Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(7):682-688. doi:10.7150/ijms.25393 This issue Cite
Research Paper
Chronic Alcohol Consumption Inhibits Autophagy and Promotes Apoptosis in the Liver
Mario Menk ![]() , Jan Adriaan Graw, Deniz Poyraz, Nadine Möbius, Claudia D. Spies, Clarissa von Haefen
, Jan Adriaan Graw, Deniz Poyraz, Nadine Möbius, Claudia D. Spies, Clarissa von Haefen
Department of Anesthesiology and Operative Intensive Care Medicine (CCM/CVK), Charité - University Medicine Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health; Campus Virchow-Klinikum; Augustenburger Platz 1, 13353 Berlin, Germany
Received 2018-2-6; Accepted 2018-3-25; Published 2018-4-27
Abstract
Background: Chronic alcohol consumption is a major cause of liver injury. However, the molecular mechanisms by which alcohol impairs hepatocellular function and induces cell death remain unclear. Macroautophagy (hereafter called 'autophagy') is a degradation pathway involved in the survival or death of cells during conditions of cellular stress. This study examines the effect of chronic alcohol consumption on hepatocellular autophagy in an animal model.
Methods: During a 12-week period male Wistar rats were fed a Lieber-DeCarli diet containing 5% alcohol (EtOH group; n=10), or an isocaloric diet (control group; n=10). Hepatic expression of key regulatory autophagy proteins (e.g. Beclin-1, ATG-3, ATG-5, p62/SQSTM1 and LC3) were detected by real-time polymerase chain reaction and Western blot analysis. Markers of cellular stress and apoptotic cell death (e.g. HO-1, caspase-3, PARP-1 and Bcl-2) were determined, and levels of reduced and oxidized glutathione were measured.
Results: Chronic alcohol consumption caused cellular and oxidative stress in the liver. Transcriptional and translational expression of Beclin-1 and ATG-5 was significantly impaired. The protein expression of LC3-I and LC3-II was significantly increased, while the ratio of LC3I/II remained unchanged in the EtOH group compared with controls. Hepatocellular expression of p62/SQSTM1 and markers of apoptotic cell death (such as cleaved caspase-3 and cleaved PARP-1) were significantly increased in the EtOH group indicating a disrupted autophagic flux and increased rate of apoptosis in the liver.
Conclusions: In this model, chronic alcohol consumption impaired hepatocellular autophagy and induced apoptotic cell death. It appears that changes in autophagy might contribute to alcohol-induced structural and functional hepatocellular injury.
Keywords: autophagy, chronic ethanol, alcohol, liver, apoptosis
Introduction
Chronic alcohol consumption is a well-known risk factor for the development of alcoholic liver disease. Alcohol-induced hepatocellular injury is mainly caused by reactive oxygen species and local inflammation, which causes structural and functional damage of the liver and ultimately leads to increased loss of hepatic cells (1-3). Progression of alcoholic liver disease exhibits a clinical spectrum ranging from steatosis to hepatitis, inflammation, and liver cirrhosis.
Of the diverse mechanisms involved in the regulation of the survival or death of cells during cellular stress, autophagy has emerged as an important pathway. Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved cellular degradation process involving the transport and delivery of cytoplasmic protein complexes or organelles to the lysosome for the purpose of supplying substrates for energy generation or maintaining cellular homeostasis (4,5). Autophagy can be considered a temporary survival mechanism during periods of cellular stress (5). Moreover, autophagy may act as a surveillance mechanism in stressed or injured cells to remove damaged organelles that otherwise would be harmful or could trigger apoptotic cell death. Autophagy has gained increasing attention because its pathway has been implicated in several human diseases, and as a possibly new type of programmed cell death that differs from classical apoptosis (6). However, the contribution of autophagy to programmed cell death remains a matter of intense debate. There is complex cross-talk to the pathway of classical apoptosis and it is now generally accepted that inhibition of autophagy increases susceptibility to cell death (6).
