International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(4):339-351. doi:10.7150/ijms.22927 This issue Cite
Research Paper
Claudin-3 Inhibits Lung Squamous Cell Carcinoma Cell Epithelial-mesenchymal Transition and Invasion via Suppression of the Wnt/β-catenin Signaling Pathway
Juanjuan Che1, Dongsheng Yue2, Bin Zhang2, Hua Zhang2, Yansong Huo2, Liuwei Gao2, Hongchao Zhen1, Yan Yang3, Bangwei Cao1 ![]()
1. Department of Oncology, Beijing Friendship Hospital, Capital Medical University, Beijing, P.R. China;
2. Department of Lung Cancer, Lung Cancer Center, Tianjin Medical University Cancer Institute and Hospital, Tianjin, P.R. China;
3. Department of Pathology, Beijing Friendship Hospital, Capital Medical University, Beijing, P.R. China.
Received 2017-9-21; Accepted 2017-12-15; Published 2018-2-5
Citation:
Che J, Yue D, Zhang B, Zhang H, Huo Y, Gao L, Zhen H, Yang Y, Cao B. Claudin-3 Inhibits Lung Squamous Cell Carcinoma Cell Epithelial-mesenchymal Transition and Invasion via Suppression of the Wnt/β-catenin Signaling Pathway. Int J Med Sci 2018; 15(4):339-351. doi:10.7150/ijms.22927. https://www.medsci.org/v15p0339.htm
Other stylesAbstract
Altered expression of claudin-3 (CLDN3), a key cytoskeletal structural protein of the tight junctions in the epithelium, is associated with the development and metastasis of various human cancers. CLDN3 expression has been shown to be significantly associated with the prognosis of lung squamous cell carcinoma (SqCC). This study investigated the role of CLDN3 in inhibiting lung SqCC cell migration and invasion as well as the underlying molecular mechanisms. The CLDN3 levels were assessed between 20 paired lung SqCC tissues and adjacent normal tissues using quantitative real-time polymerase chain reaction (qRT-PCR) and western blot. The ectopic CLDN3 overexpression or knockdown was generated by using a plasmid carrying CLDN3 cDNA or shRNA, respectively. CLDN3 expression was significantly reduced in lung SqCC tissues vs. the adjacent normal tissues. The ectopic CLDN3 overexpression markedly inhibited the migration, invasion, and epithelial-mesenchymal transition (EMT) of lung cancer H520 cells, whereas CLDN3 knockdown had an inverse effect on SK-MES-1 cells. However, cell viability and plate colony formation assays showed that both CLDN3 knockdown and overexpression did not affect SqCC cell proliferation. Both tissue and cell data revealed that CLDN3 expression was significantly associated with the expression of the EMT biomarkers E-cadherin and Vimentin. Furthermore, CLDN3-modulated EMT and expression of the EMT markers were through regulation of the Wnt/β-catenin signaling pathway. In conclusion, this study identified reduced CLDN3 expression in lung SqCC tissues, which was associated with the progression and metastasis of lung SqCC and was attributed to EMT by activation of the Wnt pathway. Thus, CLDN3 could be further evaluated as a novel biomarker for predicting the prognosis of lung SqCC and as a target for the treatment of lung SqCC in the future.
Keywords: Lung squamous cell carcinoma, claudin-3, EMT, Wnt, metastasis
Introduction
Lung cancer is one of the most diagnosed malignancies with the highest incidence and mortality rate in the world [1]. Non-small cell lung cancer (NSCLC) accounts for approximately 85% of lung cancer cases and mainly includes three distant histological subtypes, i.e., lung adenocarcinoma (AC), lung squamous cell carcinoma (SqCC), and large cell carcinoma [2]. Etiologically, tobacco smoke is the most significant risk factor that causes lung cancer development and progression; specifically, tobacco smoke insults the airway epithelia and induces them to undergo hyperplastic and squamous cell metaplastic changes. Such changes of the airway epithelia eventually progress to low- or high-grade dysplastic lesions and malignant transformation (in situ carcinoma and invasive lung cancer) [3-5]. On the molecular level, the development of NSCLC is related to either loss of tumor suppressor genes and/or activation of oncogenes [6-7]; while during NSCLC progression, the epithelial-mesenchymal transition (EMT) is the beginning of the process of cancer metastasis [8]. Thus, further research on the molecular mechanism for lung cancer progression, especially lung SqCC, could provide a novel insight into treatment strategies.
As an epithelium-originated malignancy, lung SqCC accounts for approximately 30% of all lung cancer cases and typically occurs close to the large airways [9-10]. During lung SqCC progression, tumor cells gain the ability to migrate and invade into the surrounding tissues and distant organs, similar to most other human cancers [9-10]. EMT is an important phenomenon in cancer progression, the process of which is initiated by the disappearance of the basic structures of the epithelia, followed by epithelial cell degeneration, enhancement of migration capacity, and loss of cell polarity and tight connections [11]. As for the tight junction structure in the epithelia or endothelia, it is localized at the apical basal region of adjacent epithelial or endothelial membranes to provide a continuous intercellular seal and form the biological membrane at physical interfaces [12]. At the molecular level, there are three types of membrane proteins observed in the tight junctions: junctional adhesion molecules, occludins, and claudins (CLDNs) [13]. The junctional adhesion molecules are members of the immunoglobulin (Ig) superfamily; they are thought to mediate homotypic cell adhesion and to regulate monocyte transmigration [14]. Occludins are integral membrane proteins with four transmembrane segments [15]. A previous study has demonstrated that occludin-null embryonic stem cells have tight junctions with a normal structure and function, indicating that occludin may not have any important roles in the tight junctions [16]. In contrast, CLDNs play an important role in the EMT process; to date, there are 27 CLDN members found in the human body and more than 12 CLDN members are expressed in the lung epithelia [17]. The distal lung epithelial cells mainly express CLDN3 and CLDN4 [17]. In addition, aberrant CLDN3 expression has been associated with the development and metastasis of various human cancers, but the data are controversial; for example, CLDN3 protein is overexpressed in ovarian, breast, and prostate cancers as well as in esophageal AC [18-21]. In another study, the CLDN3 levels were higher in prostate cancer patients with a Gleason score of ≥8, compared to patients with benign prostatic hyperplasia (p = 0.012) and those with a Gleason score of 6-7 [22]. In lung AC, CLDN3 expression increases the malignant potential of lung AC cells, indicating that CLDN3 plays a role in epidermal growth factor receptor activation [23]. However, CLDN3 expression is decreased in hepatocellular carcinoma [24] as well as in esophageal SqCC, and loss of CLDN3 expression is associated with the presence of distant metastases [20]. In lung cancer, CLDN3 has been found to be increased in lung AC, but lost in lung SqCC [23, 25].
