Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(9):875-882. doi:10.7150/ijms.23074 This issue Cite
Research Paper
Morphine Induces Fibroblast Activation through Up-regulation of Connexin 43 Expression: Implication of Fibrosis in Wound Healing
Ping-Ching Wu1,2,3*, Wen-Li Hsu4*, Chun-Lin Chen5,6, Chen-Fuh Lam7, Yaw-Bin Huang6,8, Chien-Chi Huang9, Ming-Hong Lin10,11 ![]() , Ming-Wei Lin6,12
, Ming-Wei Lin6,12 ![]()
1. Department of Biomedical Engineering, National Cheng Kung University, Tainan, Taiwan
2. Institute of Oral Medicine and Department of Stomatology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University Tainan, Taiwan
3. Medical Device Innovation Center, Taiwan Innovation Center of Medical Devices and Technology, National Cheng Kung University Hospital, National Cheng Kung University, Tainan, Taiwan
4. Lipid Science and Aging Research Center, Kaohsiung Medical University, Kaohsiung, Taiwan.
5. Department of Biological Science, National Sun Yat-sen University, Kaohsiung, Taiwan
6. Center for Stem Cell Research, Kaohsiung Medical University, Kaohsiung, Taiwan
7. Department of Anesthesiology, E-Da Hospital/E-Da Cancer Hospital/I-Shou University, Kaohsiung, Taiwan.
8. School of Pharmacy, Kaohsiung Medical University, Kaohsiung, Taiwan
9. Department of Anesthesiology, National Cheng Kung University College of Medicine and Hospital, Tainan, Taiwan
10. Department of Microbiology and Immunology, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
11. Department of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
12. Department of Medical Research, E-Da Hospital/E-Da Cancer Hospital, Kaohsiung, Taiwan
*Equal contributors
Received 2017-9-28; Accepted 2018-2-2; Published 2018-6-4
Abstract
Morphine is the most effective drugs for attenuating various types of severe pain, but morphine abuse carries a high risk of systemic fibrosis. Our previous have indicated that systemic administration of morphine hinders angiogenesis and delays wound healing. Here we have explained the pathological mechanism underlying the effect of morphine on wound healing. To determine how morphine affects wound healing, we first created a wound in mice treated them with a combination of a low doses (5 mg/kg/day) and high doses (20 or 30 mg/kg/day) of morphine. An In vivo study revealed that high-dose morphine-induced abnormal myofibroblasts persist after the end of wound healing because of connexin 43 (Cx43) upregulation. High-dose morphine-induced Cx43 increased the expression levels of focal adhesion molecules, namely fibronectin and alpha-smooth muscle actin (α-SMA) through the activation of transforming growth factor (TGF)-β1 signaling. In addition, we found that Cx43 contributed to TGF-βRII/ Smad2/3 signaling for regulating the differentiation of fibroblasts into myofibroblasts during high-dose morphine exposure. In conclusion, the abnormal regulation of Cx43 by morphine may induce systemic fibrosis because of abnormal myofibroblast function.
Keywords: Morphine, Wound Healing, Fibrosis, Cx43
Introduction
Morphine is the most effective drugs for attenuating various types of severe pain. High doses of morphine are commonly used for controlling severe wound pain, such as in burn, cancerous wounds, and large surgical wounds [1]; however, complications and serious side effects of morphine use, including pulmonary fibrosis, lipid fibrosis and hepatic damage, have been reported after long-term morphine use [2, 3]. Although the pathological mechanism of high-dose morphine in inducing fibrosis remains unclear, our previous study suggested that the long-term use of high-dose morphine impaired angiogenesis, increased systemic oxidative stress, and hindered migration of endothelial progenitor cells [4]. Systemic sclerosis (SSc) is a systemic fibrosis disease that is characterized by deficient angiogenesis and increased oxidative stress with inflammatory response [5]. The symptoms of SSc cause systemic vascular resistance because of hypothermia in skin and pulmonary fibrosis [6, 7]. It implicates the high dose morphine facilitates the induction of not only pulmonary or lipid fibrosis but also systemic fibrosis.