In the liver, however, a basal level of autophagic turnover is essential for normal liver function. Hepatic autophagy can be considerably increased in response to starvation or stressed conditions, thereby contributing to cell survival and maintenance of normal liver function (7,8). Hepatocytes engage higher levels of autophagy than other types of cells (9). Therefore, it is thought that impairment of hepatic autophagy may have a considerable effect on hepatocellular function (5). The impact of alcohol consumption on the autophagic pathway in the liver is not yet completely understood. Ding and colleagues showed that autophagy is stimulated under acute administration of alcohol and, under these conditions, autophagy protected liver cells from acute alcohol toxicity and reduced hepatic steatosis (10). On the other hand, little is known about the impact of chronic alcohol consumption on hepatocellular autophagy. There is evidence that chronic alcohol may inhibit autophagic mechanisms in the liver. Lin and colleagues described suppression of autophagy in a mouse model with chronic alcohol intake over a period of 4 weeks; remarkably, alcohol-induced liver injury was worse when autophagy was inhibited (11).
The effects of chronic alcohol consumption on autophagy are partly contradictory and not well characterized; however, we hypothesized that chronic alcohol intake may modulate autophagy and apoptosis. Therefore, this study investigates whether autophagy is inhibited in response to long-term alcohol exposure and whether this contributes to hepatic cell death in a rat model of chronic alcohol consumption.
Materials and Methods
Animal model
This animal study was approved by the Committee on Use and Care of Animals at the Charité-Universitätsmedizin Berlin, Germany, and by the state animal committee (LAGeSo, Germany; approval No. G0259/08). All procedures were conducted in accordance with institutional guidelines of the Charité- Universitätsmedizin Berlin, Germany.
A total of 20 adult male Wistar rats (300-350g) (Harlan; Rossdorf, Germany) were randomly assigned to an alcohol-treated group (n=10) or to a control group (n=10). To establish a model of chronic alcohol consumption, the Lieber-DeCarli liquid diet technique was used (Lieber and DeCarli, 1989). The diet (E15784-301/AIN93G-VM) with maltose-dextrin (dextrose equivalent 8.0-9.9, R111M040) or alcohol (5% v/v) providing 36% of total calories was purchased from Ssniff (Soest, Germany) and contained EtOH 5% (v/v) for the alcohol-treated group. For the control group, an isocaloric diet with maltrose-dextrin instead of EtOH was used. Drinking water was provided ad libitum in both groups. Water intake, diet and body weight were measured daily in each animal. Rats were housed in a room with 12-h light/dark cycles.
After 12 weeks, all rats were anesthetized with isoflurane and sacrificed by cervical dislocation. Liver was removed, immediately frozen on dry ice, and stored at -80°C for subsequent processing. To examine the effects of chronic alcohol consumption on the autophagic pathway of the liver, the 10 animals in each group were examined.
Molecular Studies
Determination of total glutathione (GSH and GSSG)
Total glutathione (GSH and GSSG) was measured in liver homogenates using the thiol reagent 5,50 dithiobis-2-nitrobenzoic acid (DTNB), as described previously (12). In short, for determination of reduced glutathione (GSH) and oxidized glutathione (GSSG), livers were homogenized and treated with a mixture of metaphosphoric acid, EDTA and NaCl. After centrifugation, aliquots were taken for neutralization with disodiumhydrogen-phosphate followed by addition of DTNB. GSH was determined in a spectrophotometer at 412 nm. For determination of GSSG, 4-vinylpyridine was added. After incubation for 1 h at room temperature, GSSG was determined spectrophotometrically at 412 nm.
Quantitative real-time polymerase chain reaction (PCR)
Total cellular RNA was isolated from snap-frozen tissue by acidic phenol/chloroform extraction and DNase I treated (Roche Diagnostics, Mannheim, Germany). Then, 2 µg of RNA were reverse transcribed at 42 °C with 200 U of Moloney murine leukemia virus reverse transcriptase and 2 µM oligo d(T) 16 primer (Promega, Mannheim, Germany) in 25 µl of reaction mixture. Resulting cDNA was quantified by real-time polymerase chain reaction (RT-PCR) Master Mix (Applied Biosystems, Darmstadt, Germany) using FAM-5´ ⁄ TAMRA-3´ labeled probes for autophagy-related protein 3 (ATG-3), ATG-5, Beclin-1, Bcl-2 and HPRT (Metabion, Munich, Germany). Data represent the mean expression level ± standard deviation (standardized to HPRT expression) calculated according to the 2-∆∆CT method (13) of at least three independent measurements per cDNA (technical triplicates). The sequence of the primers used is listed in Table 1.
