.
4. Research center of neuroscience, First hospital of Jilin University, Changchun 130021, China.
5. Key Laboratory of Pathobiology, Ministry of Education, Jilin University, Changchun 130021, China.
#: These two authors contributed equally to this work.
Received 2018-2-22; Accepted 2018-5-21; Published 2018-6-14
Citation:
Gao Z, Wang H, Zhang B, Wu X, Zhang Y, Ge P, Chi G, Liang J. Trehalose inhibits H2O2-induced autophagic death in dopaminergic SH-SY5Y cells via mitigation of ROS-dependent endoplasmic reticulum stress and AMPK activation. Int J Med Sci 2018; 15(10):1014-1024. doi:10.7150/ijms.25656. https://www.medsci.org/v15p1014.htm
Other stylesAbstract
Autophagy is a catabolic process to maintain intracellular homeostasis via removal of cytoplasmic macromolecules and damaged cellular organelles through lysosome-mediated degradation. Trehalose is often regarded as an autophagy inducer, but we reported previously that it could prevent ischemic insults-induced autophagic death in neurons. Thus, we further investigated in this study whether trehalose could protect human dopaminergic SH-SY5Y cells against H2O2-induced lethal autophagy. We found pretreatment with trehalose not only prevented H2O2-induced death in SH-SY5Y cells, but also reversed H2O2-induced upregulation of LC3II, Beclin1 and ATG5 and downregulation of p62. Then, we proved that either autophagy inhibitor 3MA or genetic knockdown of ATG5 prevented H2O2-triggered death in SH-SY5Y cells. These indicated that trehalose could inhibit H2O2-induced autophagic death in SH-SY5Y cells. Further, we found that trehalose inhibited H2O2-induced AMPK activation and endoplasmic reticulum (ER) stress. Moreover, inhibition of AMPK activation with compound C or alleviation of ER stress with chemical chaperone 4-PBA obviously attenuated H2O2-induced changes in autophagy-related proteins. Notably, we found that trehalose inhibited H2O2-induced increase of intracellular ROS and reduction in the activities of CAT and SOD. Consistently, our data revealed as well that mitigation of intracellular ROS levels with antioxidant NAC markedly attenuated H2O2-induced AMPK activation and ER stress. Therefore, we demonstrated in this study that trehalose prevented H2O2-induced autophagic death in SH-SY5Y cells via mitigation of ROS-dependent endoplasmic reticulum stress and AMPK activation.
Keywords: Trehalose, Oxidative stress, Autophagy, AMPK, Endoplasmic reticulum stress
1. Introduction
Autophagy is an evolutionarily conserved and highly regulated homeostatic process by which cytoplasmic macromolecules and organelles are degraded for removal through lysosomal system [1]. It has been found that autophagy plays dual roles in regulation of cell destiny. On the one hand, autophagy makes cells survive interior or exterior stresses. Previous reports have shown that autophagy protects brain tissue against the damage caused by epilepsy or neurodegenerative diseases such as Parkinson disease and Alzheimer disease [2-4]. On the other hand, over-activated autophagy triggers non-apoptotic programmed cell death (autophagic cell death) through excessive self-digestion and degradation of essential cellular constituents [5]. Accumulating evidences demonstrate that autophagy is involved in the pathological process of brain damage caused by hypoxic-ischemia, head trauma, and transient focal ischemia [6-8]. Thus, autophagy is emerging as a protective strategy against neuronal death.
Trehalose is a natural non-reducing disaccharide comprised of two molecules of glucose and available in many organisms including yeast, fungi, and invertebrates, but not in mammals [9]. It has multiple biological functions, including inhibition of inflammation, induction of molecular chaperone, alleviation of oxidative stress and mitigation of endoplasmic reticulum stress (ER stress)[10-13]. Trehalose is also regarded as an mTOR-independent autophagy inducer given that it mitigated neuronal damage caused by pathological protein aggregation via induction of autophagy [14, 15]. However, we proved previously that it protected neurons against ischemic insults via inhibition of autophagy [16]. Thus, the role of trehalose in regulation of autophagy is still needed to be elucidated. SH‑SY5Y cells are human dopaminergic neuroblastoma cells, which are comparable to neurons with regards to their morphological, neurochemical and electrophysiological properties and have been extensively applied to evaluate neuronal injury or death in neurodegenerative disease, cerebral ischemia/reperfusion and epilepsy [17]. Considering that oxidative stress is a pathway leading to the neuronal damage caused by ischemic insults [18], we used H2O2 and SH-SY5Y cells in this study to induce autophagy, examine the effect of trehalose on autophagy and investigate its underlying mechanism.
2. Materials and Methods
2.1. Reagents
Trehalose, 3MA (3-Methyladenine), 4-PBA (4-Phenylbutyric Acid), NAC (N-Acetyl Cysteine) and H2O2 were purchased from Sigma-Aldrich Company (St. Louis, MO). Dorsomorphin (Compound C) was from Selleck Chemicals (Houston, TX). Trehalose was diluted with cell culture medium. Superoxide Dismutase (SOD) activity and Catalase assay kits were obtained from BioVision Inc. (Milpitas, CA, USA). Other reagents were from Sigma-Aldrich Company.
2.2. Cell line and culture
Human SH-SY5Y cells were obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). The cells were cultured in DMEM supplemented with 10% fetal bovine serum, 2 mmol/L glutamine (Gibco, Grand Island, NY, USA), penicillin (100 U/mL) and streptomycin (100 μg/mL), and maintained at 37 ºC and 5% CO2 in a humid environment. The medium was replaced twice each week.
2.3. Lactate dehydrogenase release cell death assay
SH-SY5Y cells (4×104) were seeded onto 96-well microplate and cultured 24 h. Lactate dehydrogenase cytotoxicity assay kit (Beyotime Biotech, Nanjing, China) was used to assay cellular death rate. According to the manufacturer's instructions, the absorbance value of each sample was read at 490nm, and cell death ratio was calculated by using the following formula: cell death ratio %= (A sample-A control / A max-A control) × 100. A sample: sample absorbance value; A control: the absorbance value of control group; A max: the absorbance value of positive group.
