Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(4):309-322. doi:10.7150/ijms.23215 This issue Cite
Review
While it is not deliberate, much of today's biomedical research contains logical and technical flaws, showing a need for corrective action
Yan He1,2 ![]() , Chengfu Yuan3
, Chengfu Yuan3 ![]() , Lichan Chen4, Yanjie Liu5, Haiyan Zhou6, Ningzhi Xu7
, Lichan Chen4, Yanjie Liu5, Haiyan Zhou6, Ningzhi Xu7 ![]() , Dezhong Joshua Liao1,2,5
, Dezhong Joshua Liao1,2,5 ![]()
1. Key Lab of Endemic and Ethnic Diseases of the Ministry of Education of China in Guizhou Medical University, Guiyang, Guizhou 550004, P. R. China
2. Molecular Biology Center, Guizhou Medical University, Guiyang, Guizhou 550004, P.R. China
3. Department of Biochemistry, China Three Gorges University, Yichang City, Hubei 443002, P.R. China
4. Hormel Institute, University of Minnesota, Austin, MN 55912, USA
5. Department of Pathology, Guizhou Medical University, Guiyang, Guizhou 550004, P.R. China
6. Clinical Research Center, Guizhou Medical University Hospital, Guiyang, Guizhou 550004, P.R. China
7. Laboratory of Cell and Molecular Biology & State Key Laboratory of Molecular Oncology, National Cancer Center/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100021, PR China
Received 2017-10-8; Accepted 2017-12-21; Published 2018-1-19
Abstract
Biomedical research has advanced swiftly in recent decades, largely due to progress in biotechnology. However, this rapid spread of new, and not always-fully understood, technology has also created a lot of false or irreproducible data and artifacts, which sometimes have led to erroneous conclusions. When describing various scientific issues, scientists have developed a habit of saying “on one hand… but on the other hand…”, because discrepant data and conclusions have become omnipresent. One reason for this problematic situation is that we are not always thoughtful enough in study design, and sometimes lack enough philosophical contemplation. Another major reason is that we are too rushed in introducing new technology into our research without assimilating technical details. In this essay, we provide examples in different research realms to justify our points. To help readers test their own weaknesses, we raise questions on technical details of RNA reverse transcription, polymerase chain reactions, western blotting and immunohistochemical staining, as these methods are basic and are the base for other modern biotechnologies. Hopefully, after contemplation and reflection on these questions, readers will agree that we indeed know too little about these basic techniques, especially about the artifacts they may create, and thus many conclusions drawn from the studies using those ever-more-sophisticated techniques may be even more problematic.
Keywords: Biotechnology, reproducibility, Artifacts, Reverse transcription, Polymerase chain reactions, microRNA, siRNA
Introduction
It has been reported that most published biomedical research findings are false [1], and 75-90% of the published studies are irreproducible [1, 2]. For example, a group of researchers at the Amgen Corporation recently reported in Nature that only 11% of published academic research was reproducible [2]. Although the exact estimations on the percentage of the false or irreproducible data vary among different studies [3-5], with some figures as low as only 50% (which to us is still way too high) [6], all relevant studies suggest that the situation is severe [7-9] and fatally threatens scientific integrity [2, 10, 11]. Moreover, it makes 85% of the research funding wasted, according to some publications, as highlighted in the 2014 Lancet series entitled “Research: increasing value, reducing waste” [1, 12-15]. This situation is ironic, as medical research aims to prevent, diagnose or cure diseases but, instead, ends up being a “patient” itself that is in a dire need of diagnosis and cure [1, 16, 17]. The causes for the tremendous inaccuracy and irreproducibility are multifaceted, and some of them have been addressed extensively in the literature [1, 4, 17-21], such as the strains from the career, tenure and research funding triangle [17, 22, 23]. In fact, these strains have created not only black markets for paper production but also “Scientific Citation Index worship”, such as in China [18, 19]. Although many organizations, including the US National Institute of Health (NIH), have established new policies [24] and initiatives [21, 25-27] and some agencies have set up incentive strategies [28] to improve the situation, yet the problem is still unrelenting. We have for years been contemplating these adverse facets of biomedical research and have attempted to diagnose the causes from such novel slants that are somewhat less frequently addressed in the literature. In our opinion, the swift progress and proliferation of biotechnology in the past three decades have greatly advanced biomedical research. However, the wide and rapid dispersion of biotechnology over the whole field of biomedical research has its dark side [22], as it leads to bounteous artifacts which in turn often lead to biased or even wrongful conclusions, making mistakes omnipresent in biomedical research. This is largely because we have not given enough thought on our study design and because we know too little about the technical details of the modern biotechnology we used. In this essay, we discourse on our musings.
