Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(5):517-527. doi:10.7150/ijms.22454 This issue Cite
Research Paper
Atorvastatin Attenuates Metabolic Remodeling in Ischemic Myocardium through the Downregulation of UCP2 Expression
Chunyan Yang1*, Dongming Zhao1,2*, Guohui Liu1, Haikuo Zheng1, Hongliang Yang1, Sibao Yang1, Ping Yang1 ![]()
1. Department of Cardiology, China-Japan Union Hospital, Jilin University, Changchun, 130033, China
2. Department of Cardiology, the affiliated hospital of Beihua University, Jilin, China
* These authors contributed equally to this work
Received 2017-8-20; Accepted 2018-2-5; Published 2018-3-8
Abstract
Uncoupling protein 2 (UCP2) is primarily expressed in the myocardium and is closely related to myocardial ischemia/reperfusion injury and myocardial metabolism. To explore the effects and the mechanisms of UCP2 on atorvastatin-mediated myocardium protection, the rat model of myocardial ischemia was established by ligation of the left anterior descending coronary arteries (LADs). The rats were divided into the sham operation (SO) group, myocardial infarction (MI) group and MI-atorvastatin group. The study that atorvastatin reduced myocardial remodeling and improved the disturbed myocardial energy metabolism after MI. Furthermore, the mechanisms of myocardial metabolic remodeling affected by atorvastatin were explored. The atorvastatin group showed a significantly decreased expression of UCP2 mRNA and protein. Furthermore, the primary rat cardiomyocytes were cultured and treated with angiotensin II (Ang II) to induce cardiomyocyte hypertrophy. The results showed that in the atorvastatin group, the surface area of the cardiomyocytes, the total protein content per unit of cells, and the expression of the UCP2 protein were significantly decreased. These data suggested that atorvastatin significantly attenuated the myocardial remodeling by downregulating the expression of UCP2 that was found to improve the myocardial energy metabolism, inhibit myocardial hypertrophy, and eventually reduce myocardial remodeling
Keywords: atorvastatin, heart failure, uncoupling proteins-2, metabolic remodeling
Introduction
Ventricular remodeling is the fundamental pathological process underlying the development of heart failure (HF), and the energy metabolism disorder that occurs after HF might be the major contributor to ventricular remodeling [1, 2]. A previous study reported that as the fetal heart was highly dependent on carbohydrates for energy production, the metabolic remodeling process could be considered as part of the return to fetal gene program, a hallmark of cardiac hypertrophy [3]. An increasing number of studies reported that myocardial metabolic remodeling is the process in which the heart loses its ability to utilize different substrates, thereby, becoming primarily dependent on the metabolism of a single substrate such as glucose or fatty acids for energy production [4, 5]. Myocardial metabolic remodeling is key to the pathogenesis of a variety of cardiac diseases such as left ventricular hypertrophy, myocardial ischemia, and diabetic cardiomyopathy [6, 7]. Myocardial energy metabolic disorders and ventricular remodeling create a vicious cycle that promotes the development and progression of HF. Therefore, the regulation of cardiac energy metabolism represents a potential treatment approach for HF.
Uncoupling proteins (UCPs), localized in the mitochondrial inner membrane, are important transporter proteins involved in energy metabolism [8]. The uncoupling protein 2 (UCP2) is primarily expressed in the myocardium [9]. The mitochondrial inner membrane protein, UCP2, functions as a proton transporter and plays a major role in energy metabolism. UCP2 exhibits a high proton transport activity. The upregulation of UCP2 activity enhances the proton transport capacity, which allows the protons to be shuttled directly into the mitochondrial matrix. The electrochemical gradient across the mitochondrial inner membrane that drives adenosine triphosphate (ATP) synthesis is altered, and free energy stored in the proton electrochemical potential is reduced. These phenotypes eventually lead to the uncoupling of oxidative phosphorylation and the reduction of ATP synthesis [10]. The mechanisms underlying the UCP2 overexpression may involve the activation of the sympathetic nervous system (SNS) and the renin-angiotensin-aldosterone system (RAAS) in response to HF [11, 12]. Thus, the UCP2 protein may serve as an indispensable link between energy metabolism and ventricular remodeling.
In recent years, the non-lipid-lowering effects of statins related to the cardiovascular field have gained increasing attention. Patel et al. demonstrated that simvastatin, a pleiotropic HMG-CoA reductase inhibitor, induces the regression of cardiac hypertrophy and fibrosis, reduces the levels of activated stress-responsive signaling kinases, and improves LV filling pressures in a transgenic rabbit model that fully recapitulates the phenotype of human hypertrophic cardiomyopathy [13]. Other studies have shown that simvastatin treatment reverses the established cardiomyocyte hypertrophy and improves left ventricular function [14, 16]. Such studies also demonstrated that statins reversed the negative changes associated with left ventricular remodeling; however, the underlying mechanisms of action of statins are not yet clarified. Thus, the present study established a model of HF after myocardial infarction (MI) was constructed in the present study to examine the effects of atorvastatin on myocardial energy metabolism and the expression of the energy metabolism-related gene UCP2 in rats suffering from HF post-MI. In addition, a model of cardiomyocyte hypertrophy was established by using in vitro cultured primary rat cardiomyocytes, and the mechanisms underlying the effects of atorvastatin on myocardial energy metabolism were explored. The present study provides a theoretical basis for understanding the pathogenesis of HF, thereby proposing novel approaches for the treatment of HF.