2.4. Measurement of intracellular ROS
SH-SY5Y cells (4×104) were seeded onto 96-well microplate and cultured 24 h, and then treated with H2O2 at indicated concentrations. The average level of intracellular ROS was evaluated by using redox-sensitive dye DCFH-DA (Beyotime Biotech, Nanjing, China). All the experimental cells were washed twice in PBS and stained in the dark for 30 min with 20μmol/L DCFH-DA. After the cells were dissolved with 1% Triton X-100, the fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength 530 nm using a fluorescence spectrometer (HTS 7000, Perkin Elmer, Boston, MA). The ROS levels were expressed as arbitrary unit/mg protein, then as the folds of control.
Other groups of SH-SY5Y cells were seeded onto a culture dish in a diameter of 3 cm and cultured 24 h. After being treated with H2O2, the cells were stained with DCFH-DA as described above, and observed under fluorescence microscope (Olympus IX71, Tokyo, Japan).
2.5. AO and MDC staining
SH-SY5Y cells (4×104) were seeded on 96-well plates. Some cells were incubated in PBS with 1μg/mL AO (Acridine orange, Sigma-Aldrich Company, St. Louis, MO) for 15 min at room temperature in the dark, others were incubated with 100 µmol/L MDC (Monodansylcadaverine, Sigma-Aldrich Company, St. Louis, MO) solution for 1 h at 37 ºC in the dark. After being washed with PBS, they were examined under a fluorescence microscope (Olympus IX71, Tokyo, Japan) at × 20 objective lens magnification.
2.6. Transfection of small interfering RNA (siRNA)
SH-SY5Y cells (2×105) cells were seeded onto a culture dish in a diameter of 10 cm. Transfection of siRNA was performed by using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instructions. The ATG5 siRNA (5′-GACGUUGGUAACUGACAAATT-3′), and scrambled siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′) were purchased from GenePharma Company (Suzhou, China). After siRNA transfection overnight, the cells were incubated with H2O2 at indicated dose for subsequent experiments.
2.7. Protein isolation
After being collected by centrifugation, SH-SY5Y cells were suspended in ice-cold lysis buffer and homogenized as described previously [16]. The homogenates were centrifuged at 10,000 g at 4 oC for 10 min to obtain the supernatants and the pellets, the protein concentrations of which were determined by using Bio-Rad protein assay kit.
2.8. Gel Electrophoresis and Western Blotting
Equal protein amounts were electrophoresed on 10% SDS gels and transferred to PVDF membranes (Millipore, Billerica, MA, USA). Membranes were blocked with 3% bovine serum albumin in TBS for 30 min at room temperature, then incubated overnight at 4 ºC with the following primary antibodies including: rabbit polyclonal autophagy LC3 (1:1000; Sigma-Aldrich, St. Louis, MO), rabbit polyclonal Beclin-1 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-p62(1:1000, Abcam, Cambridge, MA), rabbit monoclonal anti-ATG5(1:1000, Abcam, Cambridge, MA); rabbit polycolonal anti-GRP 78(1:1000, Abcam, Cambridge, MA), rabbit polyclonal anti-IRE-1(1:1000, Abcam, Cambridge, MA), rabbit polyclonal anti-ATF6(1:1000, Abcam, Cambridge, MA), rabbit monoclonal anti-Nrf2 (1:1000, Abcam, Cambridge, MA), rabbit monoclonal anti-phospho-Nrf2 (1:1000, Abcam, Cambridge, MA), rabbit polyclonal anti-PERK (1:1000, Cell Signaling Technology, Danvers, MA), rabbit polyclonal anti-phospho-PERK (1:1000, Cell Signaling Technology, Danvers, MA), and mouse monoclonal β-actin (1:2000; Santa Cruz Biotechnology). The immunoblot membranes were then incubated with horseradish-peroxidase conjugated anti-mouse (1:2000 Cell Signaling Technology), or anti-rabbit IgG (1:2000, Cell Signaling Technology) for 1 h at room temperature. The immunoreactive proteins were visualized on a Kodak X-omat LS film (Eastman Kodak Company, New Haven, CT, USA) with an enhanced chemiluminescence. Densitometry was performed with Kodak ID image analyses software (Eastman Kodak Company).
2.9. Measurement of cellular anti‑oxidative enzymes
The activity of antioxidant enzymes, superoxide dismutase (SOD) and catalase (CAT) were measured according to the manufacturer's instructions. After incubation for 3h with 500 μmol/L, H2O2 following pretreatment with or without trehalose, SH‑SY5Y cells were collected from the culture dishes using a scraper, centrifuged at 1,000 ×g for 10 min at 4˚C, and the cell pellets were washed with PBS.
For the SOD activity assay, the cells were suspended in ice‑cold lysis buffer [0.1 mmol/L Tris/HCl (pH 7.4), 0.25 mol/L sucrose, 5 mmol/L β‑mercaptoethanol and 0.1 mg/mL phenylmethylsulfonyl fluoride (PMSF); Sigma‑Aldrich], homogenized with a glass Pyrex microhomogenizer (20 strokes; Beyotime Institute of Biotechnology), and centrifuged at 1,500×g for 5 min at 4˚C. The supernatant was then collected for assaying.
For the CAT activity assay, the cells were suspended in ice‑cold assay buffer (Sigma‑Aldrich), homogenized with microhomogenizer (20 strokes), centrifuged at 10,000 ×g for 15 min at 4˚C. The supernatant was then collected for assaying.
The CAT was spectrophotometrically determined by measuring decreased absorbance at 570 nm, using the CAT assay kit, and SOD was measured spectrophotometrically by monitoring the absorbance at 450 nm using the SOD assay kit. CAT and SOD activities were expressed as U/mg protein.
2.10. Statistical analysis
All data represent at least 4 independent experiments and are expressed as mean±SD. Statistical comparisons were made using one-way ANOVA. P-values of less than 0.05 were considered to represent statistical significance.