SSc-induced fibrosis causes replacement of normal tissue architecture with excessive deposition of extracellular matrix (ECM) in response to inflammation in or damage to the skin [8]. The initial cellular moderators of SSc-induced fibrosis are collagen-producing myofibroblasts, which are activated by paracrine and autocrine signals, in response to fibroblasts injury [9]. Transforming growth factor-β (TGF-β) is the key regulator in SSc malignance. It disrupts the normal balance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases. It causes excessive synthesis of ECM and impairs ECM catabolism, thus leading to collagen accumulation and subsequent fibrosis [10]. Several molecules, such as alpha smooth muscle actin (ɑ-SMA) [11], fibronectin [11], S100A4, fibroblast specific protein 1 (FSP 1) [12] and connexins [13], are upregulated in myofibroblasts to regulate the pathological functions; either ɑ-SMA or fibronectin is involved in regulating focal adhesions in myofibroblasts, and both proteins play a crucial role in the development of fibrotic disorders [11]. S100A4 modulates cell shape and motility by interacting with components of the cytoskeleton; it is involved in the interconversions that occur between keratocytes, fibroblasts, and myofibroblasts during wound healing [12]. In addition, connexins, are believed to play a crucial role in governing and development of tissues. Connexin43 (Cx43) is the most widely expressed connexin, it is found in the endothelium and fibroblasts and is implicated in wound healing [14, 15]. Cx43 expression increases on wound closure and Cx43 levels control myofibroblasts differentiation [13]. Knockdown of Cx43 expression has recently been demonstrated to accelerate wound healing by reducing the number of α-SMA-positive myofibroblasts and inducing endothelial proliferation in vivo [16]. Long-term improvement of the rate of wound healing involved a significant reduction in the extent of granulation tissue deposition and subsequent formation of smaller and less distorted scars. Hence, studies have suggested that inhibition of Cx43 is a new therapeutic approach for prevention of fibrous membrane formation [17-20].
Our previous study revealed that morphine enhances accumulation of collagen in incisional wound tissues in a dose-dependent manner. Furthermore, in the study, the expression levels of TGF-β and MMP-2 proteins were significantly enhanced in morphine-treated mice [21]. We hypothesized that high-dose morphine induces systemic fibrosis through activation of the TGF-β signaling pathway. Cx43 causes Smad family signaling to mediate the differentiation of cardiac fibroblasts into myofibroblasts [22]. Thus, high-dose morphine activated Cx43 expression and further regulated α-SMA through the TGF-β signaling pathway, which induces myofibroblast formation. Our study indicated that high-dose morphine induces differentiation of fibroblasts into myofibroblasts through stimulation of α-SMA expression in Cx43-dependent TGF-β/Smad2/3 activation. Because the presence of α-SMA-positive myofibroblasts is a critical factor during wound healing, these findings indicate the role of Cx43 expression in morphine-induced systemic fibrosis.
Materials and Methods
Animal model
Mice (C57BL/6J, 8-10 weeks old) were obtained from the Animal Center of the National Cheng Kung University (Tainan, Taiwan). Mice were dorsal shaved and sprayed with 70% ethanol. A full-thickness incisional wound (approximately 2 cm in length) was created by a surgical scissors and crack closure on the dorsum of anesthetized animals. The wound was closed by interrupted suture using a 4º- nylon thread. Mice were randomly assigned to control or morphine-treated group and received normal saline or morphine (5 or 20 mg/kg/d, i.p.) injection for 14 consecutive days, as described in our previous study. All procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (The National Cheng Kung University College of Medicine, Tainan, Taiwan).
Cell culture
Human normal skin fibroblasts, WS1, were obtained from American Type Culture Collection (ATCC) and cultured in MEM-α with 10% FBS in a humidified 5% CO2 at 37°C.
Western Blot Analysis
Extracted protein were loaded into polyacrylamide gels and transferred onto PVDF membranes. The membranes were blocked in 5% nonfat milk containing 0.3% tween 20, and then probed with anti-Cx43 (BD Transduction Labs), anti-fibronectin (BD Transduction Labs), anti-β-actin (sigma), anti-GAPDH (Abcam), anti-α-SMA (Abcam) p-smad2/3 (Abcam), TGFβR2 (Abcam) or anti-S100A4 (Abcam) antibody at 4°C overnight. After washing, the membranes were incubated with horseradish peroxidase-linked secondary antibody, and bands were visualized using enhanced chemiluminescence system (GE Healthcare) and then exposing the blots. Protein levels were quantified by scanning densitometry (Alpha Image).
RT-PCR Analysis
Total RNA was isolated from WS1 cells with Trizol reagent (Invitrogen, Carlsbad, CA) and reversed transcribed into cDNA using Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA). After reverse transcription, amplification of mRNA was done by PCR SuperMix from Invitrogen with specific primer under the following conditions: 1 cycle of 94 °C for 3 min, 28 cycles composed of 30 sec denaturation at 94 °C, 30 sec primer annealing at 57 °C, 1 min extension at 72 °C, and followed by 72 °C for the final extension for 7 min. PCR products were analyzed on 1.5% (w/v) agarose gel containing ethidium bromide and then visualized under ultraviolet light.