Materials and methods
Materials
The protocols in animal experiments were approved by the Jilin University Ethics Committee and performed in accordance with the International Guiding Principles for Animal Research. Female Wistar rats weighing 200-220 g and newborn 1-day-old Wistar rats were provided by the Experimental Animal Center in the School of Basic Medical Sciences, Jilin University (Changchun, China). The Certificate of Conformity was SCXK-(Ji) 2003-0001. Atorvastatin for in vitro experiments was purchased from Pfizer (New York City, NY, USA). Ang II and atorvastatin for in vivo experiments were obtained from Sigma-Aldrich (St. Louis, MO, USA). Antibody against UCP2 was obtained from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA).
Establishment of the rat MI model
Female Wistar rats, with a body weight of 200-220 g were used. The rat model of heart failure after MI was established by ligation of the anterior descending coronary artery. The protocol was summarized as follows: diethylether-anesthetized rats were fixed on the operating table. The pericardium was opened to expose the heart. The left anterior descending (LAD) coronary artery was identified, and the coronary artery ligated. Subsequently, the heart was placed back into the thoracic cavity, and the blood and gas within the thoracic cavity were squeezed out. The purse-string suture was quickly pulled tight to close the chest. The entire duration of thoracotomy did not exceed 30 s. A sham operation group was also prepared, with only threading and without ligation, as the control group. Subsequently, after 4 weeks, the rats were divided into the sham operation group (SO group, normal diet, n = 6), the myocardial infarction model group (MI group, normal diet, n = 6), and the MI model + atorvastatin group (Ator group, normal diet + atorvastatin 10 mg/kg per day, n = 6), and the treatment was continued for an additional 4 weeks before evaluation of the relevant indicators. All animals received humane care and the experimental procedures were approved by the Animal Ethics Committee of Jilin University.
Measurement of hemodynamics
After drug administration, an intraperitoneal injection of 3% pentobarbital sodium solution (30 mg/kg) was administered for anesthesia. After measuring the body weight, left ventricular cannulation was established through the right common carotid artery. The pressure transducer was connected to the AP-621G carrier amplifier. The left ventricular end-systolic pressure (LVESP) and the left ventricular end-diastolic pressure (LVEDP) were recorded using the RM-6000 eight-channel physiological recorder. The LVESP signal was delivered to the differentiator to trace the maximal rate of left ventricular pressure rise (+dp/dtmax) and maximal rate of left ventricular pressure fall (-dp/dtmax).
Hematoxylin and eosin (HE) staining
HE staining was performed to evaluate the pathological and morphological changes in the myocardial tissue. The detailed procedures were as follows: the myocardium sample (approximately 2 mm thick) was collected and fixed with 4% paraformaldehyde at 4ºC for 72 h. Then, routine paraffin embedding was performed and 5 μm thick slices were ectioned. Subsequently, the sections were xylene- deparaffinized, hydrated through gradient ethanol, stained with the HE, dehydrated through gradient ethanol, cleared in xylene, and finally mountedin neutral resin. The pathological and morphological changes in the myocardial tissue were observed under an optical microscope.
Transmission electron microscopy
Small pieces of myocardial tissue (approximately 2 mm3) were excised from the apex of the heart and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer at 4 °C, followed by 3 washed in the same buffer. Then, the samples were then fixed with 1% osmium tetroxide for 2 h, dehydrated through a gradient ethanol series, and embedded in Epoxy Resin 812 (Agar Scientific-pre-mix kit 812, Agar Sci entific), and polymerized overnight at 60°C. Resin-embedded blocks were cut into 60-80 nm ultrathin sections with an ultramicrotome (LKB-III, Sweden). The ultrathin sections were placed on carbon-coated nickel grids and examined with a JEM-1200EX transmission electron microscope to observe the changes in the myocardial ultrastructure.
Lactic acid, free fatty acid (FFA), and the adenosine triphosphatase (ATPase) activities assay
The serum lactate levels, FFA concentrations, sodium-potassium-ATPase (Na+-K+-ATPase) activities, calcium-magnesium-ATPase (Ca2+-Mg2+-ATPase) activities, and the erythrocyte membrane ATPase activities were detected by colorimetry. Lactic acid assay kit, Non-esterified free fatty acids assay kit, Na+-K+-ATPase assay kit, and Ca2+-Mg2+-ATPase assay kit were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The experimental steps were in strict accordance with the manufacturer's instructions.
Enzyme-linked immunosorbent assay (ELISA)
In the myocardial tissue homogenates, the levels of ATP were detected by ELISA. Briefly, 50 mg myocardial tissue was homogenized in 0.5 mL Tris-EDTA buffer (pH7.5) containing 1% Triton X-100, 50 mmol/L Tris-HCl, 150 mmol/L NaCl, 2 mmol/L EDTA, 2 mmol/L PMSF, 1 mmol/L NaF, 4 μg/mL leupeptin, and 1 μg/mL aprotinin. The extracts were centrifuged at 12000 rpm at 4 °C for 30 min and supernatants collected. The protein concentrations were determined using the protein assay kit following the manufacturer's instructions.
Semi-quantitative reverse transcription polymerase chain reaction (qRT-PCR)
The levels of UCP-2 mRNA in the no-ninfarcted area of the left ventricle were detected by qRT-PCR. Total RNA was isolated using TRIzol reagen (Invitrogen, CA, USA) according to the manufacturer's instructions. cDNA was synthesized from 2 μg of total RNA. The PCR for β-actin, UCP-2 was performed using the following primers: β-actin (359 bp, NM03114): sense 5'-GCTCGTCGTCGACAACGGCTC-3', antisense 5'-CAAACATGATCTGGGTCATCTTCTC-3'; UCP-2 (298 bp, NMAF039033): sense 5' -CAAGCGGAGGAAGGAAGG-3', antisense 5'- CAATGTTGCCCGAAA TGC-3'. The reactions were conducted at the following cycle conditions: pre-denaturation at 95 ˚C for 90 s, denaturation at 94 ˚C for 30 s, annealing at 57 ˚C for 30 s, and extension at 72 ˚C for 50 s. It was 32 cycles. The PCR extension was at 72˚C for 10 minutes. The products were separated on 1.5% agarose gels that were stained with ethidium bromide. The intensity of bands was evaluated by image analysis (ImageMaster VDS, Pharmacia Biotech).