3. Results
3.1. H2O2 triggered autophagic death in SH-SY5Y cells
We have reported previously that H2O2 induced death in SH-SY5Y cell [17], but its underlying mechanism remain unclear. Given that autophagy plays an important role in regulating cell death, we thus examined whether exterior H2O2 could induce lethal autophagy in SH-SY5Y cells. As proved by LDH release assay, we found that the death of SH-SY5Y cells resulting from 6h incubation with H2O2 at indicated concentration was significantly attenuated by pretreatment with autophagy inhibitor 3MA at 3mmol/L for 1h (Fig.1A). Then, fluorescent substances of AO and MDC were used to detect autophagy. Fluorescence microscopy revealed that, much more red spots detected by AO and stronger punctate fluorescence of MDC distributed in the cytoplasm of the SH-SY5Y cells treated with 250μmol/L H2O2 for 3h, when compared with the cells in the control group (Fig.1B). Then, western blotting proved that H2O2 induced significant upregulation of autophagic hallmark proteins LC3II, Beclin1 and ATG5, but downregulation of autophagy substrate p62/sequestosome1 (Fig.1C). In contrast, pretreatment with 3MA prevented H2O2-induced increase of LC3II, Beclin1 and ATG5, and decrease of p62 (Fig.1C).
Then, we compared H2O2-induced differences in the autophagy-related proteins between the cells transfected with SiRNA ATG5 and scrambled SiRNA and found that H2O2-induced changes of the autophagy-related proteins LC3II, Beclin-1 and p62 were all reversed in the SiRNA ATG5 group (Fig.1D). Moreover, LDH release assay showed that knockdown of ATG5 with SiRNA significantly made the cells resistant to H2O2-induced death (Fig.1E). Therefore, these results indicated that H2O2 triggered autophagic death in SH-SY5Y cells.
3.2. H2O2 induced lethal autophagy via increase of intracellular ROS
To elucidate the factors accounting for the lethal autophagy caused by H2O2, we examined H2O2-induced changes in intracellular ROS. Fluorescence microscopy showed that the green fluorescence detected by ROS probe DCFH-DA at incubation 3h was much brighter in the cells treated with either 250μmol/L or 500μmol/L H2O2, when compared with the cells in the control group (Fig.2A). Statistical analysis of the green fluorescence intensity demonstrated that 500μmol/L H2O2 induced higher levels of ROS in the SH-SY5Y (Fig.2B). In contrast, pretreatment with antioxidant NAC at 2mmol/L for 1h obviously inhibited the increase of intracellular ROS caused by H2O2 at either 250μmol/L or 500μmol/L (Fig.2B). Moreover, NAC prevented markedly H2O2-induced SH-SY5Y cell death, which was revealed by LDH release assay (Fig.2C). Further, western blotting demonstrated that H2O2-induced increases of LC3II, Beclin1 and ATG5 and reduction of p62 were all prevented in the presence of NAC (Fig.2D). Thus, these results indicated that exterior H2O2 triggered lethal autophagy in SH-SY5Y cells via increase of intracellular ROS.
Figure 1
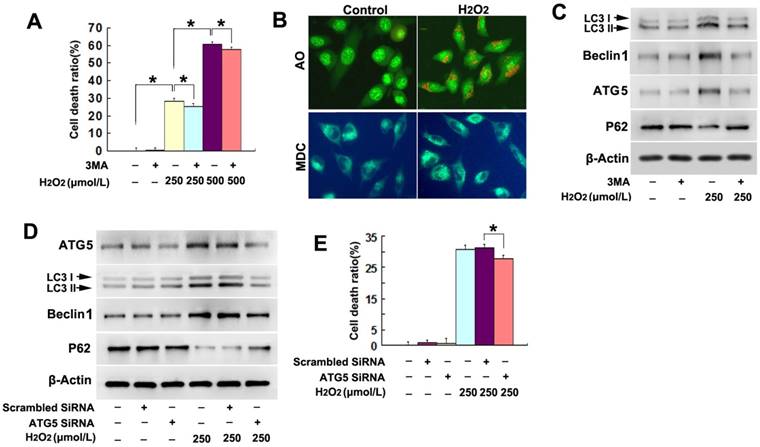
H2O2 triggered autophagic death in SH-SY5Y cells. (A) LDH release assay demonstrated that autophagy inhibitor 3MA significantly prevented H2O2-induced death in SH-SY5Y cell. (B) Fluorescence microscopy revealed that much more red spots detected by AO and stronger punctate fluorescence of MDC distributed in the cytoplasm of the SH-SY5Y cells treated with 250μmol/L H2O2 for 3h (20×). (C) Western blotting proved 3MA obviously reversed H2O2-induced upregulation of LC3II, Beclin-1 and ATG5, and downregulation of p62. (D) Both the increases of LC3II and Beclin-1 and the reduction of p62 caused by H2O2 were reversed when ATG5 was knocked down by using siRNA. (E) LDH release assay showed that knockdown of ATG5 inhibited H2O2-induced death in SH-SY5Y cells. The values are expressed as mean±SEM (n=5 per group). *: p < 0.01.
Figure 2
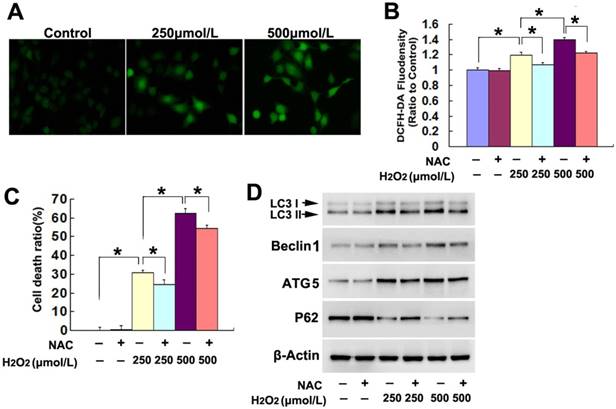
H2O2 induced lethal autophagy via increase of intracellular ROS. (A) Fluorescence microscopy showed that the green fluorescence detected by ROS probe DCFH-DA increased markedly in the cells treated 3h with H2O2, when compared with that in the control cells (20×). (B) Statistical analysis revealed that H2O2 induced concentration-dependent increase in the green fluorescence density, which was significantly alleviated in the presence of antioxidant NAC at 2mmol/L. (C) LDH release assay proved that NAC prevented H2O2-induced death in SH-SY5Y cells. (D) Western blotting demonstrated that NAC reversed H2O2-induced upregulation of LC3II, beclin1 and ATG5 and downregulation of p62. The values are expressed as mean±SEM (n=5 per group). *: p < 0.01.