Transfection of siRNA
Commercialized custom Stealth™ RNAi for human Cx43 and scrambled negative control siRNA, which does not interfere with any known mRNA, were obtained from Invitrogen (Invitrogen, Carlsbad, CA). WS1 cells were transfected with 25 nM of the siRNAs using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's protocol. Briefly, gene-specific siRNA oligomers were diluted in Opti-MEM I reduced serum medium (Opti-MEM, Invitrogen, Carlsbad, CA) and mixed with transfection reagent. After 20 min incubation at room temperature, the complexes were added to the cells. Transfected cells were incubated at 37 ºC for 48 hours and harvested. Message RNA level was evaluated using RT-PCR.
nM of the siRNAs using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's protocol. Briefly, gene-specific siRNA oligomers were diluted in Opti-MEM I reduced serum medium (Opti-MEM, Invitrogen, Carlsbad, CA) and mixed with transfection reagent. After 20 min incubation at room temperature, the complexes were added to the cells. Transfected cells were incubated at 37 ºC for 48 hours and harvested. Message RNA level was evaluated using RT-PCR.
Immunofluorescence
The unfixed frozen wound tissues segments were sectioned with a cryostat and placed on glass slides. Cut OCT-embedded tissues sections (10μm thick) were stained with analyzed immunofluorescence. Block each section with 2% BSA blocking buffer for 60 min at room temperature. Incubation for overnight at 4℃ with anti-Cx43 (1:1000, BD Transduction Labs); anti-S100A4 (1:500, Abcam). Sections were washed then second antibody incubated for 2 hrs at room temperature l with fluorescence-conjugated secondary antibodies Alexa Fluor® 488 goat anti-mouse (Invitrogen, Carlsbad, CA) and Alexa Fluor® 594 goat anti-rabbit (Invitrogen, Carlsbad, CA). Measurement of green and red fluorescence labeling by a laser scanning were imaged by confocal imaging system.
Statistical analyses
Statistical comparisons were performed by means of a Student's t-test. The statistical significance was set at P values less than 0.05.
Results
Myofibroblasts persist after the end of wound healing because of high-dose morphine treatment
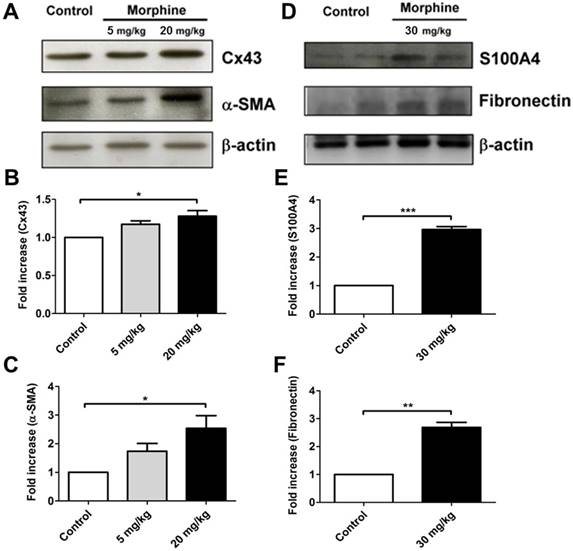
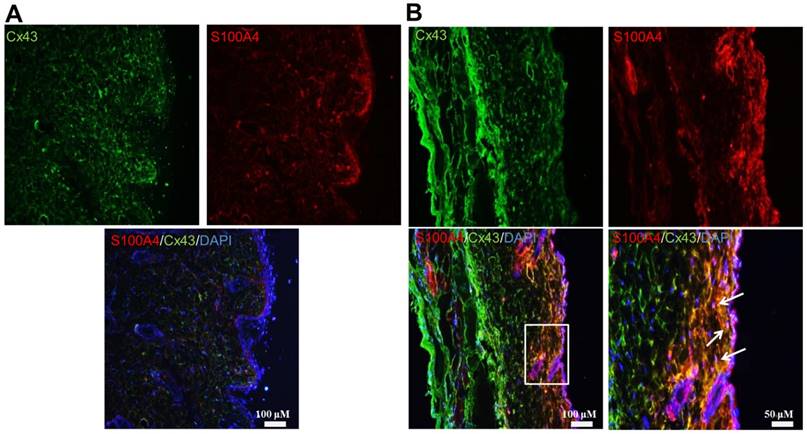
To investigate the role of morphine in regulating systemic fibrosis, we first observed wound healing in animals treated with low-dose and high-dose morphine. Systemic fibrosis, particularly SSc, is cause by excessive deposition of ECM components by myofibroblasts after injury [23]. Myofibroblasts either differentiate into fat cells or undergo apoptosis. Consequently, a scar is formed during wound healing [24, 25]. However, in SSc, the myofibroblasts continue to remodel the ECM even after the end of wound healing [25], thereby causing disease. Herein, we collected wound tissue at the end of wound healing from control and morphine-treated animals. On day 14 of wound healing, high-dose morphine increased the expression levels of Cx43, α-SMA, fibronectin and S100A4, which are key molecules in myofibroblasts presentation (Fig. 1). Our previous study indicated the accumulation of collagen as well as deficient angiogenesis in incisional wound tissue. In this study, we demonstrated that the pathological effects of high-dose morphine were observed in myofibroblasts. As shown in Fig. 2, myofibroblasts persisted after the end of wound healing with upregulation of Cx43 and S100A4 in the group treated with high-dose morphine, but not in the control group. However, thus far, no clear evidence has explained how high-dose morphine-induced pathological