Immunohistochemistry
The non-infarcted left ventricular tissue was deparaffinized, rehydrated in a gradient series of alcohol solutions, and washed two times in distilled water. The sections were incubated with endogenous peroxidase blocked in 50 μL of 3% hydrogen peroxide (H2O2) at room temperature for 10 min and washed with PBS (pH 7.4). The sections were blocked with 2% bovine serum albumin (BSA) in PBS was added and incubated at room temperature for 30 min. Subsequently, the sections were probed with UCP2 antibody at 4 °C overnight, and the protein assayed with an Ultrasensitive SP kit. The sections were counterstained with hematoxylin. The IgG from normal rabbit served as a negative control.
Western blotting
Total protein from myocardium tissues was extracted and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Then, the proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane, blocked and probed sequentially with primary antibody against UCP2, followed by the appropriate secondary antibody. After washing, the immunoreactive complexes were detected using an electrochemilumenescence reagent.
Cell culture and surface area measurement
The primary cultures of neonatal rat cardiomyocytes were prepared from the cardiac ventricles of neonatal Wistar rats as described previously []. Briefly, cardiac ventricles obtained from 1- to 2-day-old Wistar rat neonates, were gently minced and enzymatically dissociated using trypsin (pH 7.4), 0.02 mol/L PBS at 37 °C. The dissociated cells were filtered through the 200-µm mesh, collected by centrifugation, and incubated for 1.5 h at 37 °C in a cell culture flask. The non-cardiomyocytes, fibroblasts, or endothelial cells, could easily adhere to plastic surfaces than cardiomyocytes. After 1.5 h, the supernatant was collected and cells plated at a density of 5 x 105/mL in a 60-mm dish in DMEM/F-12 culture medium containing 10% fetal calf serum and antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin) for 48 h. Simultaneously, 5-Bromo-2'-deoxyuridine (BrdU, 10-4 mol/L) was added to prevent the proliferation of non-myocytes. After 48 h, the cells were seeded and serum-starved for 24 h. Subsequently, the cells were divided into the following groups: Ang II (0.1 µmol/L), atorvastatin + Ang II (10 µmol/L atorvastatin + 0.1 µmol/L Ang II), and control (only medium); according to the respective additions to the culture dishes for 24 h. Inverted phase contrast microscope was used to acquire the images of cardiomyocytes at 100× magnification. The surface area of a minimum of 25 cells per treatment group was measured and averaged to produce one N value.
Total protein content per unit of cells
The protein extracts from cardiomyocytes were prepared as follows: the cells were washed two times in ice-cold D-Hank's medium and lysed using 100 μL lysis buffer (50 mM Tris-Cl pH7.5, 150 mM NaCl, 2 mM EDTA, 1% NP-40) on ice for 60 min; then, the cells were scrapped into a centrifuge tube. Then, the cell lysates were centrifuged at 12,000 rpm at 4 °C for 30 min in a table centrifuge to remove cell debris. The cleared supernatants were transferred to fresh tubes and protein concentrations were determined by Bradford assay (Pierce, Rockford, IL, USA). BSA was used as standard protein control. Finally, the total protein content was calculated.
Statistical analysis
Statistical analyses were performed using SPSS 10.0 software. Data were presented as the mean ± standard deviation (SD). The difference between the two groups was evaluated by unpaired t-test. The differences among different groups were determined by Kruskal-Wallis test. Probability (P)-value of < 0.05 was considered as statistically significant.
Results
Atorvastatin attenuates left ventricular remodeling and improves cardiac function in rats with HF after MI
To investigate the protective role of atorvastatin in HF after MI, we initially examined the hemodynamics of different groups. Compared to the sham-operated group, rats in the model group exhibited an increase in LVEDP and a decrease in LVSP and ±dp/dt max (mmHg/s). Compared to the model group, rats in the atorvastatin-treated group showed a decreased LVEDP and a markedly elevated LVSP and +dp/dt max (mmHg/s); the differences were statistically significant (all P < 0.05) (Fig. 1A).
Atorvastatin suppressed HF in the rat with MI. (A) Hemodynamic index, including ±dP/dtmax, LVEDP, and LVSP. (B) Increased LVMI is a major indicator to determine the existence of left ventricular hypertrophy or left ventricular remodeling. The LVMI is equal to the left ventricular weight divided by the body weight (LVW/BW). (C) Representative images of hearts. Four weeks after drug administration, the heart was removed, and tissue sections stained with H&E (×200 magnification) or scanned by electron microscope (×20,000 magnification). +dP/dtmax, maximal rate of rise in the blood pressure in the ventricular chamber; -dp/dtmax, maximal rate of decline in the blood pressure in the ventricular chamber; LVEDP, left ventricular end diastolic pressure; LVSP, left ventricular systolic pressure. Data were presented as mean ± SD. *P < 0.05, **P < 0.01 vs. SO group; #P < 0.05, ##P < 0.01 vs. MI group.
