Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Gut microbiota and their...
Experimental methods of gut...
Patterns of RAS involved in DN
The interplay between kidney,...
Summary
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(8):816-822. doi:10.7150/ijms.25543 This issue Cite
Review
Intestinal dysbiosis activates renal renin-angiotensin system contributing to incipient diabetic nephropathy
Chen Chen Lu1, Kun Ling Ma1 ![]() , Xiong Zhong Ruan2, Bi Cheng Liu1
, Xiong Zhong Ruan2, Bi Cheng Liu1
1. Institute of Nephrology, Zhong Da Hospital, School of Medicine, Southeast University, Nanjing City, Jiangsu Province, China.
2. Centre for Nephrology, University College London (UCL) Medical School, Royal Free Campus, UK.
Received 2018-2-3; Accepted 2018-4-14; Published 2018-5-22
Abstract
Considerable interest nowadays has focused on gut microbiota owing to their pleiotropic roles in human health and diseases. This intestinal community can arouse a variety of activities in the host and function as “a microbial organ” by generating bioactive metabolites and participating in a series of metabolism-dependent pathways. Alternations in the composition of gut microbiota, referred to as intestinal dysbiosis, are reportedly associated with several diseases, especially diabetes mellitus and its complications. Here we focus on the relationship between gut microbiota and diabetic nephropathy (DN), as the latter is one of the major causes of chronic kidney diseases. The activation of renin angiotensin system (RAS) is a critical factor to the onset of DN, and emerging data has demonstrated a provoking and mediating role of gut microbiota for this system in the context of metabolic diseases. The purpose of the current review is to highlight some research updates about the underlying interplay between gut microbiota, their metabolites, and the development and progression of DN, along with exploring innovative approaches to targeting this intestinal community as a therapeutic perspective in clinical management of DN patients.
Keywords: Gut microbiota, diabetic nephropathy, renin-angiotensin system
Introduction
Diabetes mellitus (DM) is defined as a set of metabolic disorders that generally appears as hyperglycemia. According to the 8th international diabetes federation (IDF) diabetes atlas, there are approximately 425 million diabetic patients of 20-79 years old worldwide, and this number would increase to 693 if without measures taken. Such high incidence has also brought about increasing morbidity of diabetic nephropathy (DN), which remains as a major cause of chronic kidney diseases and ultimately leads to irreparable renal failure[1].
In last few years, considerable research has demonstrated an indispensable role of gut microbiota in sustaining metabolic homeostasis. This intestinal community exerts a variety of pathophysiologic effects and has been described as a “microbial organ”. Gut microbiota could help maintain human body homeostasis by hindering the growth of intestinal pathogens, fermenting unused energy matrix, or producing essential nutrients; however, the epithelial barriers of gut necessarily restrain the microbiota outside the circulation to avoid bacterial translocation [2] and systemic inflammation[3]. Intestinal dysbiosis, referred as alterations of the composition or metabolic status of gut microbiota, has been reported to increase permeability and augment mucosal immune responses of the gut, contributing to the progression of DM and insulin resistance[4, 5].
Renin-angiotensin system (RAS) has long been central to pathogenic and progressive changes of DN, and its effects seem more local rather than in the circulation[6]. Emerging evidence has suggested that intestinal microbiota is associated with the activation of local RAS not only in the gut, but in other vital organs as well, and in this context, especially the kidney.
Gut microbiota and their metabolites
There is approximately 100 trillion microbiota inhabiting human gut, with up to 5000 species and a high density of 1012 cells/ml of luminal contents[7]. The composition of gut microbiota in adults are probably related to long-term dietary patterns and can be referred as enterotypes, defined as a certain state of the equilibrium in different groups of gut microbiota, usually determined by the relative abundance of three predominant bacterial genera: Bacteroides, Prevotella and Ruminococcus[8]. Generally, Firmicutes and Bacteroidetes are the main phyla in the gut microbiota, taking up about 60-80% and 20-40% of the whole bacterial load; others are up to several extrinsic factors relevant to genetic makeup, diet and oral antibiotic use of early life, etc.
The metabolites of gut microbiota have been reported to exert many pathophysiologic effects, among which short chain fatty acids (SCFAs) have been increasingly recognized to be involved in both the maintenance of human health and the development of several diseases.