Sequence of oligonucleotides used for qRT-PCR.
| gene | forward primer 5´- 3´ reverse primer 5´- 3´ | Probe 5´ 6-FAM - TAMRA-3´ |
|---|---|---|
| ATG-3 | GCAGCACCATGCAGGTGAG TGGTCACTCGGTCCAGGATC | TCGTGTGCCAGCGCTGTAGCC A |
| ATG-5 | ACATCAGCATTGTGCCCCA TGTCATGCTTCGGTGTCCTG | CAGACTGAAGGCCGTGTCCTGCTCA |
| Beclin-1 | TGCGACAGTCTCTCCGTGC GGCCACTTCCAGAGCCTTTC | TGCTCCGGTCCCAGGATGCAGA |
| Bcl-2 | TGAACCGGCATCTGCACATGAACCGGCATCTGCACA AGAGGTCGCATGCTGGG | AACGGAGGCTGGGATGCCTTTGTG |
| HPRT | GGAAAGAACGTCTTGATTGTTGAA CCAACACTTCGAGAGGTCCTTTT | CTTTCCTTGGTCAAGCAGTACAGCCCC |
Abbreviations: qRT-PCR= quantitative real-time reverse transcriptase polymerase chain reaction; ATG= autophagy-related protein; Bcl-2=B cell lymphoma 2; HPRT=Hypoxanthine-guanine phosophoribosyltransferase
Immunoblotting
Rat livers were homogenized in cold lysis buffer containing 10 mM Tris/HCl, pH 7.5, 300 mM NaCl, 1%Triton X-100, 2 mM MgCl2, 5 µM EDTA, and the protease inhibitor cocktail, Complete Mini (Roche Diagnostics, Mannheim, Germany). Protein concentration of homogenates was determined using the bicinchoninic acid method (Pierce, Rockford, USA). Then, 20 µg of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transfer to a nitro-cellulose membrane (0.2 µm pore, Bio-Rad, Munich, Germany). After blocking in 5% low-fat milk solution, the membranes were incubated overnight (4 °C) with primary polyclonal rabbit anti-HO-1 antibody (1:1000, Cell Signaling, cat. no. 5141), primary polyclonal rabbit anti-Beclin-1 antibody (1:1000, Cell Signaling, cat. no. 3738), polyclonal rabbit anti-LC3A/B antibody (1:1000, Cell Signaling, cat. no. 4108), polyclonal rabbit anti-p62/ SQSTM1 antibody (1:1000, Cell Signaling, cat. no. 5114), polyclonal rabbit anti-Bcl-2 antibody (1:1000, Cell Signaling, cat. no. 2870), monoclonal rabbit anti-PARP antibody (1:1000, Cell Signaling, cat. no. 9532), polyclonal rabbit anti-cleaved caspase-3 antibody (1:1000, Cell Signaling, cat. no. 9661), in 5% non-fat dry milk in PBST. Secondary goat anti-rabbit and goat anti-mouse horseradish peroxidase-conjugated antibody (Southern Biotechnology Associates, Birmingham, AL, USA) was used at a concentration of 1:50,000. Blots were revealed with a SuperSignal West Pico Chemiluminescent Substrate detection kit (Pierce, Rockford, USA). Quantification of the respective band density was performed using the image analysis program ImageJ 1.42q (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Groups were compared using a one-way analysis of variance (ANOVA), and significance was determined using Bonferroni's correction for multiple comparisons with independent sample t-test. A two-sided p-value <0.05 was considered statistically significant. All graphics and statistical analyses were performed using the GraphPad Prism 6.0 software (La Jolla, CA, USA).