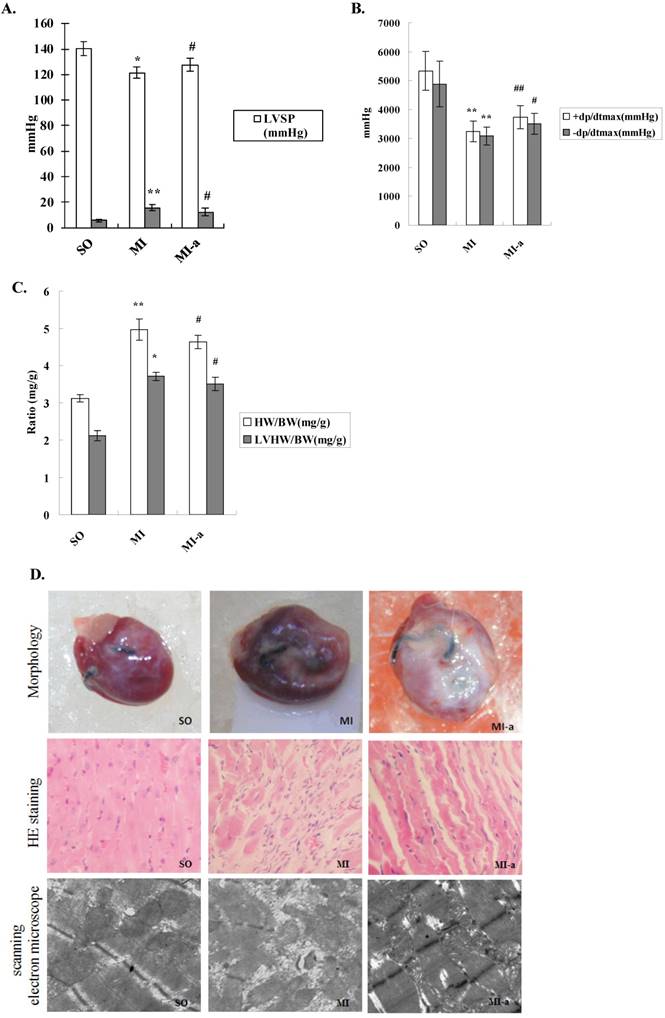
The left ventricular mass index is defined as the ratio of left ventricular weight (LVW) to body weight (BW). An increase in the left ventricular mass index is a vital indicator of left ventricular hypertrophy or left ventricular remodeling. The left ventricular mass indices were examined, and the results showed that as compared to the sham-operated group, the LVW/BW ratios were markedly increased in rats from the model group. Compared to the model group, the LVW/BW ratios were decreased in rats from the atorvastatin-treated group. The differences were statistically significant (all P < 0.05) (Fig. 1B).
The morphological indices of the myocardium were examined by HE staining and electron microscopy. Compared to the sham-operated group, the myocardial fibers in the model group were arranged in a rather disordered manner, and the cardiomyocytes in the model group showed varying degrees of degeneration and hypertrophy. In addition, excessive deposition of fibrous tissue into the extracellular matrix was observed in the model group. The myocardial interstitial fibrosis and cardiomyocyte hypertrophy were significantly reduced in the atorvastatin-treated group as compared to the model group (Fig. 1C). These results indicated that left ventricular remodeling occurred in response to HF, suggesting that atorvastatin was able to significantly attenuated the left ventricular remodeling.
Atorvastatin might attenuate deleterious ventricular remodeling by affecting energy metabolism in cardiomyocytes
Myocardial metabolic remodeling refers to the process by which impaired myocardial energy production ultimately leads to the structural and functional cardiac abnormalities. Previous studies have shown that myocardial metabolic remodeling plays a crucial role in the development and progression of HF. FFAs constitute a major class of compounds that provide energy to the body. FFA is the primary source of energy for the myocardium. To clarify the effects of atorvastatin on myocardial energy metabolism after HF, we first examined the serum lactate and FFA levels and the concentrations were found to be significantly increased in the model group as compared to the sham-operated group (P<0.001). In addition, the serum lactate levels and FFA concentrations were significantly reduced in the atorvastatin-treated group compared to the model group (P < 0.01) (Fig. 2A). These results indicated that atorvastatin improved the myocardial energy metabolism after HF.
Atorvastatin might attenuate deleterious ventricular remodeling by affecting energy metabolism in cardiomyocytes. (A) The serum lactate levels and FFA concentrations. (B) Levels of ATP in myocardial tissue. The levels of ATP were detected by ELISA. (C) ATPase activities in the serum and the erythrocyte membrane. Na+-K+-ATPase activities, Ca2+-Mg2+-ATPase activities, and erythrocyte membrane ATPase activities were detected by colorimetry. The data are presented as the means ± SD. **P < 0.01 vs. SO group; #P < 0.05, ##P < 0.01 vs. MI group.
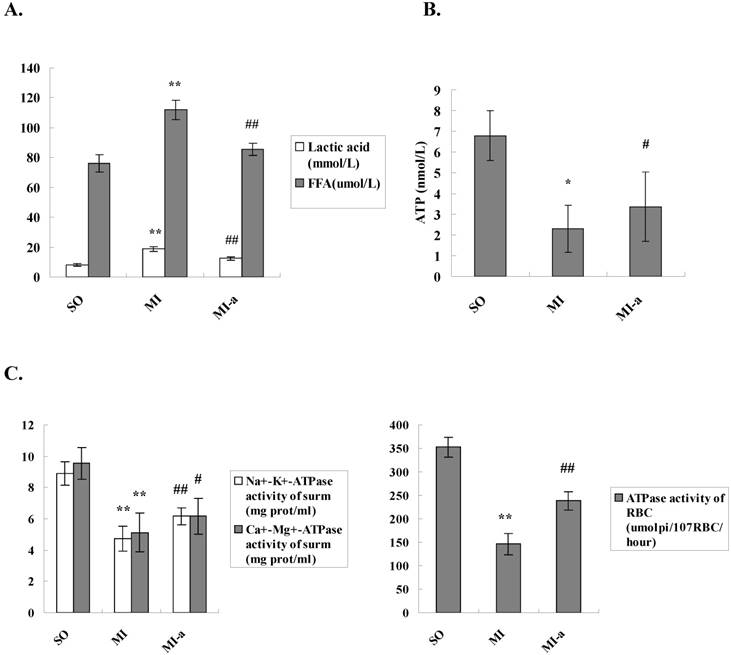
ATP is the only energy source that is used directly by the myocardium. In the present study, the ATP levels in the myocardium were examined to determine the effects of atorvastatin on myocardial energy metabolism after HF. The results showed that the ATP content in the non-infarcted myocardium was significantly decreased in the model group as compared to the sham-operated group (P < 0.01). In contrast, the ATP content in the non-infarcted myocardium was significantly elevated in the atorvastatin-treated group as compared to the model group (P < 0.05) (Fig. 2B).