myofibroblasts persist after the end of wound healing, despite the side effects of morphine, namely hypothermia or pulmonary fibrosis. Cx43, which regulates differentiation of fibroblasts into myofibroblasts, is upregulated at the end of wound healing under high-dose morphine treatment. In our study, Cx43 was potentially involved in high-dose morphine-induced systemic fibrosis. Furthermore, the results suggested that high-dose morphine-induced pathological myofibroblasts persisted after the end of wound healing because of Cx43 upregulation.
Morphine increased the expressions of Cx43 and focal adhesion markers in vivo. Western blot analysis for expressions of (A) Cx43, and α-SMA obtained from the wound tissue homogenates isolated from the control and morphine-treated (5 and 20 mg/kg/day) mice. Expression levels of Cx43 and α-SMA were quantified as shown in (B) and (C) respectively. (*, p < 0.05) (D) Expression levels of S100A4 and fibronectin were obtained from the control and morphine-treated (30 mg/kg/day) mice. Wound tissues were obtained on day 14 after creation of incisional wound. The expressions of S100A4 and fibronectin were quantified and are shown in (E) and (F), respectively. (**, p < 0.01; ***, p < 0.001)
Myofibroblasts persisted after the end of wound healing in high-dose morphine treated mice. Wound tissues were obtained from the control and morphine-treated (30 mg/kg/day) mice on day 14 after creation of incisional wounds. Immunofluorescence staining in the wound tissue of (A) control group and (B) morphine-treated group; Cx43 and S100A4 exhibited conjugated green fluorescence and red fluorescence, respectively, which were co-localized and merged with yellow color in skin tissue after morphine treatment. The boxed region is magnified in the merged region arrows indicate the position of myofibroblasts.
Cx43 caused high dose morphine-induced expression of focal adhesion molecules in the WS-1 cell
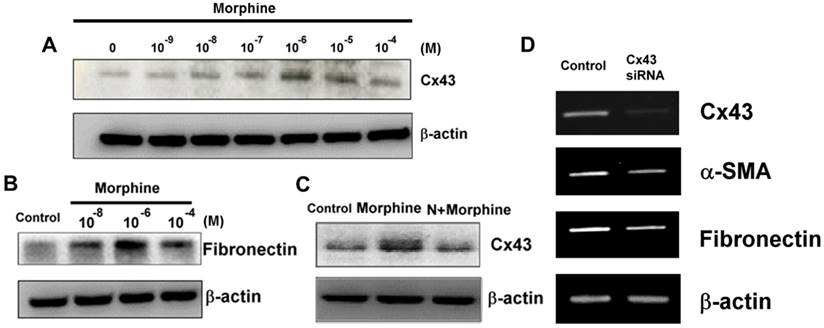
We further explored the effects of Cx43 on the differentiation of pathological fibroblasts into myofibroblasts under high-dose morphine treatment. As shown in Fig. 3A, morphine directly facilitates induction of Cx43 expression in WS-1 cells, and this phenomenon is restrained by naloxone (Fig. 3C), an antagonist that reverses the adverse effects of morphine [26]. Focal adhesion molecules, namely fibronectin and α-SMA (data not shown), were also upregulated because of high-dose morphine treatment (Fig. 3B). These findings implied that high-dose morphine induced the differentiation of fibroblasts into myofibroblasts by increasing the levels of focal adhesion molecules. Notably, knockdown of Cx43 exhibited low expression levels of fibronectin and α-SMA (Fig. 3D), thus indicating that morphine-induced-Cx43 expression may regulate the levels of focal adhesion molecules through several mechanisms. Cx43 is involved in modulating TGF-β signaling, which stimulates the differentiation of cardiac fibroblasts into myofibroblasts [22]. High-dose morphine-induced-Cx43 expression activated TGF-β signaling and increased fibronectin and α-SMA levels. Our previous study revealed that high-dose morphine significantly enhances accumulation of TGF-β and MMP-2 during wound healing in mice and increases wound tensile strength [27]; hence, Cx43 is crucial for the regulation of high-dose morphine-induced pathological effects of SSc in fibroblasts. Our findings indicated that pathological myofibroblast formation after the end of wound healing is caused by the promotion of focal adhesion molecules by high-dose morphine-induced Cx43.