The roles of SCFAs are not only restricted to the intestine, but to participating in a series of biological activities at extra-intestinal locations as well. Gijs den Besten et al have found that intake of dietary fiber could improve glucose homeostasis which was associated with elevated SCFAs from the intestine to other organs rather than with the fecal SCFA concentrations[9]. Furthermore, disturbance of the microbiota resulting from antibiotics reportedly led to a thinner colon and protective mucus layer, indicating that elevated shift of SCFAs from the gut lumen would get into the surrounding blood vessels[10], which identifies the effect of gut microbiota in gut architecture and intestinal barrier. The composition of the microbiota decides the levels and ratios of their metabolites, among which short-chain fatty acids (SCFAs) are vital components[11]. Dysbiosis of the gut microbiota might reduce the production of SCFAs and depredate epithelial barrier integrity, leading to the disorder of energy and immune homeostasis.
Different microbial phyla might produce homogeneous metabolites[12]. As the primary products generated from gut microbial fermentation of dietary fiber[13], acetate and propionate are produced by the bacteria of Bacteroidetes at high levels, while high amounts of butyrate come from the Firmicutes phylum[14]. Butyrate and propionate are mostly purged by colon or liver, whereas acetate could enter the circulation or reach other tissues beyond the gut[15].
Butyrate-producing bacteria is essential to assure intestinal homeostasis. For instance, Firmicutes F. prausnitzii and Eubacterium rectale, both belong to Clostridium cluster which is one of the most abundant bacteria in a healthy colonic community. Patients with type 2 diabetes have shown a moderate difference of gut microbiota, specifically manifested as decreasing butyrate-producing bacteria and higher amounts of opportunistic pathogens compared to normal people[16, 17]. There has also been evidence of anti-diabetic effects from a sort of butyrate-producing clostridium via the upregulation of butyrate production and increasing SCFA receptors in the gut[18].
It was observed that under conditions of excessive fat oxidation like diabetes, the production of endogenous acetate predominates. The alternation of gut microbiota is probably responsible for increased production of acetate[19], which might also modulate acetate absorption[20], as germ-free mice had negligible acetate in their serum and tissues[19, 21]. Because of the role gut microbiota along with their metabolites play in host homeostasis and the fact that dietary interventions exert an important effect on the composition of gut microbiota, proper manipulation of microbial constituent has been assumed as a plausible approach for prevention and therapy of diseases. Marques F.Z. et al [22] have detected that a great deal of fiber consumption could modify gut microbial populations and elevate the abundance of acetate-producing bacteria. Moreover, supplementation of acetate could attenuate intestinal dysbiosis, which is assessed by a fluctuant ratio of Firmicutes to Bacteroidetes. Besides, acetate also markedly reduced glomerular and tubulointerstitial fibrosis. Coincidentally, compared to the controls treatment with acetate-producing bacteria could improve the renal injuries of mice after acute kidney injury[23], probably via the regulating of inflammatory processes and epigenetic modifications in the kidney by SCFAs.
Emerging evidence has suggested that SCFAs could leave the intestine via some transporters[24] and bind to corresponding receptors like G protein-coupled receptors (GPCRs) after being produced by the gut microbiota. GPR43 and Olfactory receptor (Olfr) 78 are both reportedly functional receptors for SCFAs[25, 26], and participate in physiological pathways in response to signals from the microbiota.
Compared with wild-type mice, Gpr43-/- mice have elevated fecal SCFA and plasma acetate concentrations, along with an increase of SCFA-producing bacteria in the gut, all of which were blunted by antibiotic treatment or under germ free conditions[27]. Miles Fuller et al have pointed out that treatment with antibiotics promotes GPR43-independent improvement in glucose tolerance, and reconstitution of the gut microbiota could withdraw such a benefit[28].
Olfr78 was originally discovered in the afferent arteriole of renal juxtaglomerular apparatus (JGA), at which renin is stored until finally released into the circulation[29]. It has been found that Olfr78, which responds solely to acetate and propionate, could mediate the release of renin induced by SCFAs[30].