In addition, the ATPase activities in the serum and erythrocyte membrane were examined. The results showed that the serum Na+-K+-ATPase and Ca2+-Mg2+-ATPase activities, as well as the erythrocyte membrane ATPase activities, were dramatically decreased in the rats from the model group as compared to the sham-operated group (P < 0.01-0.001). Compared to the model group, the serum Na+-K+-ATPase and Ca2+-Mg2+-ATPase activities and the erythrocyte membrane ATPase activities were significantly elevated in the atorvastatin-treated group (P < 0.05-0.01) (Fig. 2C). These results further supported that atorvastatin improved the myocardial energy metabolism after HF, which might be key mechanism underlying the improvement of left ventricular remodeling by atorvastatin.
Atorvastatin affect energy metabolism in cardiomyocytes after HF via the downregulation of UCP2 expression
UCP2 is a major transporter protein involved in energy metabolism. UCP2 is localized in the mitochondrial inner membrane. The upregulation of UCP2 activity leads to a reduction in ATP synthesis. In order to elucidate the role of UCP2 in atorvastatin-regulated myocardial energy metabolism after HF, we first analyzed the expression of UCP2 mRNA in the myocardium in all groups of rats by PCR. The results showed that as compared to the sham-operated group, the expression of UCP2 mRNA was markedly increased in the non-infarcted region of the left ventricle in the model group (P < 0.01). Furthermore, the UCP2 mRNA expression was significantly reduced in the atorvastatin-treated group as compared to the model group (P < 0.05) (Fig. 3A). These results indicated that atorvastatin might improve myocardial energy metabolism after HF via the inhibition of UCP2 expression.
Atorvastatin affects energy metabolism in cardiomyocytes after HF via downregulation of UCP2 expression. (A) The expression of UCP2 mRNA by qRT-PCR. The total RNA of non-infarcted left ventricular tissue was extracted using TRIzol, and qRT-PCR was performed to determine the levels of UCP2 mRNA. The data are presented as the means ± SD. **P < 0.01 vs. SO group; #P < 0.05, ##P < 0.01 vs. MI group. (B) The expression of UCP2 in rat cardiac tissues by immunohistochemistry (×200 magnification). (C) Western blotting was performed to determine the expression of the UCP2 protein. Protein extract was prepared from myocardial tissue, separated by SDS-PAGE, and immunoblotted sequentially using the anti-UCP2 antibody. The graph shows the densitometric quantification of the UCP2 protein relative to β-actin that was as an internal control. The data are presented as the means ± SD. **P < 0.01 vs. SO group; #P < 0.05, ##P < 0.01 vs. MI group.
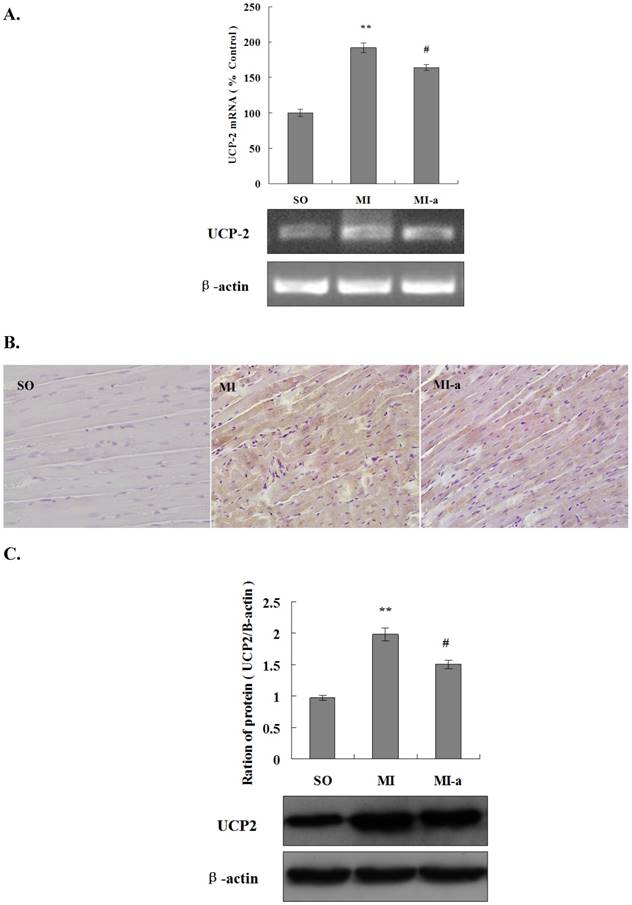
In the present study, UCP2 protein expression in the myocardium was analyzed in all groups by immunohistochemistry. The results showed that as compared to the sham-operated group, the expression of the UCP2 protein was significantly increased in the non-infarcted region of the left ventricle in the model group. Furthermore, the expression of the UCP2 protein was significantly reduced in the atorvastatin-treated group as compared to the model group (Fig. 3B).
To quantify the UCP2 protein expression in the myocardium in all groups of rats, a Western blot analysis was performed. These results were consistent with those of PCR and the immunohistochemistry (Fig. 3C), indicating that the downregulation of UCP2 expression might be one of the major factors contributing to the atorvastatin-induced improvement of myocardial energy metabolism after HF.