High-dose morphine stimulated α-SMA expression levels depending on Cx43 expression levels. Treatment of WS-1 cells with morphine increased the expression levels of (A) Cx43 and (B) fibronectin. (C) Morphine (10-4 μM)-induced Cx43 expression was reversed by naloxone (N, 10-4 μM). (D) Knockdown of Cx43 expression attenuated mRNA levels of Cx43, α-SMA and fibronectin in the WS-1 cells.
High dose morphine-induced fibroblasts to differentiation through the Cx43-dependent TGF-βRII signaling pathway
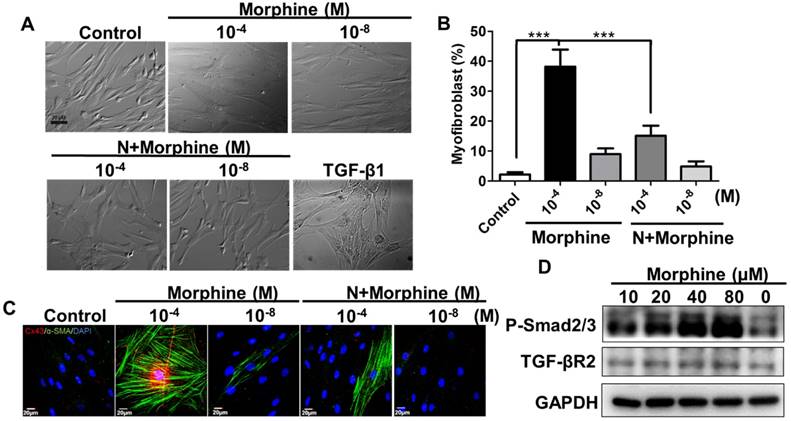
Next, we tested whether high-dose morphine-induced differentiation of fibroblasts into myofibroblasts through the Cx43-activated TGF-β signaling pathway. As previously discussed, myofibroblast formation after the end of wound healing under high-dose morphine treatment, implied that high dose morphine treatment facilitated the differentiation of fibroblasts into myofibroblasts. A comparison of the percentages of myofibroblasts between high-dose (10-4 M) and low dose (10-8 M) morphine treatments revealed that high-dose morphine treatment increased the differentiation of fibroblasts into myofibroblasts by approximately 40% (Fig. 4A, 4B) and upregulated Cx43 and α-SMA expression levels (Fig. 4C). However, co-treatment with naloxone attenuated the ratio of high-dose morphine-induced-fibroblast differentiation to 17%, and reduced the expression levels of Cx43 and α-SMA (Fig. 4C). To determine whether Cx43-regulated TGF-β signaling to promote fibroblast differentiation, we analyzed the protein levels of phospho-Smad2/3 and TGF-βR2. Our previous study explored the upregulation of TGF-β1 in incisional wound tissue under high-dose morphine exposure [27]. When TGF-β1 binds to the TGF-βRII recruiting TGF-βRI receptor, Smad2/3 is translocated to the receptor complex, phosphorylated, and incorporated to form a heteromeric complex with Smad4 [28]; this Smads complex translocates to the nucleus and binds to the Smad-binding elements to activate expression of downstream genes, such as α-SMA, S100A4 or fibronectin [29-31]. In our study, we found that morphine increased the level of phospho-Smad2/3 and TGF-βRII in a dose dependent manner (Fig. 4D). Taken together, high-dose morphine-induced-Cx43 resulted in TGF-βRII/ Smad2/3 signaling to regulate differentiation of fibroblasts into myofibroblasts.