Experimental methods of gut microbiota
Research on gut microbiota mainly apply interventions or foster certain animals that have a significant impact on the composition of intestinal microbiota, including fecal microbiota transplantation (FMT), antibiotic treatment and germ-free mice (see in Figure 1).
Experimental methods of gut microbiota. (a) fecal microbiota transfer (FMT); (b) germ- free mice and gnotobiotic mice which are implanted with a certain sort of microbiota; (c) antibiotic treatment via oral gavage or free drinking.
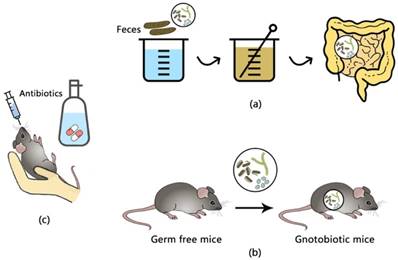
FMT is referred to infusing a suspension of feces from a donor into the intestine of a receptor, and it firstly came to be known as an effective treatment for recurrent Clostridium difficile infection[31]. The substantial change of fecal microbiota of the recipient would shift dramatically to those of the donor after the transplantation[32], and such changes have therapeutical perspectives as there has been evidence that intestinal infusions of microbiota from lean controls enhanced the insulin sensitivity of recipients with metabolic syndrome, additionally with elevated levels of butyrate-producing bacteria[33].
Germ-free (GF) animals are animals that have no microorganisms and raised within sterile isolators and have free access to autoclaved feeding materials, in order to insulate them from viral or bacterial agents[34]. When known strains of bacteria or microbiota are implanted into a GF animal, it is usually defined as a gnotobiotic animal[35], which are used to investigate the impact of certain microbial flora or genic function under the existence of known gut microbiota.
Antibiotic treatment via free drinking or oral gavage is a common approach to directly observe the influence of specific gut flora on the host as it can be more easily implemented. Although antibiotics eliminates gut microbiota and correspondingly reduce fecal SCFAs which are expected to decrease circulating SCFAs, it has been reported in wild-type mice that antibiotics, vice versa, increased the concentrations of plasma SCFAs and acetate levels[28].
Patterns of RAS involved in DN
RAS exerts both general and local functions: circulatory RAS regulates blood pressure and fluid homeostasis, while local RAS operates within tissues or organs. The kidney is unique in owing all RAS members within renal tubule, interstitium and even intracellular distribution[36].
As the major effector of RAS, angiotensin II (Ang Ⅱ) is mostly generated via the cleavage of Ang І by angiotensin converting enzyme (ACE), which is abundant in the endothelial cells of renal vasculature of rats and the renal tubules of humans. Stimulated by renin, Angiotensinogen (AGT) locally produced from proximal tubular cells would form Ang І, which can also be delivered to the kidney.
High glucose could stimulate the production of Ang Ⅱ, the activities of which possibly contribute to diabetic nephropathy through: higher glomerular capillary pressure and permeability, excessive mesangial matrix accumulation induced by TGF-β and inhibited matrix degradation, insulin resistance, etc[37].
Multiple intrarenal activities of Ang II are induced via binding to Ang II type 1 (AT1R) receptors which are localized on kidney arterioles, glomerular mesangial cells and the membrane of proximal tubular cells[38]. The Ang II - AT1R couple imposes effects including constricting both afferent and efferent arterioles, promoting mesangial cells contraction and decreasing medullary blood flow[37]. The renoprotective effects of ACE inhibition are partly through a decrease of glomerular capillary pressure[39], and activation of AT1R in the glomerulus is sufficient to accelerate renal injury and inflammation[40]. The extensive apply of both angiotensin-converting enzyme inhibitor (ACEI) and angiotensin receptor blockade (ARB) in treatment of DN suggests that Ang II is an irritating factor of progressive renal injuries[41].
Discovery of new components in recent years has broadened the perception beyond classic RAS. Accumulating hints have shown that ACE2-regulated Ang1-7 production represents a more effective target for the renal RAS than the circulation[6].