Our previous data have shown that CLDN3 protein was expressed in 67 of 103 (67%) completely resected SqCC tissues and that stage I SqCC tissues had a significantly higher CLDN3 expression than stage IIIA tissues [25]. Moreover, CLDN3 expression was associated with the expression of E-cadherin but inversely correlated with the expression of β-catenin and Vimentin. The reduced CLDN3 expression was significantly associated with lung SqCC advanced tumor-node-metastasis (TNM) stages, lymph node metastasis, and disease recurrence [25]. Therefore, the aims of this study were to analyze the differential expression of CLDN3 mRNA and protein in lung SqCC vs. the adjacent normal tissues and then to investigate the effects of CLDN3 overexpression and knockdown on the regulation of lung SqCC EMT phenotypes and gene expression as well as the underlying molecular mechanisms.
Materials and methods
Ethics statement
The protocol of this study was approved by the Research Ethics Committees of Beijing Friendship Hospital (Beijing, China) and the Cancer Institute and Hospital of Tianjin Medical University (Tianjin, China). This study was conducted in accordance with ethics standards and the Declaration of Helsinki as well as national and international guidelines. The patients provided their consent in writing before providing their residual tissue specimens for this prospective study.
Patients
Patients with stage I-IIIA lung SqCC who underwent a complete tumor resection and systematic node dissection of the hilar and mediastinal lymph nodes at the Cancer Institute and Hospital of Tianjin Medical University or Beijing Friendship Hospital were entered into this study. In addition, we used a set of 20 matched pairs of lung SqCC and adjacent normal lung tissues. After patient consent, they were all enrolled into our current study and provided both tumor and adjacent normal tissue specimens that were banked after surgery. Patients receiving neo-adjuvant chemotherapy or radiation therapy, or having a second primary cancer diagnosed within 5 years, were excluded from this study. The histology of each tumor was diagnosed by two certified pathologists. The TNM stage was determined according to the International System for Staging Lung Cancer.
Cell lines and culture
The lung SqCC H1703, H2170, SK-MES-1, and H520 cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). The H1703, H2170, and H520 cell lines were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium containing 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA), and SK-MES-1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM)/F12 containing 10% FBS.
Western blot
The total cellular protein was extracted from cells and tissue specimens using a radioimmunoprecipitation assay buffer (BioRad, Hercules, CA, USA), and the concentrations of each sample were determined using the Bradford Protein Assay (BioRad). Protein samples of 50 µg each were separated by electrophoresis in 8-12% sodium dodecyl sulfate-polyacrylamide gels and transferred onto polyvinylidene fluoride membranes (Bio-Rad). Primary antibodies used to detect protein expression included anti-CLDN3 (1:1000, Abcam, Cambridge, MA, USA), anti-E-cadherin (1:500, Cell Signaling Technology, Danvers, MA, USA), anti-Vimentin (1:1000, Cell Signaling Technology), anti-β-catenin (1:1000, Cell Signaling Technology), anti-c-Myc (1:1000, Cell Signaling Technology), anti-cyclinD1 (1:1000, Cell Signaling Technology), and anti-tubulin (1:3000, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
The mRNA levels of CLDN3, E-cadherin, and Vimentin in cell lines as well as in 20 paired SqCC tissues and matched nontumorous tissues were analyzed using qRT-PCR. The total RNA was isolated using the Trizol Reagent (Invitrogen) and reversely transcribed into complementary DNA (cDNA) with 1 µg of total RNA using the PrimeScript RT reagent kit (Takara, Dalian, China), according to the manufacturers' instructions. After that, 2 µg of cDNA was used as the template for qPCR amplification of different gene transcripts using SYBR Premix Ex Taq II (Takara). GAPDH was used as an internal reference. The primers used in this study were as follows: CLDN3, 5ʹ-GTCCGTCCGTCCGTCCG-3ʹ and 5ʹ-GCCCAGCACGGCCAGC-3ʹ; E-cadherin, 5ʹ-AGCCCCGCCTTATGATTCTCTG-3ʹ and 5ʹ-TGCCCCATTCGTTCAAGTAGTCAT-3ʹ; Vimentin, 5ʹ-GAGTCCACTGAGTACCGGAGAC-3ʹ and 5ʹ-TGTAGGTGGCAATCTCAATGTC-3ʹ; GAPDH, 5ʹ-GAAGGTGAAGGTCGGAGTC-3ʹ and 5ʹ-GAAGATGGTGATGGGATTTC-3ʹ. The qPCR conditions were set to 95 °C for 3 min and then 45 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 45 s. The relative mRNA level was normalized to that of the endogenous control GAPDH and calculated based on the 2-ΔΔCt method. All reactions were performed in triplicate and repeated at least once.
RNA interference and transfection
Lentivirus carrying short hairpin RNA (shRNA) to knockdown CLDN3 was obtained from Gene Chem Inc. (Shanghai, China). Two of three CLDN3-specific shRNA constructs (CLDN3 shRNA sequence 1, 5ʹ-ACCGCAAGGACTACGTCTA-3ʹ) and one “nonspecific target” shRNA construct (the company did not disclose the DNA sequence), used as a scrambled negative control, were transduced separately into 293T cells for 24 h in the presence of hexadimethrine bromide to improve the transduction efficiency. Next, the growth medium containing viral particles was replaced with fresh complete growth medium containing 10 µg/mL puromycin (this concentration was chosen because it was found to be the minimum amount to cause complete 293T cell death after a 5-day incubation). Next, CLDN3 knockdown was confirmed using western blot analysis of CLDN3 expression compared with that of the nontarget shRNA control cells.
CLDN3 cDNA and transfection
The lentivirus carrying CLDN3 cDNA (CLDN3-H520) and a negative control lentivirus (CLDN3-H520PCDH), obtained from The Beijing Genomics Institute (Beijing, China), were used to infect lung cancer cells in a 24-well plate for 24 h at a multiplicity of infection of 100, according to the manufacturer's instructions. Cells were incubated at 37 °C in a humidified incubator containing 5% CO2 overnight. To reduce cell toxicity, the transfection mixture was replaced with fresh complete growth medium 24 h later, and the cells were continuously incubated for an additional 72 h. Then, the cells were observed under a fluorescence microscope (Olympus, Tokyo, Japan) to determine the infection efficiency, and CLDN3 expression was assessed using western blot.
Tumor cell wound healing assay
NSCLC cells after CLDN3 cDNA or shRNA, or negative viral control infections (H520-PCDH, H520-PCDH-CLDN3, SK-MES-1-control, and SK-MES-1-shRNA) were seeded into a 6-well plate and grown overnight. On the next day, three linear wounds were created using a 20-µL pipette tip on cells reaching approximately 90% or more confluency. Cells were washed with ice-cold phosphate-buffered saline (PBS) and further cultured for 24 h in RPMI-1640 (Gibco) or DMEM-F12 (Gibco) containing 2% FBS (Gibco) in a humidified incubator containing 5% CO2 at 37 °C. Photographs of the wounded area were taken immediately after making the scratch (0 h time-point) and at 24 h and 48 h to monitor the wounding healing capacity of the cells. The experiments were performed in triplicate and repeated at least once. The data were summarized as the mean ± standard deviation.