In many lines of research, study designs need a deeper philosophical meditation
Many lines of biomedical experiments are designed using a “standard operating procedure (SOP)” that seems logical and thoughtful but, after careful examination, one will find the SOP rife with flaws, such as lacking relevance to cells or to humans. These flaws remind us that we need to give our study design more considerations from a philosophical viewpoint, or sometimes just from “first principles” [29], so that our studies are more relevant to the cell, the human, or the clinic and hence become more meaningful. A few of these lines of problematic study design are given below as examples to justify our claim:
Ectopic expression of a complementary DNA (cDNA), by delivering it into cells in culture with a transfection approach or into cells in animals with a transgenic technique, has become a SOP in biomedical research to scout functions of genes. For a given mRNA variant of a given gene, this SOP has indeed brought us some detail about its function. However, as we have already explicated previously [30], it also keeps us from knowing the true function of the gene inside the cell. This is because a cell will decide which one(s) of its multiple RNA transcripts, which one(s) of its mature mRNA variants, noncoding RNAs or small regulatory RNAs, as well as what ratios among these mature RNAs, it should produce in a particular physiological or pathological situation [30-34]. Forcing a cell to express the particular cDNA (i.e. a particular mRNA) of our interest is virtually depriving the cell of its right to make its own decision, which can only give us disinformation about the gene. Actually, it may provide us disinformation about the particular mRNA per se as well, since function of a particular mRNA variant is usually elicited via its particular ratios to other variants [30]. This is a philosophical issue; we forget that we are compelling the cell to express a certain amount of a particular mRNA variant we want, but not what the cell wants, whereas our aim is actually to learn what and how a cell does. The function of an mRNA variant we learn from an ectopic expression situation may never happen in reality when the cell is free from our control. Moreover, it is also a simple philosophical conclusion that we cannot construct the function of the gene simply by adding together the function of individually expressed cDNA variants, because the function of the gene relies on a collective expression of different mRNAs, noncoding RNAs and short regulatory RNAs at the particular ratios carefully tailored by the cell for the particular physiological or pathological situation [30]. We need to be wary of utilizing cDNA and, instead, should more often use genomic DNA (gDNA), which may partly, but certainly not fully, compensate for the abovementioned constraints with individual cDNAs. After receiving a gDNA construct, a cell will decide for itself how to transcribe the gDNA, how to splice the transcript(s), and how to make small regulatory RNAs from the intron sequences after splicing, etc., in the particular situation. At least for many relatively small genes, delivery of a gDNA into cells is already technically feasible.
Studies on determining the efficacy and specificity of chemotherapeutic drugs in Petri dishes routinely pair cancer cell lines with normal (actually immortalized) cell lines that are derived from the same tissue as the cancer cells. If the to-be-tested drug hits the cancer cells hard with little damage to the normal cells, the drug is interpreted to have a good cancer-specificity, as it spares the normal cells. This design has become a SOP because those studies that skip such normal cells usually get rejections from journals, since most of us are not as lucky as Einstein, Watson and Crick who could eschew the peer-reviewed procedure [35-38]. At first glance this SOP is logical, but pondering it over more deeply, oncologists will find that it has little clinical relevance, because in most cases the normal cells worried about by them are not those derived from the same tissue as the cancer. For instance, when treating breast or prostate cancers, oncologists care little about whether normal breast or prostate epithelia are also hit or not. What they worry about the most is whether the drug also hits bone marrow cells, thus decreasing the white blood cell count, whether mural cells in the gastric-intestinal system are also hit, thus causing vomiting, nausea and diarrhea, whether hair follicle cells are also hit, leading to alopecia, whether epidermal basal cells are also hit, thus thinning the skin and in turn causing pruritus, etc. [39, 40]. In a nutshell, it is those highly proliferating normal cells in the body that are of concern and thus should be included for comparison, but not the normal cells of the same tissue origin as the cancer [39, 40]. In addition, what has hardly been done and is better to do is to include hepatocytes and renal epithelial cells in the normal cell panel, since the liver and kidneys as major metabolic organs are also common targets for xenobiotics like chemo drugs. In our opinion, partly because cells in many tissues or organs are much less sensitive to chemo drugs than the abovementioned highly-proliferating ones that are not tested in the in vitro studies, many drug candidates that seem to be promising in Petri dishes have later failed in animal studies or clinical trials.