High-dose morphine induced differentiation of fibroblasts to myofibroblasts through activation of the TGF-βRII/ Smad2/3 signaling pathway. (A) Morphine (10-4 μM) elicited differentiation of fibroblasts into myofibroblasts after 3 days of treatment; TGF-β1-induced myofibroblasts were used as a positive control. (B) Quantification of differentiation of fibroblasts into myofibroblasts in (A); naloxone significantly reduced the number of myofibroblasts differentiated because of morphine. (***, p < 0.001) (C) Immunofluorescence analysis exhibited high-dose morphine-induced expressions and distribution of Cx43 (red) and α-SMA (green), which were restrained through treatment with naloxone. (D) Morphine increased TGF-β receptor type-II (TGF-βR II) and phospho-Smad2/3 (p-Smad2/3) expression levels in a dose dependent manner.
Discussion
Our previous study demonstrated that systemic administration of high-dose morphine accelerates collagen accumulation in cutaneous tissues, thus increasing the tensile strength of incisional wounds [21]. The present results suggest that the pathological mechanism of the effects of high-dose morphine on incisional wounds involve the presence of myofibroblasts, which are differentiated from fibroblasts through the Cx43 activated TGF-βRII/ Smad2/3 signaling pathway. Cx43 plays a role in regulating wound closure, and controls myofibroblast differentiation; however, upregulation of Cx43 maintains myofibroblast existence after the end of wound healing (Fig. 1). The preservation of myofibroblasts implied that high-dose morphine facilitied induction of systemic fibrosis during wound healing. Furthermore, high dose morphine induced-Cx43 may be involved in the progression of epithelial mesenchymal transition (EMT), which is regulated by the TGF-β signaling pathway [32]. In this study, high-dose morphine upregulated Cx43 expression, thereby contributing to the modulation not only fibroblast differentiation but also endothelial cell function in wound repair. As shown in Fig. 1 and Fig 2, wound tissue analysis revealed that high-dose morphine also promoted Cx43 expression throughout the entirety of the skin. Thus, an increase Cx43 expression may promote EMT through transition of endothelial cells toward mesenchymal phenotypes (smooth muscle-like phenotype). EMT is a crucial contributor to vascular development and remodeling. MMP-2 has been proposed to play a vital role in the EMT process through removal of vascular endotelial-cadherin. Our future work will explain the mechanisms underlying morphine-induced fibrosis and EMT by assessing the tensile strength of wounds in morphine-treated mice to estimate th eextent of morphine-induced fibrosis.
Chronical morphine application can contribute to delayed wound healing [33]. High-dose morphine probably contributes to endothelial cell dysfunction. Gap junctions are crucial for regulating and coordinating vascular function through direct intercellular communication [34]. Overexpression of Cx43 interrupt intercellular communication by increasing the level of focal adhesion molecules, such as α-SMA or fibronectin, following upregulation of the endothelial marker CD31, which interferes with vascular development [35]. Notably, serum levels of soluble CD31in patients with SSc are higher than those of healthy controls [36], thus confirming that deficient angiogenesis is associated with dysfunctional endothelial cells in systemic fibrosis diseases. Furthermore, morphine-induced delay of wound healing may also be associated with endothelial cell growth arrest. TGF-β signaling promotes the differentiation of endothelial cells into smooth muscle-like cells and disrupts the proliferation of endothelial cells. Furthermore, cell-cell contact itself appears to be responsible for restraining endothelial proliferation and thus maintaining a mature endothelial monolayer in a non-proliferative state; however, upregulation of the cell-cell communication, such as Cx43, downregulates endothelial cell proliferation [37], and potentially impairs angiogenesis by dysfunctional endothelia cells.
TGF-β induces tissue fibrosis through myofibroblasts pathology and Smads signaling [38]. Recent studies have demonstrated that the cooperative cascades of p38 and PI3K/AKT are involved in TGF-β1-induced Cx43 expression [39] and that Cx43 contributes to the TGF-β/Smad2/3 signaling pathway to regulate gap junctional communication [22]. Thus, morphine induces TGF-β1 and Cx43 expression, thereby potentially providing a positive-feedback network that involves Smads signaling. Moreover, an abnormal balance between matrix metalloproteinases and tissues causes collagen accumulation. These findings suggest that high-dose morphine or chronic morphine use stimulates differentiation fibroblasts into myofibroblasts through Cx43/TGF-β1 signaling pathway, thereby presenting a high risk of potentiating systemic fibrosis.