3.3. AMPK and ER stress were both involved in H2O2-induced lethal autophagy
Considering that AMP activated protein kinase (AMPK) pathway regulates the autophagy occurrence [19], we thus investigated its role in H2O2-induced lethal autophagy. As shown by western blotting analysis, H2O2 upregulated the protein levels of both AMPK and phospho-AMPK in a concentration dependent manner (Fig. 3A). However, pretreatment with NAC obviously mitigated H2O2-induced upregulation in the protein levels of AMPK and phospho-AMPK (Fig. 3A). This indicated that AMPK might be related to H2O2-induced autophagy. Then, the cells were treated with AMPK inhibitor compound C at 20μmol/L for 1h and then incubated with H2O2 at indicated concentrations for 6h. LDH release assay proved that, the death of SH-SY5Y cells caused by H2O2 at either lower or higher dosage was significantly prevented in the presence of compound C (Fig. 3B). Further, western blotting showed that compound C not only obviously inhibited AMPK phosphorylation of (Fig. 3C), but also reversed the increase of LC3II, ATG5 and Beclin1 and decrease of p62 in the cells stressed with H2O2 (Fig.3D). Thus, this indicated that AMPK activation is a pathway responsible for H2O2-induced autophagic death in SH-SY5Y cells.
Given that endoplasmic reticulum (ER) stress is also a pathway leading to autophagy, we examined the role of ER stress in H2O2-induced autophagy. As demonstrated by western blotting, H2O2 induced dosage-dependent upregulation of ER stress-related proteins, including GRP78, IRE-1, ATF6, PERK and phospho-PERK (Fig. 4A). In contrast, administration of antioxidant NAC markedly mitigated H2O2-induced increases of these ER stress-related proteins (Fig. 4A). This indicated that ER stress might be associated with H2O2-induced autophagy. Thus, the cells were treated with ER stress inhibitor 4-PBA at 5mmol/L for 1h prior to incubation with H2O2. We found 4-PBA not only significantly inhibited H2O2-induced death in SH-SY5Y cells (Fig. 4B), but also obviously attenuated the upregulation of ER stress-related proteins caused by H2O2 (Fig. 4C). Additionally, either the increase of LC3II, ATG5 and Beclin1 or the reduction of p62 resulting from H2O2 treatment was prevented in the presence of 4-PBA (Fig. 4D). Thus, these results indicated that ER stress was also involved in H2O2-induced autophagic death in SH-SY5Y cells.
3.4. Trehalose inhibited H2O2-induced autophagic death
To examine whether trehalose could prevent SH-SY5Y cell death induced by H2O2, the cells were pretreated with 5mmol/L trehalose for 48h as reported previously [16] and then incubated 6h with H2O2 at indicated concentration. LDH release assay showed that trehalose significantly attenuated SH-SY5Y cell death caused by H2O2 at either 250μmol/L or 500μmol/L (Fig.5A). Moreover, western blotting proved that trehalose inhibited H2O2-induced upregulation of LC3II, Beclin-1 and ATG-5 and downregulation of p62 (Fig.5B). Thus, these results indicated that trehalose prevented H2O2-induced autophagic death in SH-SY5Y cells.
Figure 3
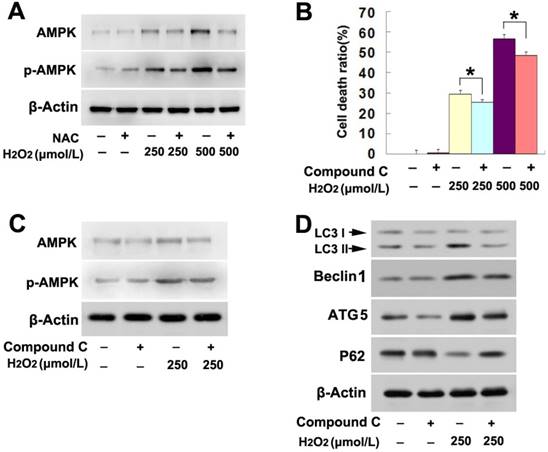
AMPK activation was involved in H2O2-induced lethal autophagy. (A) Western blotting showed that H2O2-induced concentration-dependent upregulation of AMPK and phosphorylated AMPK were both inhibited by antioxidant NAC. (B) H2O2-induced death was prevented in the SH-SY5Y cells pretreated 1h with AMPK inhibitor compound C at 20μmol/L. (C and D) Western blotting proved that compound C prevented H2O2-induced changes in the autophagy hallmark proteins , as well as upregulation and phosphorylation of AMPK. The values are expressed as mean±SEM (n=5 per group). *: p < 0.01.
Given that AMPK activation and ER stress were both involved H2O2-induced autophagic death in SH-SY5Y cells, we examined the effect of trehalose on H2O2-induced activation of AMPK and ER stress. As demonstrated by western blotting, trehalose prevented the upregulation of AMPK and phospho-AMPK, as well as alleviated the increased protein levels of GRP78, IRE-1, ATF-6, PERK and phospho-PERK caused by H2O2 treatment (Fig.5 C and D). These data indicated that trehalose prevented H2O2-induced activation of AMPK and ER stress.
3.5. Trehalose inhibited H2O2-induced increase of intracellular ROS
On the basis that inhibition of intracellular ROS with NAC prevented AMPK activation and ER stress caused by H2O2, we examined the effect of trehalose on H2O2-induced increase of intracellular ROS. When compared with that in the cells treated with H2O2 alone, pretreatment with 5mmol/L trehalose for 48h significantly inhibited H2O2-induced increase of intracellular ROS (Fig.6A). Considering nuclear factor erythroid 2-related factor 2 (Nrf2) is an important endogenous anti-oxidative proteins [20], we tested whether trehalose inhibited ROS via upregulation of Nrf2. As revealed by western blotting, trehalose alone had regulatory effect on the protein levels of Nrf2 and phospho-Nrf2 in SH-SY5Y cells. However, trehalose markedly inhibited H2O2-induced Nrf2 phosphorylation (Fig.6B). Then, we assayed the activities of SOD and CAT because they both account for clearing intracellular ROS [21]. We found that trehalose pretreatment significantly reversed H2O2-induced reduction in the activities of SOD and CAT (Fig.6C and D). Thus, these results indicated that trehalose inhibited H2O2-induced increase of intracellular ROS via maintaining the activities of SOD and CAT, not via activation of Nrf2.