Discovered as a homolog of ACE, ACE2 was initially found to be expressed in tubular epithelial cells of the kidney at high levels[42] and could ameliorate renal diseases by promoting the degradation of local Ang II[43], and deficiency of ACE2 could aggravate Ang II-induced renal fibrosis and inflammation[44]. Moreover, ACE2 has been shown protective of endothelial function[45] and its overexpression seemed to inhibit collagen production[46], both of which could retard the progression of DN.
Ang II can further be hydrolyzed by ACE2 to form Ang 1-7, which in turn signals through the Mas receptor[42] to offset the effects of Ang II binding to AT1R. It has been reported that Mas knockout mice had a relatively higher degree of glomerular hyperfiltration and fibrogenic changes, along with an upregulation of AT1R and TGF-β expression[47]. On the other hand, Ang1-7 could mediate afferent arteriolar vasodilatation and antagonize renal vasoconstrictor effects of Ang II even if devoid of any vasodilator actions by itself[48]. Therefore, this ACE2/Ang 1-7/Mas axis might negatively regulate the activity of classic RAS and probably exerting renoprotective effects[49].
Physiologically speaking, a balance between the ACE/Ang II/AT1R and ACE2/Ang 1-7)/Mas axes of the RAS is critical for renal hemodynamics. It is reported that in DN there exists a downregulation of the ACE axis whereas the ACE2 axis is stimulated[50-52], indicating of a self-protective effect in response to diabetic injuries of the kidney. Coincidentally, deletion of the ace2 gene and glomerular ACE2 overexpression reversed diabetes-related renal lesions[53, 54]. There is still controversy on the expression changes of these two axes in DN[55], the reason for such discrepancy is unclear but might relate to variable antibody sensitivity or specificity. However, novel strategies are under investigation to augment actions of the vasoprotective RAS components, particularly ACE2, in order to treat DN-relevant injuries. In diabetic models of mice, administration of ACE2 inhibitor resulted in more severe albuminuria[50, 53].
The interplay between kidney, RAS and gut microbiota
It is pointed that microbiota depletion could enhance the sensitivity to insulin and improve metabolic diseases[56], suggesting that these intestinal flora influence the host metabolic dysbiosis in a somewhat intricate manner.
From recent literature it has been highlighted a tight and coordinated connection between gut microbiota and local RAS. There has been statement that during the fermentation by probiotics, ACE inhibitory peptides can be released[57] which could produce a blood-pressure lowering effect[58].
In the early stage of DN, a well-recognized pathophysiologic feature is glomerular hyperfiltration, which results from a rise in glomerular capillary pressure[59]. In a diabetic milieu, hyperglycemia might activate the renal RAS and result in an impaired autoregulation of glomerular microcirculation, which led to distinct dilatations of afferent and efferent arterioles and expose the kidney to an increasing blood pressure[59, 60]. In this complicated process, the release of renin in the juxtaglomerular apparatus is a vital signaling mechanism in early DN. Pluznick J et al[25] have found that the SCFA receptors expressed in the renal juxtaglomerular afferent arteriole could mediate renin secretion in response to the signals from gut microbiota, which could be blunted by antibiotic treatment and in SCFA-receptor knockout mice.
RAS seems connected to GPRs as well. For example, beyond the conventional role as an intermediate of the tricarboxylic acid cycle, the accumulation of succinate in the distal nephron-collecting duct (CD), which is the predominant localization of its receptor GPR91, appears to be an important signaling through which the stored (pro)renin is released in diabetes and cells response to the stimulation of high glucose and provoke renal injuries of early DN[61]. Whether gut microbiota participates in this process remains to be investigated, though it has been demonstrated that as an end-product from the fermentation by gut microbiota, succinate can be converted to butyrate, the pathway of which has the gut symbiont Bacteroides thetaiotaomicron, C. difficile involved[62]. De Vadder F. et al have colonized GF mice with a succinate-producing bacterial species, Prevotella copri, which significantly increased the levels of succinate in the cecum, along with exerting metabolic benefits to improve glucose and insulin tolerance[63].