Results
Chronic ethanol induces oxidative and cellular stress in the liver
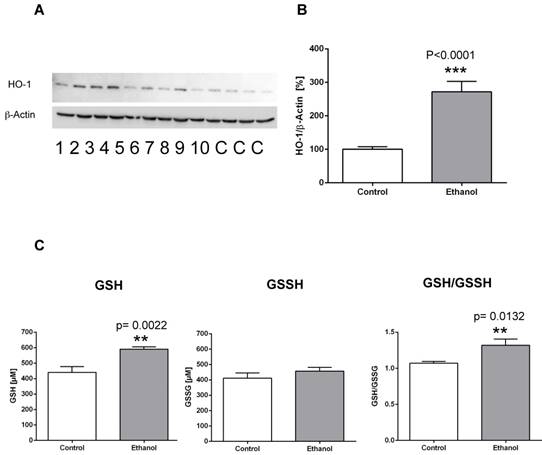
To examine the effects of chronic alcohol consumption on cellular and oxidative stress in the liver, we evaluated the expression levels of heme oxygenase-1 (HO-1) and measured levels of reduced glutathione (GSH) and oxidized glutathione (GSSH). In contrast to control animals, rats fed an alcohol-containing diet for 12 weeks showed a significantly higher expression of the stress marker protein HO-1 in the liver (Figure 1 A, B). Also, analysis of reduced and oxidized glutathione levels showed that GSH levels were significantly higher in the liver of rats after 12 weeks of alcohol consumption, whereas GSSH levels remained unchanged (Figure 1 C). Correspondingly, the GSH/GSSH ratio was significantly elevated in alcohol-fed rats, indicating oxidative stress in the liver in this chronic alcohol model.
Chronic alcohol consumption increases the expression of heme oxygenase-1 (HO-1) in the liver. (A) Representative Western blot of HO-1 in the liver of rats fed with an alcohol-containing diet for 12 weeks (ethanol, lanes 1-10) and controls (lanes C; for more clarity, protein lysates of controls have been pooled). Detection of β-actin served as loading control. This example is representative of a series of blots. (B) Quantification of bands expressed as density ratio of indicated protein/β-actin (%; control set to 100%); ***p<0.0001 compared with controls; data are presented as mean ± SEM (n=10). (C) Chronic alcohol consumption modifies levels of reduced glutathione (GSH) and oxidized glutathione (GSSG) in the liver as detected by the reaction with the thiol reagent DTNB (left/middle panel). GSH/GSSH ratio (right panel). **p<0.01 compared with controls; data are presented as mean ± SEM (n=10).
Chronic ethanol induces dysregulation of the autophagic pathway
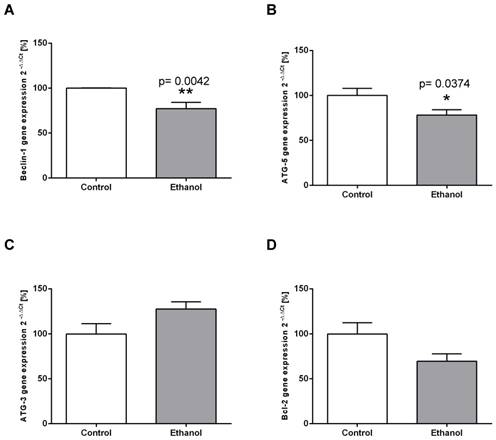
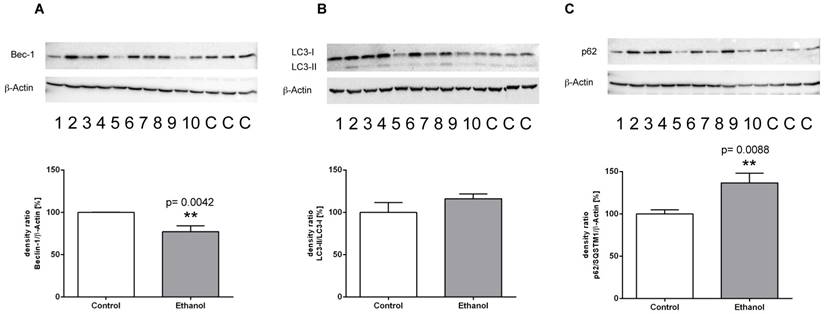
To examine whether chronic alcohol consumption interferes with the mechanisms of autophagy in the liver, we determined the expression levels of relevant mediators of the autophagic pathway on the transcriptional and/or translational level. ATG-5 and Beclin-1 are key initiation factors of the autophagosome formation. In the livers of alcohol-fed rats, the mRNA expression of both factors was significantly lower than in control animals (Figure 2 A; B). In contrast, mRNA expression levels of ATG-3 (an essential enzyme for the LC3 lipidation process) remained unchanged in both groups (Figure 2C). Chronic alcohol intake also led to dysregulation of the autophagic pathway on the protein level. Expression of Beclin-1, the main regulator of autophagy, was significantly decreased by chronic alcohol consumption, as shown by Western Blot experiments (Figure 3A). This indicates a significant inhibition of the autophagic pathway by chronic alcohol consumption. The formation of LC3-II complexes is a feature of autophagy and indicates the presence of autophagosomal membranes. At the protein level, the expression of both LC3-I and LC3-II was significantly increased in the livers of alcohol-fed rats compared to controls. However, the ratio of LC3-II/LC3-I showed no significant increase (Figure 3B). p62/SQSTM1 is a multifunctional protein and a marker of the autophagic degradation process. Compared to the control group, chronic alcohol consumption resulted in significantly increased p62/SQSTM1 levels, indicating accumulation of this degradation product in the liver cells (Figure 3C).