4. Discussion
In this study, we demonstrated that exterior H2O2 triggered autophagic death in SH-SY5Y cells via increasing intracellular ROS levels, which initiated the lethal autophagy through ROS-dependent activation of AMPK and ER stress. The protection of trehalose against H2O2-induced lethal autophagy was associated with inhibition of AMPK activation and ER stress. Moreover, trehalose mitigated intracellular ROS levels not via induction of Nrf2 phosphorylation, but maintaining the activity of CAT and SOD.
Figure 4
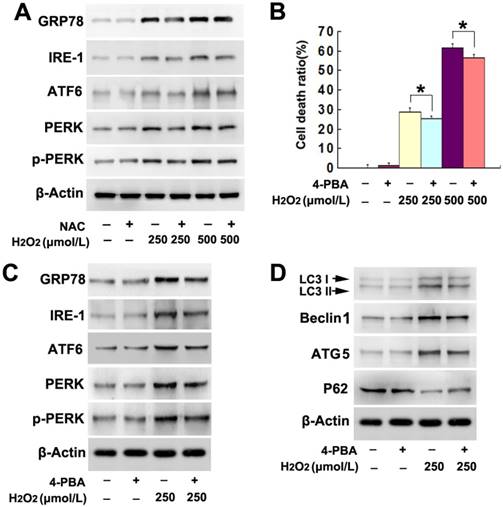
ER stress was involved in H2O2-induced lethal autophagy. (A) Western blotting showed that H2O2-induced concentration-dependent upregulation of ER stress-related proteins GRP78, IRE-1, ATF6, PERK and phosphorylated PERK, which were all prevented by NAC pretreatment. (B) H2O2-induced death was prevented in the SH-SY5Y cells pretreated 1h with chemical chaperone 4-PBA at 5mmol/L. (C and D) Western blotting demonstrated that 4-PBA mitigated H2O2-induced changes in the autophagy hallmark proteins, as well as the ER stress-related GRP78, IRE-1, ATF6, PERK and phosphorylated PERK. The values are expressed as mean±SEM (n=5 per group). *: p < 0.01.
Figure 5
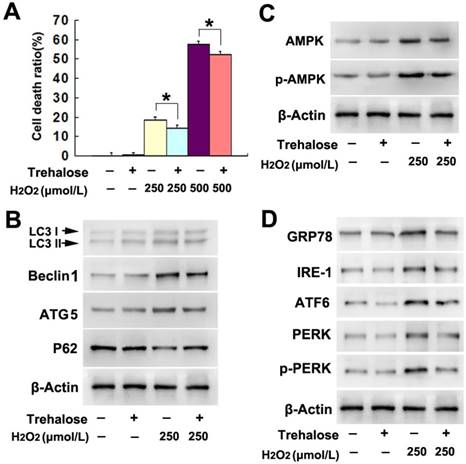
Trehalose inhibited H2O2-induced autophagic death. (A) LDH release assay showed that pretreatment 48h with trehalose at 5mmol/L significantly prevented H2O2-induced death in SH-SY5Y cells. Western blotting revealed that trehalose treatment reversed H2O2-induced changes in autophagy hallmark proteins (B), AMPK activation (C) and the upregulation of ER stress-related proteins (D). The values are expressed as mean±SEM (n=5 per group). *: p < 0.01.
Figure 6
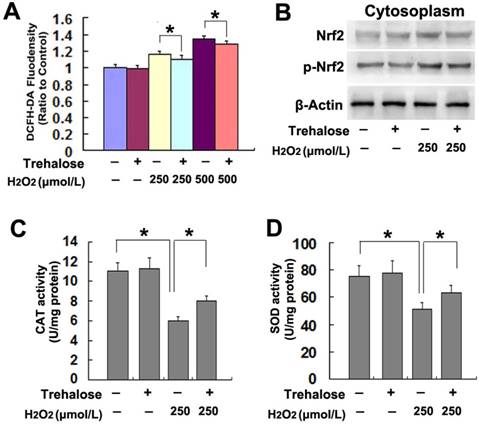
Trehalose inhibited H2O2-induced increase of intracellular ROS. (A) Statistical analysis showed that trehalose pretreatment prevented H2O2-induced increase of intracellular ROS. (B) Western blotting demonstrated that trehalose did not inhibit the upregulated expression of Nrf2 and phosphorylated Nrf2 caused by H2O2. (C and D)Enzyme activity assay revealed that trehalose suppressed H2O2-induced reduction in CAT and SOD. The values are expressed as mean±SEM (n=5 per group). *: p < 0.01.
Oxidative stress characterized with increase of intracellular reactive oxygen species (ROS) are believed to be involved in neuronal damage caused by head trauma and cerebral ischemia [22, 23]. ROS have been proven to trigger autophagic death in various types of cells including cardiomyocytes, vascular endothelial cells, renal tubular cells, neurons and glial cells [24-28]. H2O2 is often used to establish in vitro model of oxidative stress, given that it could not only diffuse easily into and out of cells and tissues, but also induce overproduction of intracellular ROS via targeting mitochondria [29]. It was also reported that treatment with exterior H2O2 resulted in mitochondrial permeability transition pore opening in SH-SY5Y cells [17]. Further, it was proved that hydroxyl radicals could be induced within the mitochondria of SH-SY5Y cells that were incubated with exterior H2O2 for 5min, and became apparent at incubation 30min [30]. Although autophagy, apoptosis and necrosis were all involved in H2O2-induced SH-SY5Y cell death [30, 31], Castino et al reported that autophagy preceded H2O2-induced apoptosis and necrosis in SH-SY5Y cells [30]. Thus, autophagy plays a crucial role in regulation of H2O2-induced cell death. We found in this study that either autophagy inhibitor 3MA or genetic knockdown of ATG5 prevented cell death caused by H2O2. Additionally, mitigation of intracellular ROS with antioxidant NAC inhibited H2O2-induced upregulation of autophagy hallmark proteins LC3II, Beclin-1, ATG-5 and downregulation of autophagy substrate p62. Thus, intracellular ROS contributed to exterior H2O2 induced autophagic death in SH-SY5Y cells.