Atorvastatin might inhibit Ang II-induced cardiomyocyte hypertrophy via the downregulation of UCP2 expression
The in vivo animal experiments described above indicated that atorvastatin might improve the energy metabolism in cardiomyocytes after HF, and consequently, attenuates ventricular remodeling through the downregulation of UCP2 expression. Myocardial hypertrophy is a characteristic of myocardial remodeling. In order to substantiate the results described above, primary rat cardiomyocytes were cultured in vitro. The cardiomyocyte hypertrophy was induced by treating the cells with Ang II, and the effect of atorvastatin on myocardial hypertrophy was investigated.
The surface area of cardiomyocytes in all experimental groups was measured using an inverted phase contrast microscope. The results showed that the cell surface area was significantly increased in the Ang II group as compared to the control group (P<0.01). In addition, coarse and thick pseudopodia were observed on the surfaces of cells in the Ang II group. The surface area of the cardiomyocytes was significantly reduced in the atorvastatin group as compared to the Ang II group (P < 0.01) (Fig. 4A). These results indicated that atorvastatin inhibited the Ang II-induced myocardial hypertrophy and attenuated the adverse myocardial remodeling.
Atorvastatin inhibited Ang II-induced cardiomyocyte hypertrophy through the downregulation of UCP2 expression. (A) Cell surface area measurement. Inverted phase contrast microscope was used to obtain the cardiomyocyte images at ×100 magnification. The surface area of a minimum of 25 cells per treatment group was measured and averaged to generate one N value. The data are presented as the means ± SD. **P < 0.01 vs. control group, ##P < 0.01 vs. Ang II group. (B) Total protein content per unit of cells. The protein extracts from cardiomyocytes were prepared, and the total protein content per unit cells calculates. The data are presented as the means ± SD. **P < 0.01 vs. control group, ##P < 0.01 vs. Ang II group. (C) Western blotting was performed to determine the UCP2 expression. Proteins were extracted from cardiomyocytes, separated by SDS-PAGE, and immunoblotted sequentially using the anti-UCP2 antibody. The graph shows the result of densitometric quantification of the UCP2 protein relative to β-actin utilized as an internal control. The data are presented as the means ± SD. **P < 0.01 vs. control group; #P < 0.05, ##P 0.01 vs. Ang II group.
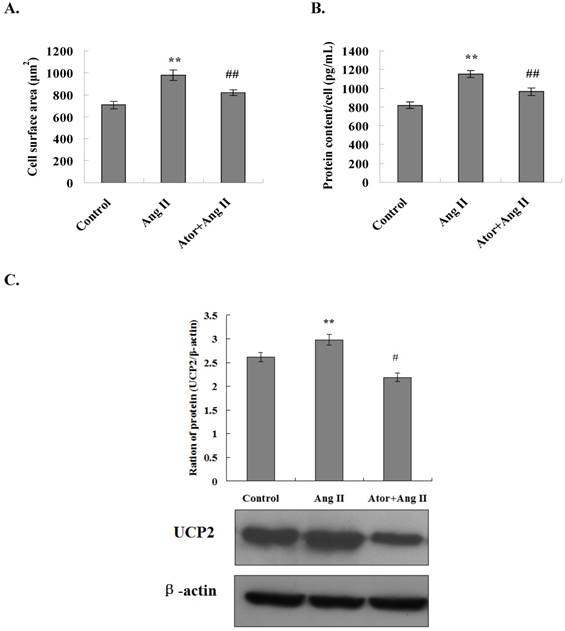
The total protein content per unit of cardiomyocytes was also examined in all experimental groups. The results showed that the total protein content per unit of cells increased considerably in the Ang II group as compared to the control group with a statistically significant difference (P < 0.01). Moreover, the total protein content per unit of cells decreased significantly in the atorvastatin group as compared to the Ang II group (P < 0.01) (Fig. 4B). These observations indicated that atorvastatin inhibits Ang II-induced myocardial hypertrophy and attenuates adverse myocardial remodeling.
Myocardial energy metabolic remodeling might eventually lead to cardiac structural remodeling. UCP2 is closely related to myocardial energy metabolism. To determine whether atorvastatin attenuates adverse myocardial remodeling by regulating the UCP2 expression, primary rat cardiomyocytes were cultured in vitro and treated with Ang II to induce cardiomyocyte hypertrophy. The expression of the UCP2 protein in cardiomyocytes in all experimental groups was examined by Western blot analysis. The results showed that the level of UCP2 protein expression was significantly increased in the Ang II group as compared to the normal control group (P < 0.01). Moreover, the UCP2 protein expression was significantly decreased in the atorvastatin group as compared to the Ang II group (P < 0.01) (Fig. 4C). These results were in agreement with those of the in vivo experiments, further indicating that atorvastatin might improve myocardial energy metabolism and eventually improve myocardial remodeling via the downregulation of UCP2 expression.
Discussion
The major finding of this study is that administration of atorvastatin during the peri-infarct period improves LV function and attenuates adverse LV remodeling after MI. This salubrious effect was associated with the improvement of myocardial energy metabolism after atorvastatin-induced MI.