Chronic alcohol consumption reduces transcriptional expression of autophagy-related proteins in the liver. Relative mRNA expression of (A) Beclin-1, (B) ATG-5, (C) ATG-3 and (D) Bcl-2 was analyzed by real-time polymerase chain reaction. HPRT served as housekeeping gene, expression of control was set to 100%. *p<0.05; **p<0.01 compared with controls; data are presented as mean ± SEM (n=10). ATG=autophagy-related gene; Bcl-2= B-cell lymphoma 2 gene.
Protein expression of (A) Beclin-1, (B) LC3-I/LC3-II and (C) p62/ SQSTM1 in livers of rats fed with an alcohol-containing diet for 12 weeks (ethanol, lanes 1-10) and controls (lanes C; for more clarity, protein lysates of controls have been pooled). Detection of β-actin served as loading control. This example is representative of a series of blots. Quantification of bands is expressed as density ratio of indicated protein/β-actin (%); control set to 100%); **p<0.01 compared with controls; data are presented as mean ± SEM (n=10).
Inhibition of autophagy promotes apoptosis
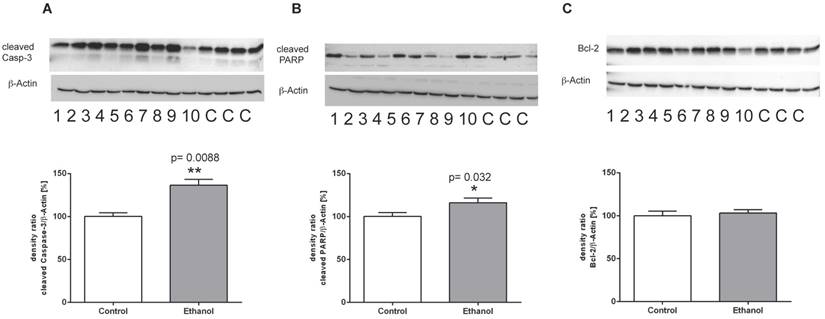
To further analyze the mechanisms of cell death in this chronic model of alcohol consumption, we determined the expression of key regulatory proteins of the apoptotic signaling pathway using Western blot analysis. Chronic alcohol consumption caused a marked induction of cleaved caspase 3 in the liver, indicating a significantly increased activity of the execution phase of cell apoptosis (Figure 4A). Also, cleavage of PARP-1 was significantly induced in liver cells of alcohol-fed animals (Figure 4B). Both proteins are markers of programmed cell death and the presence of their cleaved forms indicates activation of the apoptotic signaling pathway in liver cells of alcohol-fed rats. Along that line, expression of the anti-apoptotic Bcl-2 protein showed no significant difference between controls and alcohol-fed rats (Figure 4 C).
Protein expression of (A) (cleaved) Caspase-3, (B) (cleaved) PARP-1 and (C) Bcl-2 in livers of rats after 12 weeks of chronic alcohol consumption (ethanol; lanes 1-10) or controls (lanes C, for more clarity, protein lysates of controls have been pooled). Detection of β-actin served as loading control. This example is representative of a series of blots. Quantification of bands is expressed as density ratio of indicated protein/β-actin (%); control set to 100%; *p<0.05, **p<0.01 compared with controls; data are presented as mean ± SEM (n=10).
Discussion
This study demonstrates that, in this animal model, chronic alcohol consumption i) impairs autophagy, ii) promotes apoptotic cell death in the liver, and iii) increases cellular and oxidative stress in the liver.