Acknowledgements
The authors thank the support provided by the MOST (106-2320-B-650-002-, 106-2221-E-006-002-, 106-2119-M-006-008, 106-2119-M-038-001-, 105-2314-B-037-059-, 104-2321-B-037-003-MY3, 105-2812-8-006-002), Kaohsiung Medical University “Aim for the Top Universities Grant [KMU-TP105G00], [KMU-TP105G01] & [KMU-TP105G02], KMUDK106, and NSYSU-KMU Joint Research Project (KMU-NSYSU106, 107-I001).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rimaz S, Alavi CE, Sedighinejad A, Tolouie M, Kavoosi S, Koochakinejad L. Effect of gabapentin on morphine consumption and pain after surgical debridement of burn wounds: a double-blind randomized clinical trial study. Archives of trauma research. 2012;1:38-43
2. Gottlieb LS, Trey C. The effects of fluorinated anesthetics on the liver and kidneys. Annual review of medicine. 1974;25:411-29
3. Bekheet SH. Morphine sulphate induced histopathological and histochemical changes in the rat liver. Tissue Cell. 2010;42:266-72
4. Lam CF, Chang PJ, Huang YS, Sung YH, Huang CC, Lin MW. et al. Prolonged use of high-dose morphine impairs angiogenesis and mobilization of endothelial progenitor cells in mice. Anesthesia and analgesia. 2008;107:686-92
5. Shaw TJ, Kishi K, Mori R. Wound-associated skin fibrosis: mechanisms and treatments based on modulating the inflammatory response. Endocrine, metabolic & immune disorders drug targets. 2010;10:320-30
6. Bakal K, Danckers M, Denson JL, Sauthoff H. Therapeutic hypothermia after cardiac arrest in a patient with systemic sclerosis and Raynaud phenomenon. Chest. 2015;147:e27-e30
7. Herzog EL, Mathur A, Tager AM, Feghali-Bostwick C, Schneider F, Varga J. Review: interstitial lung disease associated with systemic sclerosis and idiopathic pulmonary fibrosis: how similar and distinct? Arthritis & rheumatology. 2014;66:1967-78
8. Jimenez SA. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN rheumatology. 2013;2013:835948
9. Castelino FV, Varga J. Emerging cellular and molecular targets in fibrosis: implications for scleroderma pathogenesis and targeted therapy. Current opinion in rheumatology. 2014;26:607-14
10. Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of Systemic Sclerosis. Frontiers in immunology. 2015;6:272
11. Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ. Diverse origins of the myofibroblast-implications for kidney fibrosis. Nature reviews Nephrology. 2015;11:233-44
12. Ryan DG, Taliana L, Sun L, Wei ZG, Masur SK, Lavker RM. Involvement of S100A4 in stromal fibroblasts of the regenerating cornea. Investigative ophthalmology & visual science. 2003;44:4255-62
13. Paw M, Borek I, Wnuk D, Ryszawy D, Piwowarczyk K, Kmiotek K. et al. Connexin43 Controls the Myofibroblastic Differentiation of Bronchial Fibroblasts from Asthmatic Patients. American journal of respiratory cell and molecular biology. 2017
14. Ko K, Arora P, Lee W, McCulloch C. Biochemical and functional characterization of intercellular adhesion and gap junctions in fibroblasts. American journal of physiology Cell physiology. 2000;279:C147-57
15. Yeh HI, Lai YJ, Chang HM, Ko YS, Severs NJ, Tsai CH. Multiple connexin expression in regenerating arterial endothelial gap junctions. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:1753-62
16. Nakano Y, Oyamada M, Dai P, Nakagami T, Kinoshita S, Takamatsu T. Connexin43 knockdown accelerates wound healing but inhibits mesenchymal transition after corneal endothelial injury in vivo. Investigative ophthalmology & visual science. 2008;49:93-104
17. Kretz M, Euwens C, Hombach S, Eckardt D, Teubner B, Traub O. et al. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. Journal of cell science. 2003;116:3443-52
18. Coutinho P, Qiu C, Frank S, Wang CM, Brown T, Green CR. et al. Limiting burn extension by transient inhibition of Connexin43 expression at the site of injury. British journal of plastic surgery. 2005;58:658-67
19. Qiu C, Coutinho P, Frank S, Franke S, Law LY, Martin P. et al. Targeting connexin43 expression accelerates the rate of wound repair. Current biology: CB. 2003;13:1697-703