Tumor cell Transwell migration assay
To assess the tumor cell migration capacity, we utilized the 24-well Boyden chamber with an 8-μm pore size polycarbonate membrane (Corning, NY, USA). In brief, cells after CLDN3 cDNA or shRNA lentiviral infection were seeded into the upper chamber at a density of 1 × 105 per well in 200 μL of serum-free medium, while 600 μL of growth medium containing 10% FBS was added into the lower chamber. After that, the cells were grown for 24 h. The cells that remained on the upper surface of the membrane were removed using a cotton swab, whereas the cells that migrated to the lower surface of the membrane were fixed in methanol and stained with a three-step staining set (Thermo, London, UK). The cells in ten randomly selected microscopic fields (200× magnification) of each membrane were counted. The experiments were performed in triplicate and repeated at least once. The data were summarized as the mean ± standard deviation.
Tumor cell viability 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Tumor cell viability after gene manipulation was assayed using the MTT assay. In particular, cells were seeded into a 24-well cell culture plate at a density of 5 × 104 per well and grown overnight. On the next day, the cells were infected with CLDN3 cDNA or shRNA, or control lentivirus for 24 h. After that, the growth medium was replaced with fresh complete medium, and the cells were incubated for 1-5 days. At the end of each experiment, 10 µL of MTT (Solarbio, Beijing, China) per well at a final concentration of 0.5 mg/mL was added to the cell culture, and the cells were incubated for an additional 4 h. The medium was then replaced with 800 µL of dimethyl sulfoxide. After mixing well, the optical absorbance was measured at 490 nm using an enzyme-linked immunosorbent assay microplate reader (Bio-Rad). The relative cell viability was calculated using the following formula: relative cell viability = (mean experimental absorbance / mean control absorbance) × 100%.
Tumor cell plate colony formation assay
After cells were infected with CLDN3 cDNA or shRNA lentivirus for 24 h, they were seeded in 24-well plates at a density of 200 cells/well and grown for 10 days. The cells were then fixed in 4% paraformaldehyde for 15 min and stained with Giemsa stain for 30 min. Next, the cells were washed three times with water, and the number of visible colonies with 50 cells or more was counted. The relative colony formation ability was calculated as follows: (mean experimental colony number / inoculation number) × 100%.
Immunofluorescence staining
After cells were infected with CLDN3 cDNA or shRNA lentivirus for 24 h, they were seeded onto 8-well chamber slides and grown for 24 h. Next, the cells were fixed in 4% paraformaldehyde in PBS for 10 min, permeabilized in 0.5% Triton X-100 in PBS for 10 min, and then blocked in 10% normal serum in PBS for 1 h at room temperature. After that, the cells were incubated at 4 °C overnight with primary antibodies against CLDN3 (1:200, Abcam) and E-cadherin (1:200, Cell Signaling Technology), respectively. On the next day, the cells were washed with PBS three times and further incubated with Alexa 488-conjugated IgG for 1 h at room temperature. The slides were then mounted with Vectashield mounting medium containing 4ʹ,6-diamidino-2-phenylindole (DAPI) to visualize the cell nuclei (Vector Laboratories Inc., Burlingame, CA, USA), and the images were acquired at 200× magnification using a Leica SCN 400 Slide Scanner and analyzed.
Results
Association of reduced CLDN3 expression with altered EMT markers in lung SqCC tissues compared with adjacent normal tissues
In this study, we first confirmed differential CLDN3 expression in lung SqCC vs. the paired adjacent normal tissues in 20 lung SqCC cases using qRT-PCR and western blot. The baseline characteristics of these 20 patients are shown in Table 1. In brief, this study included 16 males and 4 females, 17 of whom had a history of tobacco smoking (85%) and 12 (60%) of whom were older than 60 years of age. The numbers of patients at tumor pathological stage T1, T2, and T3 were 4 (20%), 10 (50%), and 6 (30%), respectively; while 8 (40%) patients had pathological stage I, 6 (30%) patients had stage II, and 6 (30%) patients had stage IIIA cancer.
Our qRT-PCR analysis confirmed reduced CLDN3 expression in lung SqCC tissues compared with the adjacent normal tissues (Fig. 1). The western blotting data were consistent with the qRT-PCR data (Fig. 1). Moreover, our current data showed that CLDN3 expression was the lowest in stage IIIA SqCC tissues, compared with that of stage I and the adjacent normal tissues (Fig. 1).
Furthermore, our qRT-PCR data showed that a low level of CLDN3 expression was associated with a low E-cadherin level in lung SqCC tissues but a high level of Vimentin expression (p < 0.001 and p = 0.001, respectively; Fig. 1). The western blot data were consistent with the qRT-PCR data (Fig. 1).
Figure 1
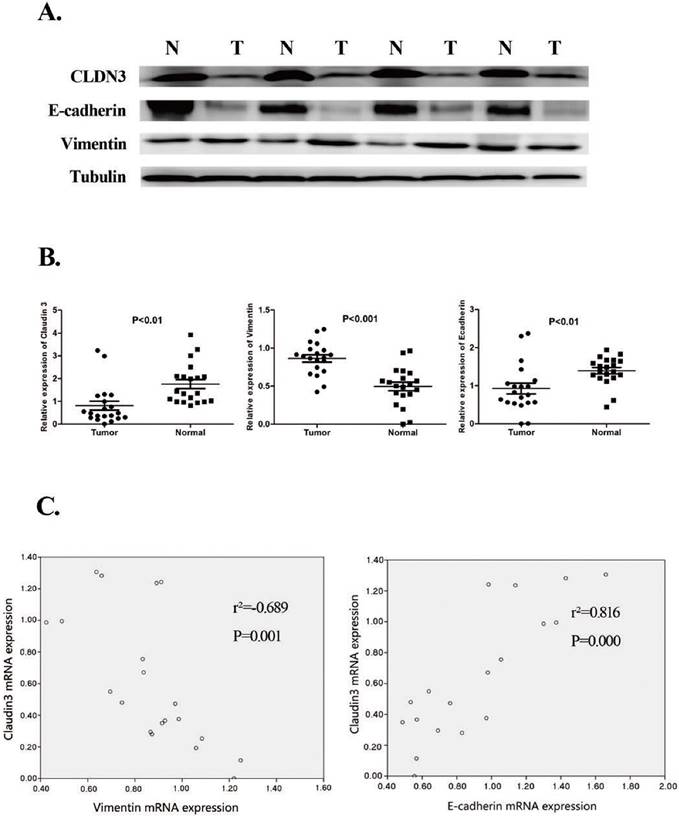
Reduced CLDN3 expression in lung SqCC tissues and association with the expression of EMT-related genes. A. Western blot. Expression of CLDN3, E-cadherin, and Vimentin proteins was detected in lung SqCC and adjacent normal lung tissues from four patients. Total tissue lysates were prepared from matched frozen normal and cancer lung tissues and subjected to western blotting. Tubulin was used as a loading control. (N, adjacent normal tissue; T, tumor tissue). The results showed that CLDN3 expression was downregulated in lung SqCC tissues, compared with normal tissues, and was associated with decreased E-cadherin and increased Vimentin expression. B. qRT-PCR. The mRNA levels of CLDN3, E-cadherin, and Vimentin in 20 paired lung SqCC and adjacent normal lung samples were determined by qRT-PCR; β-actin was used as an internal control. C. Association of CLDN3 with E-cadherin protein expression (p < 0.001) and CLDN3 with Vimentin protein expression (p < 0.01).