In research of mechanisms for carcinogenesis, our aim is simply to know how humans get cancer. However, most genetically modified animal models of carcinogenesis created by researchers are in fact new animal strains that never exist in the Mother Nature. These animals tell us “by doing so (e.g. mutating gene X or deleting gene Y) one can get cancer,” but never claim that “one gets cancer because of doing so.” This is actually a philosophical game with “putting the cart before the horse” as its essence, although it seems to just slightly deflect both the question and the answer. Playing this game advertently or inadvertently, many cancer researchers have manipulated a slew of genes and have created a sheer number of new, otherwise non-existing animal strains. In these manmade strains, the manipulated genes as the tumor-inducers coerce the target cells to manifest malignant histology, as we explained in more detail elsewhere [39, 40], thus providing us with numerous “oncogenic pathways” that can lead normal cells to malignancy. As an analogy, we can create many pathways, as many as we wish, leading from New York City to Washington DC, and we are safe in saying that Mr. Trump can take any of these pathways to DC, as long as we do not claim which particular one or ones were actually taken by him. By playing this philosophical game, many peers have secured a good career and become prominent, leaving oncologists to wonder whether any cell of any patient really took any one of the numerous manmade “oncogenic pathways”. The real situation is actually much worse, as many of the histologically malignant tumors induced in these genetically modified animals are not verifiably malignant, and not even authentically benign, and have little human relevance. This is because these tumors are the-inducer-dependent, mortal, non-autonomous, incapable of metastasizing, and curable simply by removal of the inducer or by a surgical removal [41, 42]. Unfortunately, few publications germane to this area discourse about these unfavorable but iconic features of “cancers” induced in many animal models. Instead, most tout their usefulness and human relevance.
The abovementioned animal models of carcinogenesis also require a deeper rumination from another philosophical slant: Many genetically manipulated animals engender overt histologically-malignant tumors in the target organ at 100% incidence, i.e. all animals develop tumor(s), although to us their malignancy is untenable, as expounded above and before [41, 42]. However, in many of these animal models, such as in several c-myc transgenic lines [43-46], there are only one to several tumors developed in each animal in the whole lifespan, whereas billions or even trillions of other cells in the same organ do not develop to malignancy, although all these cells received the same genetic modification as those cells that evolve to the tumors. We can have two opposite conclusions on this phenomenon: 1) The genetic manipulation is highly oncogenic because all animals develop cancer. 2) The genetic manipulation is basically not oncogenic because only one to several, out of billions or trillions, of the cells in the same organ of the same animal develop to cancer. We have been bedeviled by this dilemma for years but still have not yet figured out which of the two conclusions is correct, although all producers of those animal strains opt for the first one.
Modern technologies have complex technical details and many pitfalls
Ever since the 1980s, when RNA reverse transcription (RT) and polymerase chain reactions (PCR) quickly became readily used techniques in biomedical labs, biotechnology has been updated daily in a tight relation to these two methods, one way or another. RT-PCR and modern DNA sequencing, along with the relevant equipment and reagents, are among those techniques receiving the most plaudits, as they greatly accelerate biomedical research advancement. The following lines of technique, each of which possesses a string of new developments, are some of those that have emerged in the past three decades: 1) genetic modifications of animals, plants or microorganisms, which were made first from transgenic or gene-knockout technique, and then from a combination of both, and then from targeting/controllable transgenic/knockout technique; 2) gene expression profiling, from cDNA microarray to exon array and then to the whole genome scan; 3) gene expression knockdown using various regulatory RNAs, first with antisense and then with small interfering RNA (siRNA) or short hairpin RNA (shRNA), which was initially for individual mRNAs but later for the whole RNA repertoire using a whole shRNA library; 4) other manipulations of gene expression, such as using microRNA (miRNA) or small activating RNA (saRNA); 5) DNA/RNA sequencing, from the first to the second and then the third generation sequencing; and 6) proteomics, from the initial bottom-up LC-MS/MS (liquid chromatography and tandem mass spectrometry) to the recent top-down LC-MS/MS with more-sophisticated equipment. The list can be further elongated, and each of the listed techniques is associated with creation of a new research province and a whole scientific lexicon (like transcriptome, proteomics, chimeric RNA, circular RNA, etc.).