20. Nakano Y, Oyamada M, Dai P, Nakagami T, Kinoshita S, Takamatsu T. Connexin43 knockdown accelerates wound healing but inhibits mesenchymal transition after corneal endothelial injury in vivo. Investigative ophthalmology & visual science. 2008;49:93-104
21. Chang PJ, Chen MY, Huang YS, Lee CH, Huang CC, Lam CF. et al. Morphine enhances tissue content of collagen and increases wound tensile strength. Journal of anesthesia. 2010;24:240-6
22. Asazuma-Nakamura Y, Dai P, Harada Y, Jiang Y, Hamaoka K, Takamatsu T. Cx43 contributes to TGF-beta signaling to regulate differentiation of cardiac fibroblasts into myofibroblasts. Experimental cell research. 2009;315:1190-9
23. Darby IA, Hewitson TD. Fibroblast differentiation in wound healing and fibrosis. International review of cytology. 2007;257:143-79
24. Plikus MV, Guerrero-Juarez CF, Ito M, Li YR, Dedhia PH, Zheng Y. et al. Regeneration of fat cells from myofibroblasts during wound healing. Science. 2017;355:748-52
25. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nature reviews Molecular cell biology. 2002;3:349-63
26. Kugler J, Hug P, Doenicke A, Spatz R, Zimmermann W. [The effect of the morphine antagonist naloxone on the effect of fentanyl]. Arzneimittel-Forschung. 1978;28:1532-3
27. Chang PJ, Chen MY, Huang YS, Lee CH, Huang CC, Lam CF. et al. Morphine enhances tissue content of collagen and increases wound tensile strength. Journal of anesthesia. 2010;24:240-6
28. Assinder SJ, Dong Q, Kovacevic Z, Richardson DR. The TGF-beta, PI3K/Akt and PTEN pathways: established and proposed biochemical integration in prostate cancer. The Biochemical journal. 2009;417:411-21
29. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. The Journal of cell biology. 1993;122:103-11
30. Xie R, Schlumbrecht MP, Shipley GL, Xie S, Bassett RL Jr, Broaddus RR. S100A4 mediates endometrial cancer invasion and is a target of TGF-beta1 signaling. Laboratory investigation; a journal of technical methods and pathology. 2009;89:937-47
31. Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. The EMBO journal. 1999;18:1345-56
32. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell research. 2009;19:156-72
33. Egydio F, Tomimori J, Tufik S, Andersen ML. Does sleep deprivation and morphine influence wound healing? Medical hypotheses. 2011;77:353-5
34. Figueroa XF, Duling BR. Gap junctions in the control of vascular function. Antioxidants & redox signaling. 2009;11:251-66
35. Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. The American journal of pathology. 2003;163:1801-15
36. Sato S, Komura K, Hasegawa M, Fujimoto M, Takehara K. Clinical significance of soluble CD31 in patients with systemic sclerosis (SSc): association with limited cutaneous SSc. The Journal of rheumatology. 2001;28:2460-5
37. Larson DM, Wrobleski MJ, Sagar GD, Westphale EM, Beyer EC. Differential regulation of connexin43 and connexin37 in endothelial cells by cell density, growth, and TGF-beta1. The American journal of physiology. 1997;272:C405-15
38. Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Frontiers in pharmacology. 2014;5:123
39. Tacheau C, Fontaine J, Loy J, Mauviel A, Verrecchia F. TGF-beta induces connexin43 gene expression in normal murine mammary gland epithelial cells via activation of p38 and PI3K/AKT signaling pathways. Journal of cellular physiology. 2008;217:759-68
Author contact
![]() Corresponding authors: Ming-Wei Lin, Department of Medical Research, E-Da Hospital/E-Da Cancer Hospital, Kaohsiung 824, Taiwan. Tel: +886-7-6151100 ext.5413. E-mail: ta990074com, or Ming-Hong Lin, Department of Microbiology and Immunology, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan. Tel: +886-7-312-1101 ext. 2150 #11. E-mail: mhlinedu.tw
Corresponding authors: Ming-Wei Lin, Department of Medical Research, E-Da Hospital/E-Da Cancer Hospital, Kaohsiung 824, Taiwan. Tel: +886-7-6151100 ext.5413. E-mail: ta990074com, or Ming-Hong Lin, Department of Microbiology and Immunology, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan. Tel: +886-7-312-1101 ext. 2150 #11. E-mail: mhlinedu.tw