Trehalose is often used as an autophagy inducer to prevent the cell death resulting from neurodegenerative diseases [32]. However, we found that trehalose protected SH-SY5Y cells against OGD-induced damage via inhibition of lethal autophagy [16]. Consistently, we demonstrated in this study that trehalose inhibited H2O2-induced autophagic death in SH-SY5Y cells. AMPK is an important pathway leading to ROS-induced autophagic cell death [19]. In this study, administration of antioxidant NAC prevented H2O2-induced phosphorylation of AMPK, and inhibition of AMPK activation with its specific inhibitor compound C attenuated H2O2-induced autophagic death in SH-SY5Y cells. Thus, AMPK was responsible for H2O2-induced lethal autophagy. Different from previous reports showing that AMPK could be activated by trehalose [33,34], our data showed that trehalose inhibited H2O2-induced AMPK phosphorylation. Further, we found that trehalose significantly inhibited H2O2-induced increase of intracellular ROS, which was supported by the in vivo study showing that trehalose prevented UVB-mediated oxidative stress in corneal epithelium [35]. Moreover, trehalose was reported previously to selectively decrease intracellular H2O2 [36], which was found to be associated with maintaining the activities of CAT, SOD, and glutathione peroxidase [35]. Consistently, we proved as well that trehalose significantly inhibited H2O2-induced reduction in the activities of SOD and CAT. Although Nrf2 represents one of the most important cellular endogenous defense mechanisms against oxidative stress [37], we found that trehalose did not regulate Nrf2 phosphorylation in the cells not treated with H2O2, but attenuated H2O2-induced phosphorylation of Nrf2. Thus, the effect of trehalose against oxidative stress is not related to Nrf2 activation.
ER stress is a crucial factor leading to autophagy and could be activated by intracellular ROS [38]. Mild ER stress (unfolded protein response) exerts protection against cell damage, but sustained ER stress leads to cell death via apoptosis and autophagic death [39]. The mechanism underlying the ER stress induced by oxidative stress remains elusive, but it was found that ROS elicited ER stress response via attacking and modifying ER proteins [40]. Moreover, ROS could disrupt protein folding mechanism and enhance the production of misfolded proteins, which results in ER stress [41]. ER stress regulates cell destiny via activating three pathways including IRE1, PERK and ATF6 [38, 42]. In this study, we proved that H2O2 induced concentration-dependent upregulation of IRE1, ATF6, PERK and phospho-PERK, which were significantly alleviated when intracellular ROS was mitigated by antioxidant NAC. Thus, H2O2 induced ER stress via increase of intracellular ROS.
Mounting evidences have shown that IRE1, PERK and ATF6 have regulatory effect on autophagy [38, 42, 43]. Pharmacological inhibition of either IRE1 or PERK suppressed the autophagic death in the neurons stressed by transient ischemia and reperfusion [43]. Genetic knockdown of IRE1 or PERK was reported to block the ROS-dependent lethal autophagy initiated by ursolic acid in human U87 glioma cells [38]. Additionally, ATF6 is also involved in the ER stress-induced autophagy, because knockdown of ATF6 prevented quinocetone-induced autophagy in HepG2 cells [42]. ER stress could be inhibited by supplement of exterior chaperone [44, 45].4-PBA is a kind of chemical chaperone and often used to mitigate ER stress. In vitro study proved that 4-PBA inhibited the SK-N-MC cell death induced by oxygen glucose deprivation and re-oxygenation via inhibition of ER stress-dependent autophagy [44]. In vivo investigation demonstrated as well that the ER stress induced by lipopolysaccharide in mouse was markedly prevented by supplement of 4-PBA [45]. In this study, we found that administration of 4-PBA not only inhibited H2O2-induced ER stress, but also inhibited the autophagic death caused by H2O2. Thus, ER stress contributed to the autophagic death caused by exterior H2O2.
Trehalose is also thought to be a chaperone and chaperone inducer [9,11,46]. Simola et al reported that trehalose was required for conformational repair of heat-denatured proteins in the yeast endoplasmic reticulum [46]. Tanji et al found that treatment with trehalose increased the levels of several chaperon molecules, such as HSP90 and SigmaR1 in the brains of LBD model mice [11]. Our data in this study revealed that trehalose obviously reversed H2O2-induced upregulation of IRE1, ATF6, PERK and phosphor-PERK. Thus, the ER stress resulting from H2O2 treatment was mitigated in the presence of trehalose. GRP78 is an inducible endogenous molecular chaperone within the lumen of ER, which could inhibit ER stress when was over-expressed [47]. Interestingly, it was reported that downregulation of GRP78 significantly suppressed cadmium-induced senescence in PC12 cells via inhibition of autophagy [48]. Notably, we found in this study that trehalose did not up-regulate the expression of GRP78, but inhibited its over-expression induced by H2O2, which was similar to the effect of NAC or 4-PBA. Considering that trehalose could inhibit intracellular ROS, we thus think that trehalose inhibited H2O2-induced lethal autophagy was more closely associated with its inhibitory effect on ROS, than as a chaperone or chaperone inducer.
In summary, we demonstrated in this study that trehalose exerted protection against H2O2-induced autophagic death in SH-SY5Y cells via inhibition of ROS-dependent ER stress and AMPK activation. Therefore, trehalose is a potential medicine preventing cell death resulting from autophagy over-activation.