RT-PCR and CRISPR/CAS9-mediated gene editing emerged roughly before and after, respectively, the abovementioned technique series. DNA and RNA can be amplified, even exponentially if PCR is involved, and thus can be conveniently studied. However, most methods for studying DNA or RNA require a short sequence as a primer or a guider for targeting the object gene, which creates a huge problem since all DNA/RNA sequences are made with only four bases, i.e. A, T(U), C and G, and a short sequence will certainly have many homologies and highly-similar regions in the genome, which may be mistakenly targeted. A gene can be specific only when its sequence is long, at least kilo base-pairs in most cases, and there is no way of being specific if the sequence is short, because all genomes are sizable enough to have many identical or highly-similar short sequences. Bearing this in mind, when we use RT-PCR or CRISPR/CAS9 that requires short sequences as primers or as guiders, or use siRNAs, shRNAs or miRNAs that are short sequences themselves, we should realize that off-targets will inevitably be an issue. Therefore, we should concern more about “how can we avoid off-targeting” before we can be satisfied with “we have reached our target”. All techniques with one step using a short sequence have the off-target issue, besides many other weaknesses, constraints, pitfalls and flaws. Actually, many experts have realized and attempted to solve this issue using different strategies [47-66], including computational identifications of on- and off-target sequences [53, 56, 63, 65, 66], modification of relevant enzymes [48, 50-52], identification of optimal annealing temperature [62], enhancement of the tool RNA design [49, 55, 60], etc. These strategies can improve the on-target specificity and decrease the off-target problem, but, in our humble opinion, cannot fully solve it, especially in a high-throughput scale. As long as a short sequence made of the A, T(U), C and G is involved as a guider, a primer or a regulatory RNA, mis-annealing will likely occur, and thus a complete resolution of the off-target issue may require novel, i.e. currently-unavailable, strategies.
Many biotech companies commercialize different kits that are foolproof and convenient for researchers to use without knowing what the kits contain and what their principles are. While these kits have indeed facilitated our bench work, they also make us ignore technical details and in turn the technically derived artifacts, leading to biased or even wrongful interpretation of data and ensuing biased or slanted conclusions.
Technical flaws and spuriousness are often downplayed or forgotten, advertently or inadvertently
Many scientists have successfully established their career at a young age by introducing novel techniques into their research areas and publishing in high-impact journals, while leaving the research fields with numerous artifacts and biased or erroneous conclusions. For example, there are ample spurious sequences deposited in various chimeric RNA databases on the internet, as we have pointed out previously [67, 68]. Although many of these artifacts, biases or errors have later been discovered and even corrected by others, those who made them and benefited from them with grants, publications and promotions have hardly been chastised, because the mistakes are made due to the innate flaws of the new techniques that “were formerly unaware of”. Actually, reviewing the publications involving the aforementioned new techniques in the past decades, smart young scientists have already found, probably unintentionally, a short cut to establish their career and renown, which is to introduce a new, sophisticated technique into their research areas without bothering to learn the associated flaws, simply because many readers, including grant and manuscript reviewers, likely lack the experience and knowledge of the technical details as well. There always is a latency between the time when a new and sophisticated technique is widely dispersed and the time when many technically derived problems are widely recognized. This latency period is used, intentionally or unintentionally, by many scientists for career development, although this “trick” has hardly been spelled out in the literature. Indeed, if we review those early publications in high-impact journals that involve some type of sophisticated technology, such as cDNA microarray that establishes the expression profiling realm, or the recent deep RNA sequencing that establishes the chimeric RNA, circular RNA, and other RNA-related bailiwicks, we will find that many peers get famous in these research provinces without being affected by the innate problems of the technique that are later well realized. Actually, in our opinion, a teeming number of spurious sequences are still being produced right now from the pipeline of “deep RNA sequencing” by many researchers who are using this technique and publishing data without knowing its detail and without commenting on the spuriousness. Readers, especially those as senior authors of many publications and the toasts of their research spheres, are encouraged to ask themselves, valiantly, how much technical detail they really know when they perform and publish those studies involving such as transcriptome or an “-omics” approach. Several technical-detail-related problems are listed below as examples to justify our points described above.