Except for mediating fluid pressure, there is evidence indicating pleiotropic roles of gut microbiota in RAS-involved pathways. By testing on GF mice, Karbach S. H. et al have pointed that gut microbiota could facilitate Ang Ⅱ-induced vascular dysfunction and hypertension[64]. As one of the end products from the fermentation of complex carbohydrates by gut microbiota, sodium butyrate (NaBu) has been found to attenuated Ang II-induced expression of (pro)renin receptor (PRR) and renin, and therefore provide an improvement for Ang II-induced renal injuries[65]. Nevertheless, causality cannot be proven in observational studies and, unfortunately, experimental human data are extremely limited.
In terms of the gut itself, RAS also takes its indispensable position. B0AT1 is a transporter of neutral amino acid in the kidney, sharing the location of small intestine brush border membrane with ACE2, which couples with and stabilizes B0AT1 and the expression of which promotes the transporting activities of this transporter[66].
Knockout of ace2 reduced the circulating neutral amino acids and caused an impairment in the uptake of tryptophan. The absence of B0AT1 with ACE2 blunted the efficient absorption of tryptophan, leading to an aberrant activation of mTOR pathway and reducing secretion of antimicrobial peptides, subsequently altering the constitute of gut microbiota and conferring the intestine higher susceptibility to inflammations[67]. Administration of irbesartan, a sort of ARB, inhibited the activation of stress-induced AT1R pathway to reduce intestinal ROS accumulation and inflammation, restored expression of ACE2/B0AT1, activity of mTOR, dysbiosis and tryptophan metabolism[68]. In addition to the gut, Chen L.J. et al have also demonstrated that in Ang Ⅱ-infused ApoE knockout mice there existed an exacerbation in renal fibrosis, and treatment with human recombinant ACE2 (rhACE2) and rapamycin suppresses the activation of mTOR signaling and superoxide generation regulated by Ang Ⅱ, indicating the importance of ACE2 in maintaining the balance of Ang 1-7/Ang Ⅱ in the kidney[69].
On the other hand, uremic toxins are a great stimulator of renal RAS by increasing the expression of renin, AGT and AT1R both in vitro and in vivo. Particular uremic toxins cannot be removed by dialysis, among which indoxyl sulfate (IS) and p-cresol sulfate (PCS) are especial for their serum levels are closely linked the progress of chronic kidney diseases. Chiao-Yin Sun et al[70] have found that RAS inhibition markedly decreased TGF-β1 expression and EMT-associated transcription induced by IS and PCS in renal tubules which led to renal fibrosis. IS derives exclusively from the gut fermentation of diet-derived substance. Bacterial tryptophanases transform tryptophan to indole, the latter absorbed and processed by the host to generate IS. Manipulation of the tryptophanase from Bacteroides could reduce the amount of produced indole, hence lowering circulating IS levels[71], suggesting it is possible to target the microbiota as a possible strategy for treating renal diseases.
In addition to weaken the deleterious functions of gut microbiota on the host, therapeutic alteration of microbial composition should also concentrate on the preservation of the beneficial microbiota that are central to maintaining host homeostasis because these microbiota or their metabolites may mediate renoprotective effects[72].
Summary
As mentioned above, there exists a complex interplay between gut microbiota and components of intrarenal RAS in DN (see in Figure 2). In terms of the association with heightened inflammatory state and metabolic disturbance of gut microbiota, progression of DN renal injuries might be potentially attributed to this gut-kidney axis in which local RAS is possibly involved.
Gut microbiota and RAS are involved in DN pathogenesis. With demonstrable increases in DN-related pathological contexts, there is a series of links between gut microbiota and RAS. The fermentation of gut microbiota produces short chain fatty acids (SCFAs), which could bind to receptors located at kidneys and exert vascular-related effects. Besides, the alternations of gut microbiota or their metabolites, along with extrinsic stimulators (high glucose, uremix toxins, etc), are likely to break the balance between ACE and ACE2 axes of intrarenal RAS, thereby arouse a series of cascades which in turn exaggerate the renal injuries and promote DN progression.
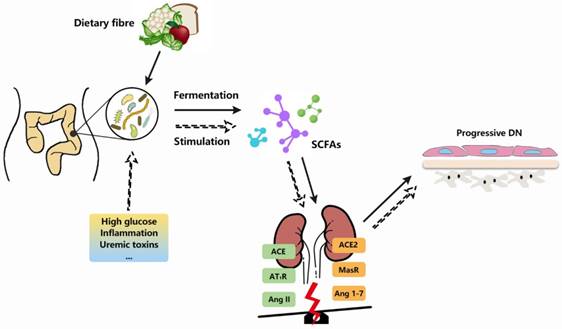
Gut microbiota and their metabolites might be able to impose a broader impact on host pathophysiology by virtue of their promiscuous nature, yet it remains a major challenge to assure their comprehensive influence and pinpoint the precise mechanisms.