Acknowledgements
This work was supported by National Nature and Science Foundation of China (81171234, 81772669, 31371125, 81571264, 81771396, 81171220, 81401068), Changbaishan Scholar Project of Jilin Province (2013026), and Scientific Research Foundation of Jilin province (20160101127JC, 20160101123JC), Pediatric Development Fund of the First Hospital of Jilin University, Foundation of Health and Family Planning Commission of Jilin Province(2010Z030), Special Fund Project of Industrial innovation of Jilin province(2017C029-1).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mariño G. Madeo F, Kroemer G. Autophagy for tissue homeostasis and neuroprotection. Curr Opin Cell Biol. 2011;23:198-206
2. Wang J, Liu Y, Li XH, Zeng XC, Li J, Zhou J. et al. Curcumin protects neuronal cells against status-epilepticus-induced hippocampal damage through induction of autophagy and inhibition of necroptosis. Can J Physiol Pharmacol. 2017;95:501-9
3. Hu X, Song Q, Li X, Li D, Zhang Q, Meng W. et al. Neuroprotective effects of Kukoamine A on neurotoxin-induced Parkinson's model through apoptosis inhibition and autophagy enhancement. Neuropharmacology. 2017;117:352-63
4. Huang M, Jiang X, Liang Y, Liu Q, Chen S, Guo Y. Berberine improves cognitive impairment by promoting autophagic clearance and inhibiting production of β-amyloid in APP/tau/PS1 mouse model of Alzheimer's disease. Exp Gerontol. 2017;91:25-33
5. Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B. et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487-98
6. Xu Y, Tian Y, Tian Y, Li X, Zhao P. Autophagy activation involved in hypoxic-ischemic brain injury induces cognitive and memory impairment in neonatal rats. J Neurochem. 2016;139:795-805
7. Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao HJ. et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54-63
8. Zheng Y, Hou J, Liu J, Yao M, Li L, Zhang B. et al. Inhibition of autophagy contributes to melatonin-mediated neuroprotection against transient focal cerebral ischemia in rats. J Pharmacol Sci. 2014;124:354-64
9. Tapia H, Koshland DE. Trehalose is a versatile and longlived chaperone for desiccation tolerance. Curr Biol. 2014;24:2758-66
10. He Q, Wang Y, Lin W, Zhang Q, Zhao J, Liu FT. et al. Trehalose alleviates PC12 neuronal death mediated by lipopolysaccharide-stimulated BV-2 cells via inhibiting nuclear transcription factor NF-κB and AP-1 activation. Neurotox Res. 2014;26:430-9
11. Tanji K, Miki Y, Maruyama A, Mimura J, Matsumiya T, Mori F. et al. Trehalose intake induces chaperone molecules along with autophagy in a mouse model of Lewy body disease. Biochem Biophys Res Commun. 2015;465:746-52
12. Echigo R, Shimohata N, Karatsu K, Yano F, Kayasuga-Kariya Y, Fujisawa A. et al. Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. J Transl Med. 2012;10:80
13. Pagliassotti MJ, Estrada AL, Hudson WM, Wei Y, Wang D, Seals DR. et al. Trehalose supplementation reduces hepatic endoplasmic reticulum stress and inflammatory signaling in old mice. J Nutr Biochem. 2017;45:15-23
14. He Q, Koprich JB, Wang Y, Yu WB, Xiao BG, Brotchie JM. et al. Treatment with Trehalose Prevents Behavioral and Neurochemical Deficits Produced in an AAV α-Synuclein Rat Model of Parkinson's Disease. Mol Neurobiol. 2016;53:2258-68
15. Perucho J, Casarejos MJ, Gomez A, Solano RM, de Yébenes JG, Mena MA. Trehalose protects from aggravation of amyloid pathology induced by isoflurane anesthesia in APP(swe) mutant mice. Curr Alzheimer Res. 2012;9:334-43
16. Li Y, Luo Y, Luo T, Lu B, Wang C, Zhang Y. et al. Trehalose Inhibits Protein Aggregation Caused by Transient Ischemic Insults Through Preservation of Proteasome Activity, Not via Induction of Autophagy. Mol Neurobiol. 2016 DOI:10.1007/s12035-016-0196-5
17. Feng C, Luo T, Zhang S, Liu K, Zhang Y, Luo Y. et al. Lycopene protects human SH-SY5Y neuroblastoma cells against hydrogen peroxide-induced death via inhibition of oxidative stress and mitochondria-associated apoptotic pathways. Mol Med Rep. 2016;13:4205-14
18. Ramos E, Patiño P, Reiter RJ, Gil-Martín E, Marco-Contelles J, Parada E. et al. Ischemic brain injury: New insights on the protective role of melatonin. Free Radic Biol Med. 2017;104:32-53
19. Li R, Zhou P, Guo Y, Lee JS, Zhou B. Tris (1, 3-dichloro-2-propyl) phosphate induces apoptosis and autophagy in SH-SY5Y cells: Involvement of ROS-mediated AMPK/mTOR/ULK1 pathways. Food Chem Toxicol. 2017;100:183-96
20. Holmström KM, Kostov RV, Dinkova-Kostova AT. The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol. 2016;1:80-91
21. Rashid K, Sinha K, Sil PC. An update on oxidative stress-mediated organ pathophysiology. Food Chem Toxicol. 2013;62:584-600
22. Lin CJ, Chen TH, Yang LY, Shih CM. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014;5:e1147
23. Wang M, Li YJ, Ding Y, Zhang HN, Sun T, Zhang K. et al. Silibinin Prevents Autophagic Cell Death upon Oxidative Stress in Cortical Neurons and Cerebral Ischemia-Reperfusion Injury. Mol Neurobiol. 2016;53:932-43
24. Wang S, Wang C, Yan F, Wang T, He Y, Li H. et al. N-Acetylcysteine Attenuates Diabetic Myocardial Ischemia Reperfusion Injury through Inhibiting Excessive Autophagy. Mediators Inflamm. 2017;2017:9257291
25. Zhang L, Wei J, Ren L, Zhang J, Wang J, Jing L. et al. Endosulfan induces autophagy and endothelial dysfunction via the AMPK/mTOR signaling pathway triggered by oxidative stress. Environ Pollut. 2017;220:843-52
26. Wang L, Mao N, Tan RZ, Wang HL, Wen J, Liu YH. et al. Ginsenoside Rg1 reduces aldosterone-induced autophagy via the AMPK/mTOR pathway in NRK-52E cells. Int J Mol Med. 2015;36:518-26
27. Yin WY, Ye Q, Huang HJ, Xia NG, Chen YY, Zhang Y. et al. Salidroside protects cortical neurons against glutamate-induced cytotoxicity by inhibiting autophagy. Mol Cell Biochem. 2016;419:53-64