Table 1
Patient Information
| Characteristics | N | % |
|---|---|---|
| Age (Years) | ||
| > 60 | 12 | 60 |
| ≤ 60 | 8 | 40 |
| Gender | ||
| Male | 16 | 80 |
| Female | 4 | 20 |
| Smoking history | ||
| Never | 3 | 15 |
| Smoker | 17 | 85 |
| T stage | ||
| T1 | 4 | 20 |
| T2 | 10 | 50 |
| T3 | 6 | 30 |
| N stage | ||
| N0 | 9 | 45 |
| N1 | 5 | 10 |
| N2 | 6 | 45 |
| TNM stage | ||
| I | 8 | 40 |
| II | 6 | 30 |
| IIIa | 6 | 30 |
Effects of CLDN3 expression or knockdown on regulation of lung SqCC cell migration in vitro
To detect the effects of CLDN3 expression on inhibition of lung SqCC cell migration in vitro, we first assessed the levels of CLDN3 protein in different lung SqCC cell lines, namely H520, SK-MES-1, H2170, and H1703, using western blot. We found that SK-MES-1 cells expressed the highest CLDN3 levels, but H520 cells expressed the lowest CLDN3 levels (Fig. 4A). Thus, we knocked down CLDN3 expression in the SK-MES-1 cell line using CLDN3 shRNA, whereas CLDN3 was overexpressed in H520 cells using CLDN3 cDNA. Our western blot data confirmed CLDN3 knockdown and overexpression in these two cell lines, respectively (Fig. 4B). Next, we performed tumor cell wound healing and Transwell assays. Our data showed that CLDN3-overexpressing H520 cells showed significantly reduced tumor cell wound healing and Transwell migration capacity; whereas CLDN3 shRNA-infected SK-MES-1 cells had upregulated tumor cell wound healing and Transwell migration capacity, in a time-dependent manner (Fig. 2). However, CLDN3 knockdown or overexpression did not affect tumor cell proliferation or colony formation in vitro (Fig. 3).
Effects of CLDN3 expression or knockdown on regulation of EMT gene expression in vitro
Both E-cadherin and Vimentin are well-established EMT biomarkers [18]. We found that CLDN3 overexpression in H520 cells induced an increase in E-cadherin expression but a decrease in Vimentin expression (Fig. 4B). In contrast, knockdown of CLDN3 expression in SK-MES-1 cells had an inverse effect on the expression of E-cadherin and Vimentin (Fig. 4 and 5). These findings indicate that CLDN3 expression was able to suppress lung SqCC cell EMT in vitro.
Effects of CLDN3 on inactivation of the Wnt/β-catenin signaling pathway
The Wnt/β-catenin pathway is the classical pathway to induce EMT and suppress E-cadherin expression [26]. Therefore, we assessed several key genes in the Wnt/β-catenin pathway, including β-catenin, c-Myc, and cyclinD1, in CLDN3-overexpressing or -knockdown lung SqCC cell lines, respectively. Our results showed that knockdown of CLDN3 expression upregulated the expression of c-Myc and cyclinD1 but downregulated the expression of β-catenin, indicating that the Wnt/β-catenin pathway was activated. In contrast, downregulation of CLDN3 reduced E-cadherin expression and promoted tumor cell EMT (Fig. 6). However, CLDN3 overexpression showed the opposite effects on the expression of these proteins (Fig. 6). Together with the phenotypic data presented in the previous sections, this study demonstrated that CLDN3 was able to inactivate the Wnt/β-catenin pathway, in turn suppressing lung SqCC cell EMT; whereas CLDN3 knockdown promoted activation of the Wnt/β-catenin pathway proteins and enhanced lung SqCC cell EMT.
Discussion
In this study, we assessed CLDN3 expression in lung SqCC tissues and cells and then determined the effects of CLDN3 overexpression and knockdown on the regulation of lung SqCC cell EMT as well as the underlying molecular mechanisms. Our data showed the following: i). Expression of CLDN3 mRNA and protein was significantly reduced in the lung SqCC tissues compared with the adjacent normal tissues; ii). Expression of CLDN3 protein in the lung SqCC and the adjacent normal tissues was closely associated with the expression of EMT-related proteins, such as E-cadherin and Vimentin, and the same was true for the lung SqCC cell lines; iii). Overexpression of CLDN3 inhibited H520 wound healing and the Transwell migration capacity; whereas knockdown of CLDN3 expression promoted SK-MES-1 cell wound healing and the Transwell migration capacity. In contrast, manipulation of CLDN3 expression did not have any effects on tumor cell proliferation or colony formation; and iv). On the molecular level, CLDN3 was able to suppress the Wnt/β-catenin pathway gene activity, which in turn inhibited tumor cell EMT. Future studies will further investigate CLDN3 as a biomarker for the early diagnosis of lung SqCC and predicting the prognosis of patients.