Despite most relevant studies are either preliminary or controversial, there still have been intriguing animal studies that have motivated the investigation of more roles gut microbiota plays in the pathogenesis and development of DN. However, results regarding their effects vary due to different experimental conditions, making the precise role of these intestinal floras related to intrarenal RAS awaits subsequent research and needs to be applied in the clinical treatment of DN.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant 81470957), the Natural Science Foundation of Jiangsu Province (BK20141343), the Jiangsu Province Six Talent Peaks Project (2015-WSN-002), the Project for Jiangsu Provincial Medical Talent (ZDRCA2016077), the Fundamental Research Funds for the Central Universities (KYCX17-0169, KYZZ15-0061), the Jiangsu Province Ordinary University Graduate Research Innovation Project (SJZZ16-004), and the Clinical Medical Science Technology Special Project of Jiangsu Province (BL2014080).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang L, Long J, Jiang W. et al. Trends in Chronic Kidney Disease in China. N Engl J Med. 2016;375:905-6
2. Spadoni I, Zagato E, Bertocchi A. et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science. 2015;350:830-4
3. Andersen K, Kesper MS, Marschner JA. et al. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. J Am Soc Nephrol. 2017;28:76-83
4. Crommen S, Simon MC. Microbial Regulation of Glucose Metabolism and Insulin Resistance. Genes (Basel). 2017;9:10
5. Grasset E, Puel A, Charpentier J. et al. A Specific Gut Microbiota Dysbiosis of Type 2 Diabetic Mice Induces GLP-1 Resistance through an Enteric NO-Dependent and Gut-Brain Axis Mechanism. Cell Metab. 2017;25:1075-90
6. Wysocki J, Ye M, Khattab AM. et al. Angiotensin-converting enzyme 2 amplification limited to the circulation does not protect mice from development of diabetic nephropathy. Kidney Int. 2017;91:1336-46
7. Chen Z, Zhu S, Xu G. Targeting gut microbiota: a potential promising therapy for diabetic kidney disease. Am J Transl Res. 2016;8:4009-16
8. Arumugam M, Raes J, Pelletier E. et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174-80
9. den Besten G, Havinga R, Bleeker A. et al. The short-chain fatty acid uptake fluxes by mice on a guar gum supplemented diet associate with amelioration of major biomarkers of the metabolic syndrome. PLoS One. 2014;9:e107392
10. Wlodarska M, Willing B, Keeney KM. et al. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect Immun. 2011;79:1536-45
11. Morris G, Berk M, Carvalho A. et al. The Role of the Microbial Metabolites Including Tryptophan Catabolites and Short Chain Fatty Acids in the Pathophysiology of Immune-Inflammatory and Neuroimmune Disease. Mol Neurobiol. 2017;54:4432-51
12. Kaiko GE, Ryu SH, Koues OI. et al. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell. 2016;165:1708-20
13. Rios-Covian D, Ruas-Madiedo P, Margolles A. et al. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front Microbiol. 2016;7:185
14. Macfarlane S, Macfarlane GT. Regulation of short-chain fatty acid production. Proc Nutr Soc. 2003;62:67-72
15. Pomare EW, Branch WJ, Cummings JH. Carbohydrate fermentation in the human colon and its relation to acetate concentrations in venous blood. J Clin Invest. 1985;75:1448-54
16. Karlsson FH, Tremaroli V, Nookaew I. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99-103
17. Qin J, Li Y, Cai Z. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55-60
18. Jia L, Li D, Feng N. et al. Anti-diabetic Effects of Clostridium butyricum CGMCC0313.1 through Promoting the Growth of Gut Butyrate-producing Bacteria in Type 2 Diabetic Mice. Sci Rep. 2017;7:7046
19. Perry RJ, Peng L, Barry NA. et al. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature. 2016;534:213-7