The autophagic response to chronic alcohol intake was assessed by analysis of the expression of specific and characteristic features of the pathway. Beclin-1 is the key regulator of autophagy. Autophagic induction via ATG-5 then mediates cleavage of LC3-I. This, in turn, generates LC3-II-complexes and autophagosomal membranes (14). Chronic alcohol consumption for 12 weeks significantly decreased Beclin-1 and ATG-5 expression in the liver on both the transcriptional and translational level. Furthermore, alcohol intake intensified markers and effectors of programmed cell-death, such as cleaved caspase-3 and cleaved PARP-1. Cleavage of PARP-1 facilitates cellular disassembly and serves as a valid marker of cells undergoing apoptosis. In addition, our results show that transcriptional and translational expression of the anti-apoptotic Bcl-2 protein remained unchanged. These results indicate that chronic exposure to alcohol not only downregulates the hepatocellular autophagic pathway, but also activates apoptotic, programmed cell death. Activation and successful processing of the autophagic pathway increases LC3-II complexes, indicating the formation of functional autophagosomes (15). Further, activation of autophagy normally reduces the protein expression of p62/SQSTM1, while pharmacological or genetic inhibition of autophagy increases intracellular p62/SQSTM1 levels (16,17). p62/SQSTM1 is a degradation product of autophagy and widely used as a marker of autophagic flux (16). In the present study, chronic alcohol consumption for 12 weeks significantly increased the expression of LC3-I, LC3-II and p62/SQSTM1 in the liver, whereas the LC3-II/LC3-I ratio (a feature of autophagic activation) remained unchanged. When p62/SQSTM1 also increases (i.e., accumulates), the autophagic flux is thought to be stopped at the process of the lysosome (17, 18). The disturbed autophagic flux induces cellular stress and, finally, apoptosis. These results support the finding that chronic alcohol consumption significantly impairs and alters autophagic mechanisms in the liver.
Only a few studies have investigated alcohol-induced changes of autophagy in the liver. For example, in mice fed with the Lieber-DeCarli diet for 4 weeks, autophagy was inhibited in a dose-dependent manner (11). Also, in a model of alcohol-fed mice, inhibition of AMPK and subsequent autophagic suppression was observed in the liver (20). In line with these findings, alcohol-induced suppression of autophagy has been demonstrated in several in vitro models. In monocytic U937, CD4 Jurkat cells, and in neuronal cells, exposure to alcohol downregulated autophagy-related proteins, such as Beclin-1 and others (21,22). In contrast to these findings, autophagy was found to be activated in cultured primary hepatocytes by acute exposure to alcohol (10,23). Oxidative stress is as a major contributing factor of this activation (24). Further, acute alcohol inhibits mTOR signaling and activates AMPK under oxidative stress conditions which, in turn, activates autophagy (25). However, these latter findings only hold true for models of acute alcohol exposure. In contrast, long-lasting chronic alcohol exposure may exert completely different effects and may eventually suppress autophagy. However, reasons for the different effects of acute versus chronic alcohol on the autophagic pathway remain to be elucidated. Autophagy is usually considered to be a temporary survival mechanism during short periods of starvation, energy depletion, or cellular stress (26). It is possible that chronic alcohol (i.e., long-lasting cellular and oxidative stress) depletes hepatocellular autophagic resources and exceeds the capacity of the autophagic pathway over time. Also, mTOR (a main inhibitor of autophagy) is activated by chronic alcohol and extensive nutrient supply (26-28). Whether or not activated mTOR signaling plays a critical role in the suppression of autophagy after chronic alcohol intake needs to be examined in future studies.
Changes in hepatocellular autophagy, such as the effects described above, might be relevant for the functionality of liver cells. Failure to eliminate potentially harmful substances by autophagic processes might disturb cellular integrity and may cause cell death, inflammation and hepatic disease (5). There is increasing evidence that autophagy might be a potential therapeutic target for hepatic diseases. Stimulation of autophagy inhibited hepatic steatosis in both acute and chronic alcoholic fatty liver disease in mice (10,11). Also, stimulation of autophagy attenuated alcohol-induced acute hepatoxicity (10). Therefore, the role of autophagy in chronic alcohol consumption needs further elucidation. Whether activation of this pathway constitutes a potential therapeutic approach in chronic alcohol disease warrants further investigation.
Conclusion
In this animal model, chronic alcohol consumption impairs hepatocellular autophagy and induces apoptotic cell death. Changes in autophagy might contribute to alcohol-induced structural and functional hepatocellular injury.