Figure 2
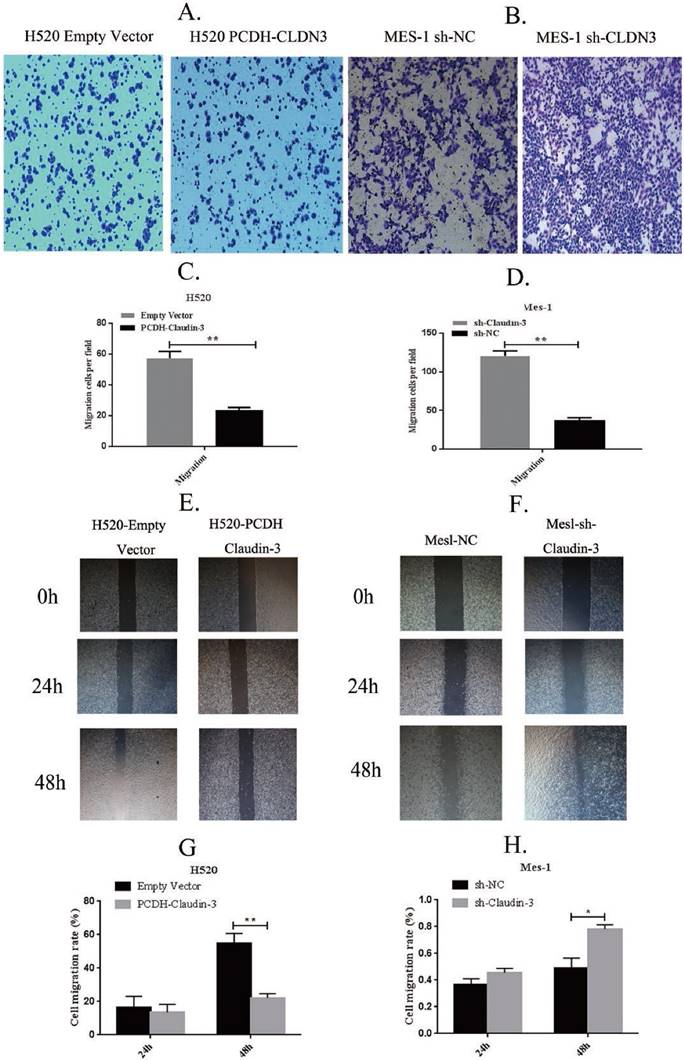
Effects of CLDN3 overexpression or knockdown on the regulation of lung SqCC cell wound healing and Transwell migration. A and B. Transwell migration assay. Ectopic CLDN3 expression significantly inhibited the H520 cell Transwell migration capacity, whereas knockdown of CLDN3 expression significantly enhanced SK-MES1 cell migration. C and D. Quantified data of A and B, respectively (**p < 0.01 using the Student's t-test). E and F. Wound healing assay. Ectopic CLDN3 expression significantly inhibited the H520 cell wound healing capacity, whereas knockdown of CLDN3 expression significantly enhanced SK-MES1 cell wound healing. G and H. Quantified data of E and F, respectively (**p < 0.01 using the Student's t-test).
Figure 3
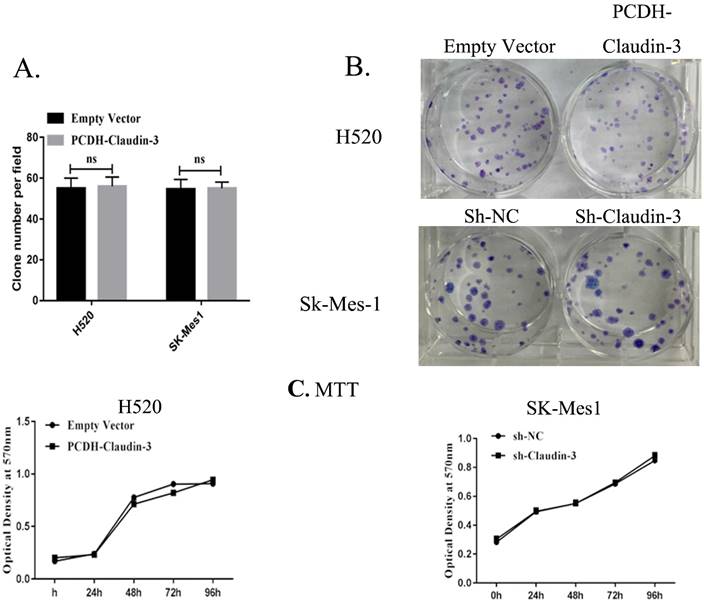
Effects of CLDN3 overexpression or knockdown on the regulation of lung SqCC cell proliferation and colony formation. A and B. Colony formation assay. Representative photographs of anchorage-dependent colonies that were stained with crystal violet. The bar graphs show that the number of cell colonies formed was not significantly different between H520-PCDH and H520-CLDN3 cells, or between SK-MES1-CON and SK-MES1-shRNA cells (p > 0.05 using the Student's t-test). C. Cell viability MTT assay. CLDN3 overexpression and knockdown in H520 and SK-MES1 cells, respectively, did not have any visible effects on cell viability.
Figure 4
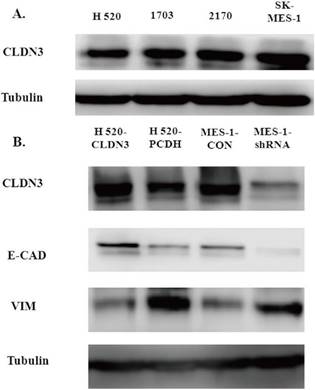
Effects of CLDN3 overexpression or knockdown on the expression of EMT-related biomarkers. A. Western blot. CLDN3 expression was detected in lung SqCC cell lines, i.e., SK-MES1 cells expressed the highest CLDN3 level, H1703 and H2170 cells expressed moderate levels of CLDN3, and H520 cells expressed the lowest level of CLDN3. B. Western blot. CLDN3 overexpression altered the expression of the EMT markers, including E-cadherin and Vimentin in H520 cells, whereas CLDN3 knockdown promoted Vimentin expression but decreased E-cadherin expression.
Figure 5
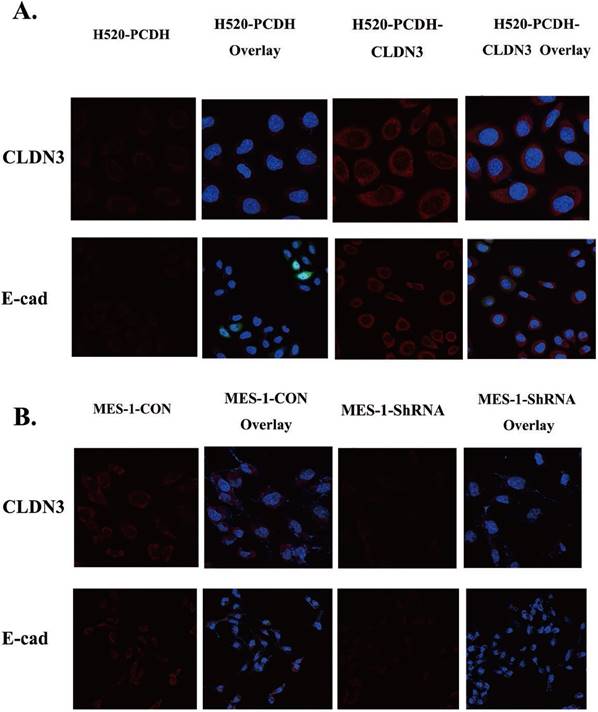
Effects of CLDN3 overexpression or knockdown on the regulation of lung SqCC cell EMT proteins. A. Immunofluorescence staining. Both CLDN3 and E-cadherin proteins appeared in red in H520 cells stably infected with CLDN3 cDNA or control virus. B. Immunofluorescence staining. Both CLDN3 and E-cadherin proteins appeared in red in SK-MES-1 cells stably infected with control shRNA or CLDN3 shRNA. Two representative fields are shown; the transfected cells had dramatic co-upregulation of CLDN3 and membrane E-cadherin, compared to control cells. DAPI (blue) was used to stain the nuclei of the cells shown in panels A and B.