Ventricular remodeling is now recognized as the fundamental pathological process underlying the development and progression of chronic HF[17]. However, the ventricular remodeling process is rather complex. Katz et al. proposed two basic mechanisms that might contribute to the development of abnormal myocardial function in HF: abnormal energy metabolism and abnormal gene expression[18]. Dysfunctional myocardial metabolism post-HF promotes the development and progression of HF. Consequently, the progression of HF increases the frequency of myocardial metabolic abnormalities. The HF progression and myocardial metabolic abnormalities promote each other, creating a self-perpetuating vicious cycle. Therefore, the regulation of myocardial energy metabolism could be a prospective target for the treatment of HF. In the present study, a rat model of HF post-MI was established, and atorvastatin intervention performed. The analysis of morphological and hemodynamic indices showed that atorvastatin significantly improved the conditions of HF after MI. These results were consistent with those of previous studies[19]. To further explore the mechanisms underlying atorvastatin-mediated improvement in HF after MI, in-depth studies were focusing on myocardial energy metabolism.
Myocardial energy metabolism is closely related to HF[20-22]. After MI, mitochondrial morphology and function are impaired, and mitochondria exhibit a limited ability to supply energy for normal myocardial activity. As a result, ATP synthesis is reduced, the myocardial contractile function is declined, and cardiac output decreased. In addition, myocardial diastolic dysfunction occurs and myocardial compliance decreases, ultimately leading to HF[23-24]. In 2004, Van Bilsen et al. proposed that myocardial metabolic remodeling[3] is characterized by the disorders of carbohydrate and lipid metabolism in cardiomyocytes triggering the changes in cardiac energy metabolic pathways. This causes the blockage of myocardial energy production, leading to abnormalities in cardiac structure and function. The study further revealed that abnormalities in myocardial energy metabolism are important contributing factors to myocardial remodeling. In order to understand whether atorvastatin improves HF outcomes through myocardial energy metabolism, the indices of myocardial energy metabolism (lactate, FFA, ATP, Na+-K+-ATPase, Ca2+-Mg2+-ATPase, and erythrocyte membrane ATPases) in all groups of rats were assessed in the present study. The results showed that atorvastatin significantly improves myocardial energy metabolism after MI, indicating that atorvastatin inhibits the myocardial remodeling and restores the cardiac function.
UCPs are localized in the mitochondrial inner membrane. UCPs are essential transporter proteins involved in energy metabolism[25-26]. UCP2 is primarily expressed in the myocardium. Activation of the sympathetic nervous system (SNS) and the Renin angiotensin aldosterone system (RAAS) after HF results in the overexpression of UPC2 and upregulation of its activity[27]. The upregulation of UCP2 activity enhances the proton transport that allows direct shuttling of the protons into the mitochondrial matrix. The electrochemical gradient across the mitochondrial inner membrane that drives ATP synthesis is altered. Thus, the free energy stored in the proton electrochemical potential is reduced, which eventually leads to the uncoupling of oxidative phosphorylation and a decline in ATP synthesis[28]. To further investigate the mechanisms underlying the improvement of atorvastatin-induced myocardial energy metabolism, the expression of UCP2 was examined. The results showed that UCP2 expression was significantly increased following HF, and atorvastatin significantly downregulated the UCP2 expression. The in vivo animal experiments indicated that UCP2 overexpression following HF might be a significant contributing factor towards myocardial energy metabolism abnormalities and myocardial remodeling. Atorvastatin improved the energy metabolism in cardiomyocytes after HF and consequently attenuated the adverse myocardial remodeling by downregulating the expression of UCP2.
Myocardial hypertrophy is one of the manifestations of myocardial remodeling[29]. The characteristic pathological changes in myocardial hypertrophy at the cellular level include increased cardiomyocyte volume, enhanced rates of protein synthesis per unit of cells, and altered sarcomere structure. To verify the results described above, primary rat cardiomyocytes were cultured in vitro. Ang II was applied to the cells to induce cardiomyocyte hypertrophy, and the effect of atorvastatin on myocardial hypertrophy was investigated. These results showed that the surface area of cardiomyocytes and the total protein content per unit of cells were significantly increased in the Ang II group. Conversely, the surface area of the cardiomyocytes and the total protein content per cell were significantly reduced in the atorvastatin group. These results indicated that atorvastatin inhibited the Ang II-induced myocardial hypertrophy and attenuated the deleterious myocardial remodeling. Furthermore, to determine whether atorvastatin improved the myocardial energy metabolism and consequently attenuated the adverse myocardial remodeling through regulation of UCP2 expression, the expression levels of UCP2 protein in cardiomyocytes were examined in all the experimental groups. The results showed that the level of UCP2 protein expression in cardiomyocytes was significantly increased in the Ang II group and significantly decreased in the atorvastatin group, which was consistent with that observed in the in vivo experiments. These results further indicated that atorvastatin improves myocardial energy metabolism and consequently attenuates the adverse myocardial remodeling through the downregulation of UCP2 expression.
In summary, myocardial energy metabolic remodeling following HF plays an essential role in the development and progression of HF. The present study found that atorvastatin improved myocardial energy metabolism, attenuated the deleterious ventricular remodeling, and suppressed HF through the downregulation of UCP2 expression. Therefore, this study provided the theoretical and experimental foundation for the clinical application of atorvastatin in the treatment of HF.