20. Wichmann A, Allahyar A, Greiner TU. et al. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe. 2013;14:582-90
21. Hoverstad T, Midtvedt T. Short-chain fatty acids in germfree mice and rats. J Nutr. 1986;116:1772-6
22. Marques FZ, Nelson E, Chu PY. et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation. 2017;135:964-77
23. Andrade-Oliveira V, Amano MT, Correa-Costa M. et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J Am Soc Nephrol. 2015;26:1877-88
24. Huang W, Zhou L, Guo H. et al. The role of short-chain fatty acids in kidney injury induced by gut-derived inflammatory response. Metabolism. 2017;68:20-30
25. Pluznick J, Protzko R, Gevorgyan H. et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA. 2013;110:4410-5
26. Kim MH, Kang SG, Park JH. et al. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145:396-406
27. Kimura I, Ozawa K, Inoue D. et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829
28. Fuller M, Li X, Fisch R. et al. FFA2 Contribution to Gestational Glucose Tolerance Is Not Disrupted by Antibiotics. PLoS One. 2016;11:e0167837
29. Pluznick JL. Renal and cardiovascular sensory receptors and blood pressure regulation. Am J Physiol Renal Physiol. 2013;305:F439-44
30. Pluznick J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes. 2014;5:202-7
31. Brandt LJ, Aroniadis OC. An overview of fecal microbiota transplantation: techniques, indications, and outcomes. Gastrointest Endosc. 2013;78:240-9
32. Weingarden A, Gonzalez A, Vazquez-Baeza Y. et al. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome. 2015;3:10
33. Vrieze A, Van Nood E, Holleman F. et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913-6
34. Al-Asmakh M, Zadjali F. Use of Germ-Free Animal Models in Microbiota-Related Research. J Microbiol Biotechnol. 2015;25:1583-8
35. Reyniers JA. THE PURE-CULTURE CONCEPT AND GNOTOBIOTICS. Ann N Y Acad Sci. 1959;78:3-16
36. Navar LG, Inscho EW, Majid SA. et al. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76:425-536
37. Carey RM, Siragy HM. The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrinol Metab. 2003;14:274-81
38. Kobori H, Nangaku M, Navar LG. et al. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251-87
39. Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol. 2010;6:319-30
40. Crowley SD, Vasievich MP, Ruiz P. et al. Glomerular type 1 angiotensin receptors augment kidney injury and inflammation in murine autoimmune nephritis. J Clin Invest. 2009;119:943-53
41. Roscioni SS, Heerspink HJ, de Zeeuw D. The effect of RAAS blockade on the progression of diabetic nephropathy. Nat Rev Nephrol. 2014;10:77-87
42. Perlot T, Penninger JM. ACE2 - from the renin-angiotensin system to gut microbiota and malnutrition. Microbes Infect. 2013;15:866-73
43. Batlle D, Wysocki J, Soler MJ. et al. Angiotensin-converting enzyme 2: enhancing the degradation of angiotensin II as a potential therapy for diabetic nephropathy. Kidney Int. 2012;81:520-8
44. Liu Z, Huang XR, Chen HY. et al. Deletion of Angiotensin-Converting Enzyme-2 Promotes Hypertensive Nephropathy by Targeting Smad7 for Ubiquitin Degradation. Hypertension. 2017;70:822-30
45. Zhang YH, Zhang YH, Dong XF. et al. ACE2 and Ang-(1-7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm Res. 2015;64:253-60
46. Grobe JL, Der Sarkissian S, Stewart JM. et al. ACE2 overexpression inhibits hypoxia-induced collagen production by cardiac fibroblasts. Clin Sci (Lond). 2007;113:357-64
47. Pinheiro SV, Ferreira AJ, Kitten GT. et al. Genetic deletion of the angiotensin-(1-7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int. 2009;75:1184-93
48. Danilczyk U, Penninger JM. Angiotensin-converting enzyme II in the heart and the kidney. Circ Res. 2006;98:463-71
49. Harris RC. Podocyte ACE2 protects against diabetic nephropathy. Kidney Int. 2012;82:255-6