Figure 6
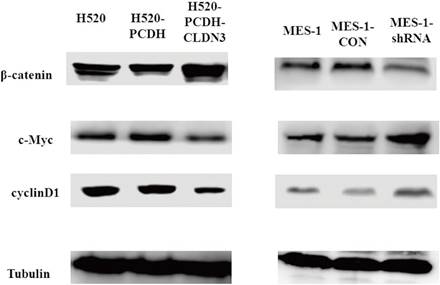
Effects of CLDN3 overexpression or knockdown on the regulation of the Wnt/β-catenin pathway activity. A. Western blot. Lentivirus-carrying CLDN3 cDNA or control-infected H520 cells showed that CLDN3 overexpression significantly inhibited the expression of the Wnt/β-catenin pathway proteins. B. Western blot. Lentivirus-carrying CLDN3 shRNA or negative control shRNA-infected cells significantly promoted the expression of the Wnt/β-catenin pathway proteins.
CLDN3 is specifically and highly expressed in the intestine, liver, kidney, testis, lung, colon, prostate, and uterus [13, 27] and is one of the major transmembrane protein components of the tight junctions in human epithelial and endothelial cells. It provides a continuous intercellular seal between the epithelia and endothelia, forming a biological membrane at physical interfaces and regulating the paracellular transport of water, solutes, and immune cells [28]. Moreover, CLDN3, together with CLDN1 and CLDN5, plays an important role in airway tight junctional permeability [29]. Aberrant CLDN3 expression has been associated with the development and metastasis of human cancers. For example, previous studies have shown that CLDN3 expression is reduced in hepatocellular carcinoma [24] and in esophageal SqCC [20]. However, another study has reported that the CLDN expression pattern is significantly different not only between lung SqCC and lung AC but also between lung SqCC and lung AC with lepidic variants and that CLDN3 expression is higher in lung AC but not in lung SqCC [30]. Our previous study also showed that CLDN3 expression is significantly reduced in stage IIIA lung SqCC compared with stage I lung SqCC (42.9% vs. 76.5%) and that CLDN3 expression is an independent predictor for the overall survival of lung SqCC patients [25]. Indeed, our current data further support these previous studies and indicate that CLDN3 expression is organ-specific and that loss of CLDN3 expression is associated with lung SqCC development and progression.
We found that the expression of CLND3 in lung SqCC tissues was downregulated, whereas a higher expression of CLDN3 in SqCC was associated with a favorable prognosis. In contrast, Zhang et al. have reported that CLND3 expression in AC tissues is upregulated and that CLDN3 overexpression is associated with a poor prognosis [23]. Therefore, we speculated that differential CLDN3 expression in different histological types of lung cancer is related to the origin and development process of tumor cells. Lung SqCC arises from the segmental and subsegmental bronchial mucosal epithelium developed through squamous metaplasia, dysplasia, and preinvasive carcinoma stages; whereas lung AC arises from the bronchial mucosal epithelium and glandular epithelium, and the majority of them originate from smaller bronchial epithelial cells. Moreover, previous studies have shown that lung AC mainly originates from type II alveolar epithelial cells, whereas lung AC and SqCC are derived from other epithelial cell components [31]. CLDN3 is primarily expressed by type II alveolar epithelial cells [32].
Functionally, loss of CLDN3 expression downregulated the mRNA and protein levels of E-cadherin but induced the inhibitory phosphorylation of glycogen synthase kinase-3β and activation of the β-catenin signaling pathway [33], all of which are associated with tumor cell EMT. EMT is an important biological process during embryonic development and cancer metastasis, i.e., epithelium-derived carcinoma cells acquire the ability to migrate and invade into the surrounding tissues or organs. In other words, through EMT, tumor cells gain the ability to break through the basement membrane to invade into the surrounding tissues or to metastasize to distant organs [11]. Previous studies have demonstrated that tumor cell EMT is associated with invasion, metastasis, and prognosis of various cancers [34-36]. In lung cancer, EMT is the key point for cancer progression and metastasis [37-39]. For example, Zhou et al. have shown that aberrant expression of E-cadherin and Vimentin was associated with the lymph node metastasis of 312 stage I-IIIA NSCLC patients [37]. Our own study also revealed that loss of E-cadherin expression was associated with the lymph node metastasis of lung SqCC and that loss of E-cadherin expression but increased β-catenin expression was associated with lung SqCC recurrence. Other previous studies of lung SqCC cell lines have shown that the migration ability of H2170 and H1703 cells was significantly upregulated after downregulation of E-cadherin expression [38, 39]. In our current study, we found that reduced CLDN3 expression was associated with loss of E-cadherin expression in lung SqCC specimens but inversely associated with Vimentin expression, compared with the paired adjacent normal tissues. However, the migration capacity of H520 cells was restrained after upregulation of E-cadherin expression through CLDN3 overexpression, whereas the migration ability of SK-MES-1 cells was induced after downregulation of E-cadherin expression using CLDN3 shRNA transduction. Our current data were consistent with previous studies [38, 39].
In our current study, we further explored the mechanism of CLDN3 in regulating lung SqCC cell EMT. We detected the expression of Wnt pathway-related proteins, including β-catenin, c-Myc, and cyclinD1. Our data showed that CLDN3 expression could prevent lung SqCC H520 cells from migration and invasion as well as EMT progression by inhibition of Wnt pathway activation, whereas CLDN3 knockdown had the opposite effects on lung SqCC SK-MES-1 cells. Our current findings are consistent with and support the data of Jiang et al. on hepatocellular carcinoma [24]. In addition to the Wnt pathway, other mechanisms by which CLDN3 regulates EMT might be involved. For example, CLDN3 could heterotypically interact with other CLDNs, including CLDN1, CLDN2, and CLDN5 [40-42]. CLDN3 protein contains a C-terminal PDZ-binding motif that is reported to be required for interaction with zonula occludens (ZO)-1 and ZO-2, which belong to the membrane-associated guanylate kinase family. ZO-1 and ZO-2 tether CLDN3 into the tight junction strands by providing a bridge to cortical actin [43-46] (Fig. 7). Their interaction plays an essential role in maintaining the integrity of the tight junctions and further influences the EMT. Thus, our current study is just a proof-of-principle study, and further studies are needed to clarify how CLDN3 suppresses lung SqCC cell EMT, but not in other cancers. Overall, future studies of CLDN3 should focus on the interaction of CLDN3 with other CLDNs or other tight junction proteins as well as the organ specificity of these CLDNs. Lastly, we will further investigate the loss of CLDN3 expression as a biomarker for predicting the prognosis of lung SqCC.