Acknowledgements
This study was supported by grants provided by the National Natural Science Foundation of China (No. 81400298, 81570360), the Project of Science and Technology of Jilin Province (No. 20150101150JC, 20170622012JC), the Program for Development and Reform Commission of Jilin Province (No. 2016C026), the fund for the Jilin Provincial Health and Family Planning Commission (No. 20152001), and the Bethune program of Jilin university (No. 2015303).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tuomainen T, Tavi P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp Cell Res. 2017 Mar 24. pii: S0014-4827(17)30178-7
2. Dalal JJ, Mishra S. Modulation of myocardial energetics: An important category of agents in the multimodal treatment of coronary artery disease and heart failure. Indian Heart J. 2017;69:393-401
3. van Bilsen M1, Smeets PJ, Gilde AJ. et al. Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovasc Res. 2004;61:218-226
4. Peterzan MA, Lygate CA, Neubauer S. et al. Metabolic remodelling in hypertrophied and failing myocardium: a review. Am J Physiol Heart Circ Physiol. 2017;313(3):H597-H616
5. De Jong KA, Lopaschuk GD. Complex Energy Metabolic Changes in Heart Failure With Preserved Ejection Fraction and Heart Failure With Reduced Ejection Fraction. Can J Cardiol. 2017;33:860-871
6. Phillips D, Ten Hove M, Schneider JE. et al. Mice over-expressing the myocardial creatine transporter develop progressive heart failure and show decreased glycolytic capacity. J Mol Cell Cardiol. 2010;48:582-90
7. Robert J. Gropler, Rob S.B. Beanlands, Vasken Dilsizian,E. Douglas Lewandowski, Flordeliza S. Villanueva, Maria Cecilia Ziadi. Imaging Myocardial Metabolic Remodeling. J Nucl Med. 2010;51:88S-101S
8. Ruiz-Ramírez A, López-Acosta O, Barrios-Maya MA. et al. Cell Death and Heart Failure in Obesity: Role of Uncoupling Proteins. Oxid Med Cell Longev. 2016;2016:9340654
9. Valouskova E, Modriansky M. Modulation of UCP2 expression by p38-a link to cardioprotection. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2008;152:3-7
10. Boss O, Hagen T, Lowell BB. Uncoupling proteins 2 and 3: potential regulators of mitochondrial energy metabolism. Diabetes. 2000;49:143-56
11. Chan S. H, Wu, C. W, et al. Transcriptional upregulation of brain-derived neurotrophic factor in rostral ventrolateral medulla by angiotensin II: significance in superoxide homeostasis and neural regulation of arterial pressure. Circ Res. 2010;107:1127-39
12. Guo P, Nishiyama A, Rahman M. et al. Contribution of reactive oxygen species to the pathogenesis of left ventricular failure in Dahl salt-sensitive hypertensive rats: effects of angiotensin II blockade. J Hypertens. 2006;24:1097-104
13. Patel R, Nagueh SF, Tsybouleva N. et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function a transgenic rabbit model of human hypertrophic cardimyopathy. Circulation. 2001;104:317-24
14. Zhang J, Cheng X, Liao YH, et a1. Simvastatin regulates myocardial cytokine expression and improves ventricular remodeling in rats after acute myocardial infarction. Cardiovasc Drugs Ther. 2005;19:13-21
15. Zou C, Qi H, Liu ZH. et al. Simvastatin activates the PPARγ-dependent pathway to prevent left ventricular hypertrophy associated with inhibition of RhoA signaling. Tex Heart Inst J. 2013;40:140-7
16. Brunetti ND, Correale M, Totaro A. et al. Lower cardiovascular mortality with atorvastatin and rosuvastatin vs simvastatin: Data from "moderate-intensity" statin users in an observational registry on chronic heart failure (Daunia Heart Failure Registry). Int J Cardiol. 2015;194:23-7
17. Reis Filho JR, Cardoso JN, Cardoso CM. et al. Reverse Cardiac Remodeling: A Marker of Better Prognosis in Heart Failure. Arq Bras Cardiol. 2015;104:502-6
18. Katz A. The myocardium in congestive heart failure. Am. J. Cardiol. 1989;63:12A-16A
19. Song XJ, Yang CY, Liu B. et al. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int. J. Med. Sci. 2011;8:564-572
20. Gupte AA, Hamilton DJ, Cordero-Reyes AM, et a1. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet. 2014;7:266-76
21. Biesemann N, Mendler L, Wietelmann A. et al. Myostatin regulates energy homeostasis in the heart and prevents heart failure. Circ Res. 2014;115:296-310
22. Dinicolantonio JJ, Niazi AK, McCarty MF. et al. L-carnitine for the treatment of acute myocardial infarction. Rev Cardiovasc Med. 2014;15:52-62
23. Daniels MC, Naya T, Rundell VL. et al. Development of contractile dysfunction in rat heart failure: hierarchy of cellular events. Am J Physiol Regul Integr Comp Physiol. 2007;293:R284-92
24. Williams D, Venardos KM, Byrne M. et al. Abnormal mitochondrial L-arginine transport contributes to the pathogenesis of heart failure and rexoygenation injury. PLoS One. 2014;9:e104643
25. Siu AH, Ko KM. Herba Cistanche extract enhances mitochondrial glutathione status and respiration in rat hearts, with possible induction of uncoupling proteins. Pharm Biol. 2010;48:512-7
26. Li N, Wang J, Gao F. et al. The relationship between uncoupling protein 2 expression and myocardial high energyphosphates content in abdominal aorta constriction induced heart failure rats. Zhonghua Xin Xue Guan Bing Za Zhi. 2009;37:1108-12
27. Li P, Guo X, Lei P. et al. PI3K/Akt/uncoupling protein 2 signaling pathway may be involved in cell senescence and apoptosis induced by angiotensin II in human vascular endothelial cells. Mol Biol Rep. 2014;41:6931-7
28. Toda C, Diano S. Mitochondrial UCP2 in the central regulation of metabolism. Best Pract Res Clin Endocrinol Metab. 2014;28:757-64
29. Oka T, Akazawa H, Naito AT. et al. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114:565-71
Author contact
![]() Corresponding author: Dr. Ping Yang, Department of Cardiology, China-Japan Union Hospital, Jilin University, Changchun 130033, China. Tel: +86-431-84995091; Fax: +86-431-84995091. E-mail: pyangedu.cn.
Corresponding author: Dr. Ping Yang, Department of Cardiology, China-Japan Union Hospital, Jilin University, Changchun 130033, China. Tel: +86-431-84995091; Fax: +86-431-84995091. E-mail: pyangedu.cn.