50. Soler MJ, Wysocki J, Ye M. et al. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007;72:614-23
51. Ye M, Wysocki J, William J. et al. Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol. 2006;17:3067-75
52. Ye M, Wysocki J, Naaz P. et al. Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice: a renoprotective combination? Hypertension. 2004;43:1120-5
53. Tikellis C, Bialkowski K, Pete J. et al. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018-25
54. Nadarajah R, Milagres R, Dilauro M. et al. Podocyte-specific overexpression of human angiotensin-converting enzyme 2 attenuates diabetic nephropathy in mice. Kidney Int. 2012;82:292-303
55. Reich HN, Oudit GY, Penninger JM. et al. Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney Int. 2008;74:1610-6
56. Suarez-Zamorano N, Fabbiano S, Chevalier C. et al. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med. 2015;21:1497-501
57. Dave LA, Hayes M, Montoya CA. et al. Human gut endogenous proteins as a potential source of angiotensin-I-converting enzyme (ACE-I)-, renin inhibitory and antioxidant peptides. Peptides. 2016;76:30-44
58. Gonzalez-Gonzalez C, Gibson T, Jauregi P. Novel probiotic-fermented milk with angiotensin I-converting enzyme inhibitory peptides produced by Bifidobacterium bifidum MF 20/5. Int J Food Microbiol. 2013;167:131-7
59. Tonneijck L, Muskiet MH, Smits MM. et al. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J Am Soc Nephrol. 2017;28:1023-39
60. Jandeleit-Dahm K, Cooper ME. Hypertension and diabetes: role of the renin-angiotensin system. Endocrinol Metab Clin North Am. 2006;35:469-90
61. Peti-Peterdi J. High glucose and renin release: the role of succinate and GPR91. Kidney Int. 2010;78:1214-7
62. Ferreyra JA, Wu KJ, Hryckowian AJ. et al. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe. 2014;16:770-7
63. Kovatcheva-Datchary P, Nilsson A, Akrami R. et al. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015;22:971-82
64. Karbach SH, Schonfelder T, Brandao I. et al. Gut Microbiota Promote Angiotensin II-Induced Arterial Hypertension and Vascular Dysfunction. J Am Heart Assoc. 2016;5:e003698
65. Wang L, Zhu Q, Lu A. et al. Sodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin-angiotensin system. J Hypertens. 2017;35:1899-908
66. Kowalczuk S, Broer A, Tietze N. et al. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008;22:2880-7
67. Hashimoto T, Perlot T, Rehman A. et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487:477-81
68. Yisireyili M, Uchida Y, Yamamoto K. et al. Angiotensin receptor blocker irbesartan reduces stress-induced intestinal inflammation via AT1a signaling and ACE2-dependent mechanism in mice. Brain Behav Immun. 2017;69:167-79
69. Chen LJ, Xu YL, Song B. et al. Angiotensin-converting enzyme 2 ameliorates renal fibrosis by blocking the activation of mTOR/ERK signaling in apolipoprotein E-deficient mice. Peptides. 2016;79:49-57
70. Sun CY, Chang SC, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS One. 2012;7:e34026
71. Devlin AS, Marcobal A, Dodd D. et al. Modulation of a Circulating Uremic Solute via Rational Genetic Manipulation of the Gut Microbiota. Cell Host Microbe. 2016;20:709-15
72. Tang WH, Kitai T, Hazen SL. Gut Microbiota in Cardiovascular Health and Disease. Circ Res. 2017;120:1183-96
Author contact
![]() Corresponding author: Kun Ling Ma, Institute of Nephrology, Zhong Da Hospital, School of Medicine, Southeast University, NO. 87, Ding Jia Qiao Road, Nan Jing City, Jiangsu Province, China, 210009. Tel: 0086 25 83262442; Fax: 0086 25 83262442; E-mail: Kun Ling Ma: klma05com
Corresponding author: Kun Ling Ma, Institute of Nephrology, Zhong Da Hospital, School of Medicine, Southeast University, NO. 87, Ding Jia Qiao Road, Nan Jing City, Jiangsu Province, China, 210009. Tel: 0086 25 83262442; Fax: 0086 25 83262442; E-mail: Kun Ling Ma: klma05com