Acknowledgements
The authors wish to thank Laraine Visser-Isles for English language editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lumeng L, Crabb DW. Alcoholic liver disease. Curr Opin Gastroenterol. 2000;16(3):208-18
2. Lieber CS. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34(1):9-19
3. Wang L, Khambu B, Zhang H, Yin XM. Autophagy in alcoholic liver disease, self-eating triggered by drinking. Clin Res Hepatol Gastroenterol. 2015;39(Suppl 1):S2-6
4. Deter RL, De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. 1967;33(2):437-49
5. Czaja MJ, Ding WX, Donohue TM, Friedman SL, Kim JS, Komatsu M. et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9(8):1131-58
6. Liu Y, Schiff M, Czymmek K, Tallóczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121(4):567-77
7. Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47(5):1773-85
8. Christian P, Sacco J, Adeli K. Autophagy: Emerging roles in lipid homeostasis and metabolic control. Biochim Biophys Acta. 2013;1831(4):819-24
9. Mortimore GE, Hutson NJ, Surmacz CA. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc Natl Acad Sci U S A. 1983;80(8):2179-83
10. Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W. et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139(5):1740-52
11. Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X. et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58(5):993-9
12. Sifringer M, Brait D, Weichelt U, Zimmerman G, Endesfelder S, Brehmer F. et al. Erythropoietin attenuates hyperoxia-induced oxidative stress in the developing rat brain. Brain Behav Immun. 2010;24(5):792-9
13. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402-8
14. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67-93
15. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313-26
16. Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A. et al. P62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603-14
17. Komatsu M1, Waguri S, Koike M, Sou YS, Ueno T, Hara T. et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149-63
18. Tian Z, Wang C, Hu C, Tian Y, Liu J, Wang X. Autophagic-lysosomal inhibition compromises ubiquitin-proteasome system performance in a p62 dependent manner in cardiomyocytes. PLoS One. 2014;9(6):e100715
19. Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33(4):517-27
20. You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of amp-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127(6):1798-808
21. von Haefen C, Sifringer M, Menk M, Spies CD. Ethanol enhances susceptibility to apoptotic cell death via down-regulation of autophagy-related proteins. Alcohol Clin Exp Res. 2011;35(8):1381-91
22. Prock TL, Miranda RC. Embryonic cerebral cortical progenitors are resistant to apoptosis, but increase expression of suicide receptor disc-complex genes and suppress autophagy following ethanol exposure. Alcohol Clin Exp Res. 2007;31(4):694-703
23. Thomes PG, Trambly CS, Thiele GM, Duryee MJ, Fox HS, Haorah J, Donohue TM. Proteasome activity and autophagosome content in liver are reciprocally regulated by ethanol treatment. Biochem Biophys Res Commun. 2012;417(1):262-7
24. Thomes PG, Ehlers RA, Trambly CS, Clemens DL, Fox HS, Tuma DJ, Donohue TM. Multilevel regulation of autophagosome content by ethanol oxidation in hepg2 cells. Autophagy. 2013;9(1):63-73
25. Sid B, Verrax J, Calderon PB. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem Pharmacol. 2013;86(2):200-9
26. Rabanal-Ruiz Y, Otten EG, Korolchuk VI. MTORC1 as the main gateway to autophagy. Essays Biochem. 2017;61(6):565-84
27. Potz BA, Lawandy IJ, Clements RT, Sellke FW. Alcohol modulates autophagy and apoptosis in pig liver tissue. J Surg Res. 2016;203(1):154-62
28. Kim YC, Guan KL. MTOR: A pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25-32
Author contact
![]() Corresponding author: Dr. med. Mario Menk, Department of Anesthesiology and Operative Intensive Care Medicine, Campus Charité Mitte and Campus Virchow-Klinikum, Charité - Universitätsmedizin Berlin, Augustenburger Platz 1, D-13353 Berlin, Germany. Telephone: +49-30-450551002; Fax: +49-30-450551909; Email: mario.menkde
Corresponding author: Dr. med. Mario Menk, Department of Anesthesiology and Operative Intensive Care Medicine, Campus Charité Mitte and Campus Virchow-Klinikum, Charité - Universitätsmedizin Berlin, Augustenburger Platz 1, D-13353 Berlin, Germany. Telephone: +49-30-450551002; Fax: +49-30-450551909; Email: mario.menkde