Figure 7
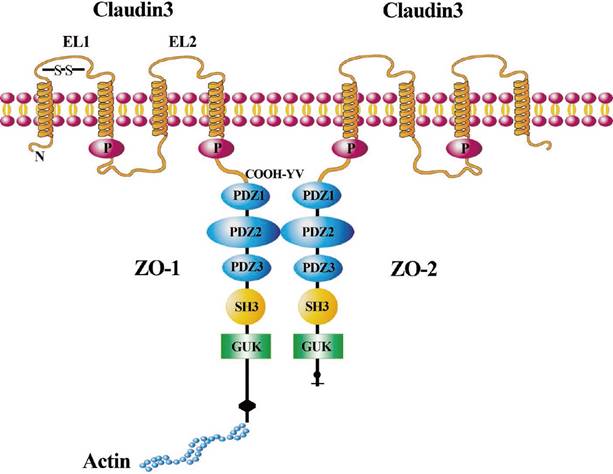
Illustration of CLDN3 signaling in cells. CLDN3 is a tetraspanin transmembrane protein with both the N- and C-terminal domains oriented toward the cytoplasm. The two extracellular loop (EL) domains are shown across the cell membrane. CLDN3 combines with the PDZ-binding motif of ZO-1/ZO-2 through the “COOH-YV” sequence. CLDN3 is linked via the scaffolding ZO-1 to proteins of the cytoskeleton such as actin. ZO-1 could connect with ZO-2 through other PDZ-binding motifs.
Abbreviations
AC: adenocarcinoma; CLDN: claudin; EMT: epithelial-mesenchymal transition; NSCLC: non-small cell carcinoma; RT-PCR: quantitative real-time polymerase chain reaction; shRNA: short hairpin RNA; SqCC: squamous cell carcinoma; ZO: zonula occludens.
Acknowledgements
We would like to thank Mr. Lianming Zhang for his assistance with the immunofluorescence staining experiments. We also would like to thank Mr. Yi Yang for his assistance with the confocal fluorescence microscopy experiments.
Funding
This study was supported by grants from the Research Foundation of Beijing Friendship Hospital, Capital Medical University (#yyqdkt 2014-11 to Juanjuan Che) and the National Natural Science Foundation of China (#81501983 to Dongsheng Yue).
Authors' contributions
Bangwei Cao designed and supervised the study and experiments, analyzed the data, and co-wrote the manuscript. Juanjuan Che developed the methodologies, performed the experiments, analyzed the data, and co-wrote the manuscript. Dongsheng Yue analyzed the data and co-wrote the manuscript. Bin Zhang developed the methodologies. Bin Zhang provided assistance with the cell culture and virus transfection experiments. Yansong Huo, Hua Zhang, Liuwei Gao, and Hongchao Zhen performed the experiments and analyzed the data. Yan Yang performed the histopathological analysis. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Tissues were collected from the Beijing Friendship Hospital and Tianjin Medical University Cancer Institute and Hospital. The study protocol was approved by the Research Ethics Committees of Beijing Friendship Hospital and the Cancer Institute and Hospital of Tianjin Medical University. Written informed consent was obtained from all participants.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Isla D, Majem M, Viñolas N, Artal A, Blasco A, Felip E. et al. A consensus statement on the gender perspective in lung cancer. Clin Transl Oncol. 2017;19:527-35
2. Ambrogio C, Nadal E, Villanueva A, Gómez-López G, Cash TP, Barbacid M. et al. KRAS-driven lung adenocarcinoma: combined DDR1/Notch inhibition as an effective therapy. ESMO Open. 2016;1:e000076
3. Auerbach O, Hammond EC, Garfinkel L. Changes in bronchial epithelium in relation to cigarette smoking, 1955-1960 vs. 1970-1977. N Engl J Med. 1979;300:381-5
4. Halpern MT, Gillespie BW, Warner KE. Patterns of absolute risk of lung cancer mortality in former smokers. J Natl Cancer Inst. 1993;85:457-64
5. Lam S, Mac Aulay C, Hung J, LeRiche J, Profio AE, Palcic B. Detection of dysplasia and carcinoma in situ with a lung imaging fluorescence endoscopedevice. J Thorac Cardiovasc Surg. 1993;105:1035-40
6. Cooper WA, Lam DC, ƠToole SA, Minna JD. Molecular biology of lung cancer. J Thoracic Dis. 2013;5:S479-90
7. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367-80
8. Malek R, Wang H, Taparra K, Tran PT. Therapeutic Targeting of Epithelial Plasticity Programs: Focus on the Epithelial-Mesenchymal Transition. Cells Tissues Organs. 2017;203:114-27
9. Yuan P, Kadara H, Behrens C, Tang X, Woods D, Solis LM. et al. Sex determining region Y-Box 2 (SOX2) is a potential cell-lineage gene highly expressed in the pathogenesis of squamous cell carcinomas of the lung. PLoS One. 2010;5:e9112
10. Hong WK, Bast RC, Hait WN. et al. Holland-Frei Cancer Medicine (8th ed). New York, USA: McGraw-Hill Ryerson. 2010
11. Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol. 2017;11:28-39
12. Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol. 1995;269:G467-75
13. Heiskala M, Peterson PA, Yang Y. The roles of claudin superfamily proteins in paracellular transport. Traffic. 2001;2:93-8
14. Martìn-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P. et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117-27
15. Balda MS, Flores-Maldonado C, Cereijido M, Matter K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J Cell Biochem. 2000;78:85-96
16. Saitou M, Fujimoto K, Doi Y, Itoh M, Fujimoto T, Furuse M. et al. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol. 1998;141:397-408
17. Koval M. Claudin heterogeneity and control of lung tight junctions. Annu Rev Physiol. 2013;75:551-67
18. Romani C, Cocco E, Bignotti E, Moratto D, Bugatti A, Todeschini P. et al. Evaluation of a novel human IgG1 anti-claudin3 antibody that specifically recognizes its aberrantly localized antigen in ovarian cancer cells and that is suitable for selective drug delivery. Oncotarget. 2015;6:34617-28
19. Heerma van Voss MR, van Diest PJ, Smolders YH, Bart J, van der Wall E, van der Groep P. Distinct claudin expression characterizes BRCA1-related breast cancer. Histopathology. 2014;65:814-27
20. Montgomery E, Mamelak AJ, Gibson M, Maitra A, Sheikh S, Amr SS. et al. Overexpression of claudin proteins in esophageal adenocarcinoma and its precursor lesions. Appl Immunohistochem Mol Morphol. 2006;14:24-30
21. Walther W, Petkov S, Kuvardina ON, Aumann J, Kobelt D, Fichtner I. et al. Novel Clostridium perfringens enterotoxin suicide gene therapy for selective treatment of claudin-3- and -4-overexpressing tumors. Gene Ther. 2012;19:494-503
22. Worst TS, von Hardenberg J, Gross JC, Erben P, Schnölzer M, Hausser I. et al. Database- augmented Mass Spectrometry Analysis of Exosomes Identifies Claudin 3 as a Putative Prostate Cancer Biomarker. Mol Cell Proteomics. 2017;16:998-1008