There have been ample publications on iPS (induced pluripotent stem) cells that show us their bewitching potential in regenerative medicine, such as for tissue/organ repair or transplantation. However, few of these papers put in enough words the unfavorable facets of these cells, such as their high chance to evolve into malignancy [69-72], although this is very reasonable to all pathologists, because it is basic pathological knowledge that cancer cells resemble embryonic cells in cellular morphology. In fact, for this reason, pathology textbooks use a set of embryological phraseologies to describe neoplasms, such as “well differentiated”, “poorly differentiated”, “undifferentiated”, etc.
The CRISPR/CAS9 technology has recently been widely used to edit genes in both cultured cells and in vivo, despite the abovementioned off-target problem that is known to most experts but probably not to other biologists. Because the guider sequences have too many identical or highly-similar sequences in the genome, using the current version of this technology to knock out a particular gene resembles, in our opinion, using a machine gun to snipe a kidnapper among many hostages. All published studies just claim “the mission is complete” without mentioning whether or not any innocent ones are also hit. There are more off-target innocent sequences in gene knockout with CRISPR/CAS9 than in mRNA knockdown with short regulatory RNAs, because a large portion of a genome is intergenic region and because on average about 91% of a precursor transcript will be lopped off as introns during RNA splicing [73]. Moreover, gene knockout is often achieved by editing the target gene's 5'-region only, but not the entire gene, which raises a few serious issues that have barely been addressed so far: First, it is largely unknown whether the remaining intact part of the gene, which usually is still very lengthy, is still able to express shorter mRNA variant(s), as has been questioned for some estrogen receptor alpha knockout and CDK4 knockout animals [74-78]. In our opinion, in many cases, the “knockout” not only deletes the wild-type mRNA and protein but also alters the ratios among different mRNA and noncoding RNA variants of the target gene (Fig 1). Second, it is unclear whether the editing-created new recombinant locus or loci (including the ones formed due to off-targeting) form new gene(s) in a way similar to the formation of fusion genes in cancer cells [30, 67, 68]. Third, and more complicatedly, whether all other regulatory RNAs (including miRNAs and antisense RNAs) and other genes encoded by the locus or loci are also affected, especially those encoded by the opposite strand of DNA, since many loci are highly crowded habitats of genes and regulatory RNAs that are encoded by both strands of the DNA double helix, as shown in figure 2. There are a sheer number of unannotated open reading frames in the human, mouse and rat genomes (Fig 2), and whether they are also affected has never been addressed in any published studies involving gene editing, to our knowledge.
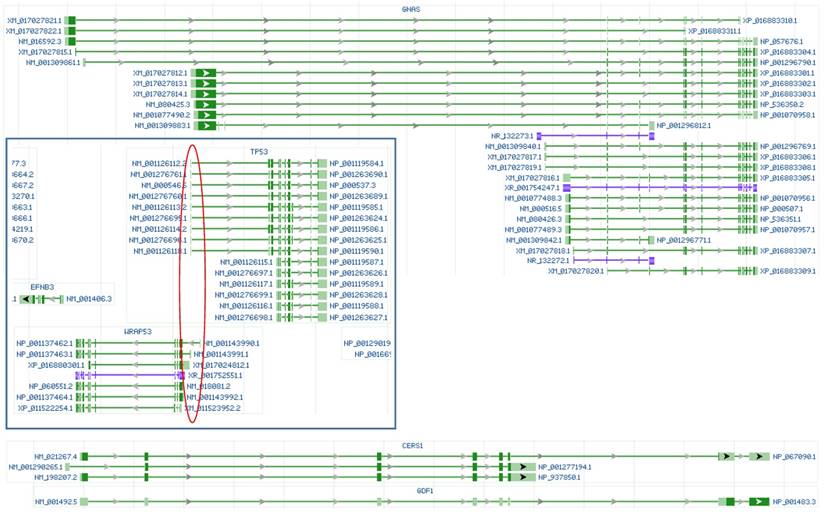
Illustrations of RNAs from the human GNAS, TP53 and WRAP53 (in box) genes, as well as the CERS1 and GDF1 genes copied from the NCBI database. Although editing the 5' region is a common practice in gene knockout technology, Knockout of GNAS by editing its 5' region may not be able to delete its short mRNA and noncoding RNA variants (in blue color). Knockout of TP53 by editing its 5' region may not be able to delete its short mRNAs and may also knock out the WRAP53 gene encoded by the opposite strand of the DNA double helix, as the first exons of both genes are in the same region (red circle). The CERS1 and GDF1 genes locate at the same genomic locus and are transcribed from the same initiation site, with their RNAs sharing some exons; therefore, knocking out either gene will also delete the other.