28. Gorojod RM, Alaimo A, Porte Alcon S, Pomilio C, Saravia F, Kotler ML. The autophagic-lysosomal pathway determines the fate of glial cells under manganese- induced oxidative stress conditions. Free Radic Biol Med. 2015;87:237-51
29. Barbouti A, Doulias PT, Nousis L, Tenopoulou M, Galaris D. DNA damage and apoptosis in hydrogen peroxide-exposed Jurkat cells: Bolus addition versus continuous generation of H(2)O(2). Free Radic Biol Med. 2002;33:691-702
30. Castino R, Bellio N, Follo C, Murphy D, Isidoro C. Inhibition of PI3k class III-dependent autophagy prevents apoptosis and necrosis by oxidative stress in dopaminergic neuroblastoma cells. Toxicol Sci. 2010;117:152-62
31. Ye J, Han Y, Chen X, Xie J, Liu X, Qiao S. et al. L-carnitine attenuates H2O2-induced neuron apoptosis via inhibition of endoplasmic reticulum stress. Neurochem Int. 2014;78:86-95
32. Meng Y, Yong Y, Yang G, Ding H, Fan Z, Tang Y. et al. Autophagy alleviates neurodegeneration caused by mild impairment of oxidative metabolism. J Neurochem. 2013;126:805-18
33. Mayer AL, Higgins CB, Heitmeier MR, Kraft TE, Qian X, Crowley JR. et al. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci Rep. 2016;6:38586
34. DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE. et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal. 2016;9:ra21
35. Cejková J, Ardan T, Cejka C, Luyckx J. Favorable effects of trehalose on the development of UVB-mediated antioxidant/pro-oxidant imbalance in the corneal epithelium, proinflammatory cytokine and matrix metalloproteinase induction, and heat shock protein 70 expression. Graefes Arch Clin Exp Ophthalmol. 2011;249:1185-94
36. Lin CF, Kuo YT, Chen TY, Chien CT. Quercetin-Rich Guava (Psidium guajava) Juice in Combination with Trehalose Reduces Autophagy, Apoptosis and Pyroptosis Formation in the Kidney and Pancreas of Type II Diabetic Rats. Molecules. 2016;21:334
37. Peng S, Hou Y, Yao J, Fang J. Activation of Nrf2-driven antioxidant enzymes by cardamonin confers neuroprotection of PC12 cells against oxidative damage. Food Funct. 2017;8:997-1007
38. Shen S, Zhang Y, Zhang R, Tu X, Gong X. Ursolic acid induces autophagy in U87MG cells via ROS-dependent endoplasmic reticulum stress. Chem Biol Interact. 2014;218:28-41
39. Xiang C, Wang Y, Zhang H, Han F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis. 2017;22:1-26
40. Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Nishi T. et al. Oxidative damage to the endoplasmic reticulum is implicated in ischemic neuronal cell death. J Cereb Blood Flow Metab. 2003;23:1117-28
41. Chong WC, Shastri MD, Eri R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int J Mol Sci. 2017;18:E771
42. Zhou Y, Zhang S, Dai C, Tang S, Yang X, Li D. et al. Quinocetone triggered ER stress-induced autophagy via ATF6/DAPK1-modulated mAtg9a trafficking. Cell Biol Toxicol. 2016;32:141-52
43. Feng D, Wang B, Wang L, Abraham N, Tao K, Huang L. et al. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J Pineal Res. 2017:62 DOI: 10.1111/jpi.12395
44. Tung WF, Chen WJ, Hung HC, Liu GY, Tung JN, Huang CC. et al. 4-Phenylbutyric Acid (4-PBA) and Lithium Cooperatively Attenuate Cell Death during Oxygen-Glucose Deprivation (OGD) and Reoxygenation. Cell Mol Neurobiol. 2015;35:849-59
45. Zeng M, Sang W, Chen S, Chen R, Zhang H, Xue F. et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett. 2017;271:26-37
46. Simola M, Hänninen AL, Stranius SM, Makarow M. Trehalose is required for conformational repair of heat-denatured proteins in the yeast endoplasmic reticulum but not for maintenance of membrane traffic functions after severe heat stress. Mol Microbiol. 2000;37:42-53
47. Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J Biol Chem. 1997;272:4327-34
48. Wang T, Yuan Y, Zou H, Yang J, Zhao S, Ma Y. et al. The ER stress regulator Bip mediates cadmium-induced autophagy and neuronal senescence. Sci Rep. 2016;6:38091
Author contact
![]() Corresponding author: Jianmin Liang, M.D., Ph.D., E-mail: liangjmedu.cn. Address: Department of Pediatrics Neurology, First Hospital, Jilin University. 71 Xinmin Avenue, Changchun 130021, Jilin Province, P.R.China.
Corresponding author: Jianmin Liang, M.D., Ph.D., E-mail: liangjmedu.cn. Address: Department of Pediatrics Neurology, First Hospital, Jilin University. 71 Xinmin Avenue, Changchun 130021, Jilin Province, P.R.China.
Citation styles
APA
Gao, Z., Wang, H., Zhang, B., Wu, X., Zhang, Y., Ge, P., Chi, G., Liang, J. (2018). Trehalose inhibits H2O2-induced autophagic death in dopaminergic SH-SY5Y cells via mitigation of ROS-dependent endoplasmic reticulum stress and AMPK activation. International Journal of Medical Sciences, 15(10), 1014-1024. https://doi.org/10.7150/ijms.25656.
ACS
Gao, Z.; Wang, H.; Zhang, B.; Wu, X.; Zhang, Y.; Ge, P.; Chi, G.; Liang, J. Trehalose inhibits H2O2-induced autophagic death in dopaminergic SH-SY5Y cells via mitigation of ROS-dependent endoplasmic reticulum stress and AMPK activation. Int. J. Med. Sci. 2018, 15 (10), 1014-1024. DOI: 10.7150/ijms.25656.
NLM
Gao Z, Wang H, Zhang B, Wu X, Zhang Y, Ge P, Chi G, Liang J. Trehalose inhibits H2O2-induced autophagic death in dopaminergic SH-SY5Y cells via mitigation of ROS-dependent endoplasmic reticulum stress and AMPK activation. Int J Med Sci 2018; 15(10):1014-1024. doi:10.7150/ijms.25656. https://www.medsci.org/v15p1014.htm
CSE
Gao Z, Wang H, Zhang B, Wu X, Zhang Y, Ge P, Chi G, Liang J. 2018. Trehalose inhibits H2O2-induced autophagic death in dopaminergic SH-SY5Y cells via mitigation of ROS-dependent endoplasmic reticulum stress and AMPK activation. Int J Med Sci. 15(10):1014-1024.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.