23. Zhang L, Wang Y, Zhang B, Zhang H, Zhou M, Wei M. et al. Claudin-3 expression increases the malignant potential of lung adenocarcinoma cells: role of epidermal growth factor receptor activation. Oncotarget. 2017;8:23033-47
24. Jiang L, Yang YD, Fu L, Xu W, Liu D, Liang Q. et al. CLDN3 inhibits cancer aggressiveness via Wnt-EMT signaling and is a potential prognostic biomarker for hepatocellular carcinoma. Oncotarget. 2014;5:7663-76
25. Che J, Yang Y, Xiao J, Zhao P, Yan B, Dong S. et al. Decreased expression of claudin-3 is associated with a poor prognosis and EMT in completely resected squamous cell lung carcinoma. Tumour Biol. 2015;36:6559-68
26. WangT Wang M, Fang S Wang Q, Fang R Chen J. Fibulin-4 is associated with prognosis of endometrial cancer patients and inhibits cancer cellinvasion and metastasis via Wnt/β-catenin signaling pathway. Oncotarget. 2017;8:18991-19012
27. Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A. 1999;96:511-6
28. Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol. 1995;269:G467-75
29. Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG. Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1166-78
30. Moldvay J, Fábián K, Jäckel M, Németh Z, Bogos K, Furák J. et al. Claudin-1 Protein Expression Is a Good Prognositic Factor in Non-Small Cell Lung Cancer, but only in Squamous Cell Carcinoma Cases. Pathol Oncol Res. 2017;23:151-56
31. Oghiso Y, Yamada Y. Immunohistochemical study on cellular origins of rat lung tumors induced by inhalation exposures to plutonium dioxide aerosols as compared to those by X-ray irradiation. J Radiat Res. 2002;43:301-11
32. Mitchell LA, Overggard CE, Ward C, Margulies SS, Koyal M. Differential effects of claudin-3 and claudin-4 on alveolar epithelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2011;301:L40-9
33. Shang X, Lin X, Alvarez E, Manorek G, Howell SB. Tight junction protein claudin-3 and claudin-4 control tumor growth and metastases. Neoplasia. 2012;14:974-85
34. Liu H, Ma Y, He HW, Zhao WL, Shao RG. SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal transition by promoting the autophagy-linked lysosomal degradation of CDH1/E-cadherin in hepatoma cells. Autophagy. 2017;13:900-13
35. Yang X, Han M, Han H, Wang B, Li S, Zhang Z. et al. Silencing Snail suppresses tumor cell proliferation and invasion by reversing epithelial-to-mesenchymal transition and arresting G2/M phase in non-small cell lung cancer. Int J Oncol. 2017;50:1251-60
36. Liu F, Gu LN, Shan BE, Geng CZ, Sang MX. Biomarkers for EMT and MET in breast cancer: An update. Oncol Lett. 2016;12:4869-76
37. Zhou L, Yu L, Wu S, Feng Z, Song W, Gong X. Clinicopathological significance of KAI1 expression and epithelial-mesenchymal transition in non-small cell lung cancer. World J Surg Oncol. 2015;13:234
38. Yue D, Li H, Che J, Zhang Y, Tolani B, Mo M. et al. EMX2 is a predictive marker for adjuvant chemotherapy in lung squamous cell carcinomas. PLoS One. 2015;10:e0132134
39. Yue D, Li H, Che J, Zhang Y, Tseng HH, Jin JQ. et al. Hedgehog/Gli promotes epithelial-mesenchymal transition in lung squamous cell carcinomas. J Exp Clin Cancer Res. 2014;33:34
40. Daugherty BL, Ward C, Smith T, Ritzenthaler JD, Koval M. Regulation of heterotypic claudin compatibility. J Biol Chem. 2007;282:30005-13
41. Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999;147:891-903
42. Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG. Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1166-78
43. Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68:403-29
44. Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell. 2005;16:3919-36
45. Bruewer M, Hopkins AM, Hobert ME, Nusrat A, Madara JL. RhoA, Rac1, and Cdc42 exert distinct effects on epithelial barrier via selective structural and biochemical modulation of junctional proteins and F-actin. Am J Physiol Cell Physiol. 2004;287:C327-35
46. Van Itallie CM, Fanning AS, Bridges A, Anderson JM. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell. 2009;20:3930-40
Author contact
![]() Corresponding author: Bangwei Cao, PhD, Department of Oncology, Beijing Friendship Hospital, Capital Medical University, 95 Yong-an Road, Xi-cheng District, Beijing, 100050, P.R. China; Tel: 0086-10-63139325; Fax: 0086-10-63139325; E-mail: oncologyedu.cn
Corresponding author: Bangwei Cao, PhD, Department of Oncology, Beijing Friendship Hospital, Capital Medical University, 95 Yong-an Road, Xi-cheng District, Beijing, 100050, P.R. China; Tel: 0086-10-63139325; Fax: 0086-10-63139325; E-mail: oncologyedu.cn
Citation styles
APA
Che, J., Yue, D., Zhang, B., Zhang, H., Huo, Y., Gao, L., Zhen, H., Yang, Y., Cao, B. (2018). Claudin-3 Inhibits Lung Squamous Cell Carcinoma Cell Epithelial-mesenchymal Transition and Invasion via Suppression of the Wnt/β-catenin Signaling Pathway. International Journal of Medical Sciences, 15(4), 339-351. https://doi.org/10.7150/ijms.22927.
ACS
Che, J.; Yue, D.; Zhang, B.; Zhang, H.; Huo, Y.; Gao, L.; Zhen, H.; Yang, Y.; Cao, B. Claudin-3 Inhibits Lung Squamous Cell Carcinoma Cell Epithelial-mesenchymal Transition and Invasion via Suppression of the Wnt/β-catenin Signaling Pathway. Int. J. Med. Sci. 2018, 15 (4), 339-351. DOI: 10.7150/ijms.22927.
NLM
Che J, Yue D, Zhang B, Zhang H, Huo Y, Gao L, Zhen H, Yang Y, Cao B. Claudin-3 Inhibits Lung Squamous Cell Carcinoma Cell Epithelial-mesenchymal Transition and Invasion via Suppression of the Wnt/β-catenin Signaling Pathway. Int J Med Sci 2018; 15(4):339-351. doi:10.7150/ijms.22927. https://www.medsci.org/v15p0339.htm
CSE
Che J, Yue D, Zhang B, Zhang H, Huo Y, Gao L, Zhen H, Yang Y, Cao B. 2018. Claudin-3 Inhibits Lung Squamous Cell Carcinoma Cell Epithelial-mesenchymal Transition and Invasion via Suppression of the Wnt/β-catenin Signaling Pathway. Int J Med Sci. 15(4):339-351.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.