Images copied from the NCBI database showing that several loci (14q23.3, 22q11.21, 19q13.2 and 2q21.1) of the human genome are crowded habitats of genes, including unannotated ones (those LOCs) as well as miRNAs (MIR) and antisenses (AS), located on either the plus strand (arrow to the right) or the minus strand (arrow to the left) of the DNA double helix. Note that some transcripts are even extended to the downstream gene as so-called read-through RNAs, such as the CHURC1-FNTB in the 14q23.3 as well as the MIA-RAB4B and RAB4-EGLN2 in the 19q13.2. It is likely that knocking out one of the two partner genes will also knock out the read-through gene.

RNA interference via miRNAs, siRNAs, saRNAs and antisense RNAs is a set of evolutionarily conserved mechanisms for regulation of gene expression. Some of these regulatory RNAs have evolved from a mechanism for cells to fight against infections by microorganisms [79]. While these regulatory RNAs have been developed as research tools for us to manipulate gene expression in cells, there have also been plentiful studies on detecting their expression in different cell or tissue types. Moreover, there have been a great many studies scouting their functions by manipulating their levels, such as using a plasmid or viral vector to ectopically express a regulatory RNA of interest, such as a miRNA. In natural situations, when a cell decides to use a miRNA, siRNA or saRNA to manipulate expression of a particular gene or to eliminate the RNA of an infectious microorganism (as the cell's defensive mechanism), the cell would know that the to-be-used miRNA, siRNA or saRNA has highly-similar or identical sequences in its genome, which will raise an off-target issue. The cell has means to avoid this problem, such as by compartmentalizing some “would be off-target” mRNAs in some organelles or protecting them with RNA-binding proteins, by shutting down their expression, by a combination of these approaches, or by other strategies [80, 81], for just a short spell (minutes may be sufficient). However, when we ectopically express a regulatory RNA, such as an siRNA, we are unable to utilize any of these approaches to avoid off-target issues and cannot control the effective time within a short spell.
Actually, we do not even know which genes may be mistakenly targeted, and bioinformatics, performed by many researchers [56, 58, 60, 63, 65, 66], cannot be much help due to several reasons: First, the great genomic polymorphism or heterogeneity makes a computational prediction of on- and off-target sequences inapplicable to any particular individual's genome. Second, different cell types express different genes in different situations. For instance, most cell types do not express insulin and thus do not need to worry about the insulin gene being mistakenly targeted, but the pancreatic β cells would be involved in such an effect. Third, the same gene that is mistakenly targeted may have quite different impacts in different cell types and in different physiological or pathological situations. In conclusion, we still lack an applicable approach to use various regulatory RNAs to specifically manipulate expression of the gene we are interested in, because we still lack a workaround to solve the off-target issue. This means that most, if not all, relevant data published so far are questionable, including those published by us [78, 82, 83], because none of the studies can assure that no other gene has also been manipulated mistakenly. All the pertinent publications just tell us that the object genes are manipulated as wished, which is far from enough. No wonder some recently reported data from using CRISPR/CAS9 are discrepant to those from using siRNA [84]. If one day we are able to know exactly which cell type has which genes as off-targets for which particular regulatory RNA at which particular situation, and we can shut down the potentially mis-targeted genes, compartmentalize their RNAs, or protect their RNAs with RNA-binding proteins at a specific time post transfection of our miRNA, saRNA or siRNA/shRNA, then we may be able to use them with confidence.
miRNAs are clustered into families based on sequence similarity, with the members or siblings in the same family differing from one another often by one single base only. This great similarity makes it difficult for us to specifically detect one sibling without mistakenly detecting the other(s), although there are some kits or tacks developed specifically for solving this technical issue. Most large-scale studies on miRNA detection in many samples (such as many cancer specimens) are conducted using routine approaches without involving special kits touted for their specificity, making it questionable whether other family member(s) or sibling(s) were also detected, especially when the abundance of the one in question was low. This concern on the sibling specificity of miRNAs has hardly been fully addressed in those large-scale studies using quantitative PCR. In fact, it is difficult to be certain, because there is no feasible way of knowing the sequence of mature miRNAs detected in a large number of samples.
It has been well known that, of most genes, each is expressed to multiple mRNA variants and then multiple protein isoforms via various mechanisms [30, 85, 86], as exemplified by the genes shown in figure 1. However, few publications describe which RNA variant(s) of the gene in question can be amplified with the RT-PCR primers used, and which protein isoform(s) of the gene in question can be detected by the primary antibody used in western blotting and immunohistochemical staining. Actually, in many cases of western blotting, the primary antibody detects multiple bands on the membrane. However, a routine but unspoken practice is to cut away the band(s) other than the desired one without persuasive evidence proving that the trashed band(s) are spurious. While antibody producing companies should provide more-specific antibodies [87], some antibodies that detect multiple proteins may not be less specific because there likely exist multiple protein isoforms. What is more worrisome is that, blamed for selling “not-specific-enough” antibodies, companies try hard to select and market those antibodies that recognize only a single protein isoform, usually the wild type form, and researchers prefer these “more-specific” ones as well. This “collaboration” between antibody suppliers and researchers extirpates, via a sort of “natural selection”, those antibodies that can detect more isoforms and thus provide us a more global picture about the protein products of the gene in question. Similarly, few publications involving immunohistochemical staining discuss whether the primary antibody used can detect multiple protein isoforms, and point it out clearly that there is no way of knowing which isoform(s) give rise to the staining. We have asked many peers a simple question as to “how many RNA variants and protein isoforms of your target gene are listed in the NCBI (US National Center for Bioinformation) database?” The thumping majority answered with “I don't know”. To the few who know, an ensuing question is “how many mRNA variants or protein isoforms of your target gene have been reported in the literature but not yet listed in the NCBI database?” So far, nobody we asked has an answer. Readers of this essay are encouraged to challenge themselves with these questions.
Many researchers tag a short sequence, which usually is a region of the c-Myc or histone (His) gene, to their cDNA construct, making it expressed as a fusion protein, but not exactly the protein of interest. This is because it is assumed that the extra peptide sequence is short and should not affect the biology of the protein in question. While this assumption had been preliminarily tested for a few proteins when this tagging technique was established, extension of this assumption to all other proteins may not always be tenable. Besides, today's antibody-producing technology, such as the phage display that can produce thousands of primary antibodies in vitro [88-90], has made antibodies available for most proteins. Therefore, in most cases it is gratuitous to use a Myc- or His-tag and then a Myc- or His-antibody to detect the expression of newly-identified proteins.
Many scientists have purged themselves from research by being illiterate in technical detail
Worldwide, academic career development is an elimination series, which for many biomedical scientists is split into two phases: in the first incarnation of their career, they eliminated rivals by winning in all sorts of exams, obtaining scarce faculty positions, and grabbing their first research grant(s). After they have established a lab and a research project, they aim to be prominent and thus spend more and more time in conferences and invited presentations as well as on manuscript and grant reviewing, while having less and less time for absorbing details of the daily-updated technology and accumulating hands-on experience in circumventing technical pitfalls. As the repercussion, gradually they know too little about technical detail to correctly understand and interpret the experimental data generated by their students, technicians and postdocs. In other words, they enter into the second incarnation of their career wherein they purge themselves from research, although their CV is elongating hastily with many more high-impact publications and grant awards and although they indeed become more influential. A slew of other scientists may not want to be transcendent but still have to spend most of their time on writing grant proposals, simply for surviving the research-funding gloom. Therefore, a common situation in the biomedical fraternity is that students, technicians and postdocs perform the bench work and probably also write the research reports that the professors know little about, especially pertaining to the technical details. In other words, there commonly is a disconnection or a poor connection between the data producers (the juniors) and the data interpreters (the seniors). In general, those principal investigators who attain more research funds know fewer technical details than those who have less funding, because the former have much less time than the latter on learning research methodology.
Some of us may be intrepid enough to admit that the above-described tenure of “first eliminating others and then eliminating ourselves” is virtually our own career trajectory. Indeed, we can ask ourselves how much technical detail we know about the data from our students and postdocs, pertaining to, such as, deep RNA sequencing, various “omics” related techniques, etc., especially on the aspects of artifacts and reproducibility [91, 92]. Many of us cannot even remind our students what pitfalls they should avoid when preparing samples for these sophisticated techniques, and thus completely rely on what the juniors can figure out for themselves, which usually is not much, haplessly. Readers can evaluate themselves about the technical detail and pitfalls of RT-PCR described in some perspective articles of ours [67, 68, 85, 93] and others [94], to get a sense how surprisingly complicated these commonly used methods are and how little we actually know about them. For instance, numerous RT-PCR experiments were conducted with the forward and reverse primers on the same exon and with the RNA samples without being subject to removal of gDNA residual, thus making it unclear whether it is the cDNA, gDNA, or both that are amplified [95]. Moreover, even the reference gene used for the RT-PCR is an issue in most cases, as we have explicated [95]. Given that RT and PCR are so basic and are the footing of many other sophisticated techniques but we still know so little about them, it is not surprising that slipups are omnipresent in biomedical research that involves so many more-sophisticated techniques [96]. All abovementioned issues, and not just data fabrication or other malfeasance, contribute to the poor reproducibility of publications. In fact, the nightmare does not end here, because in many cases reproducible studies become reproducible because the same misstep is made.
We need to have a broader knowledge and know more about ancient scientific literature
Many peers try hard to create jargons, such as “cancer stem cells”, “cancer cell dormancy”, “chimeric RNAs”, etc., to establish an iconic status of their findings. Some of the new terms are not precisely defined to be distinguishable from already-existing ones. For instance, a lucid demarcation has never been outlined in the literature to distinguish “cancer stem cells” [97] from “normal stem cells” and, especially, from “the ordinary cancer cells”, as we pointed out previously [42]. Many new nomenclatures are superfluous because they just describe ad hoc situations or phenomena that have been described many decades ago under other names. For example, in human cells, authentic chimeric RNAs are probably as scarce as hen's teeth, while many tagged as “chimeric RNAs” are actually derived from fusion genes or from transcriptional read-through that to us is transcription of unannotated genes (Fig 3). Since the differences between the regulation of these fusion genes or unannotated genes and that of regular genes occur only at the DNA level, it is irrational and has little significance to label these RNAs differently from other RNAs [67, 68, 98]. For another example, “cancer cell dormancy” and “oncogene addiction” are used to describe regression of tumors induced in some transgenic animals upon turning off the transgene, and their swift recurrence upon turning on the transgene again [99-102]. However, this phenomenon of the-inducer-dependency of tumors was already reported by Fishcer in 1906 and confirmed by Helmholz in 1907; they, according to Davis, Vasiliev and Cheung [103, 104], observed that painting the ears of rabbits with Scarlet Red could induce papillomas, but the tumors regressed upon discontinuation of the painting. Since 1910s, a sheer number of studies have shown that tumors induced in animals, unless they are at very advanced stages, will regress upon withdrawal of the chemical or transgene inducers, with a small number of references given herein [41, 105-127]. Moreover, a similar phenomenon of this inducer-dependency has also been reported since 1930s for tumors induced with sex steroids [128-135], as we reviewed previously [40, 41, 136, 137]. This situation also reflects disturbing facts that research and literature on scientific history are insufficient and that many researchers do not sufficiently peruse, or even just leaf through, the literature of 100 years ago, especially the literature slightly outside, but still appertaining to, their research interests.
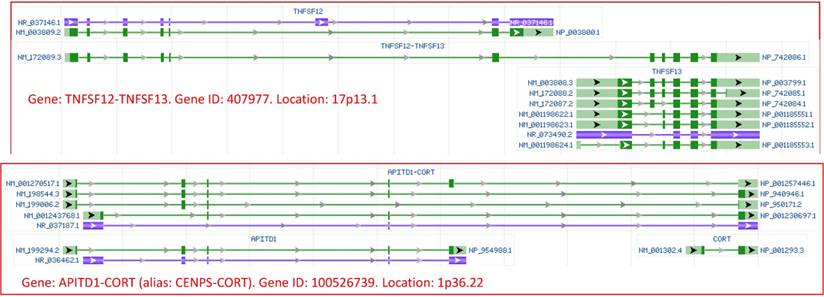
Images of transcriptional read-through derived RNAs of human origin, copied from the NCBI database. Top panel: Transcription of the TNFSF12 gene may read through its termination site and goes into the downstream gene TNFSF13, producing the TNFSF12-TNFSF13 mRNA that contains most, but not all, exons of each gene. Bottom panel: transcription of the APITD1 gene may read through its termination site and goes into the downstream gene CORT, producing several APITD1-CORT RNAs, with each RNA containing most exons of the upstream and downstream genes. Since the NCBI assigns 407877 and 100526739 as the gene identity (gene ID) for the TNFSF12-TNFSF13 and the APITD1-CORT, respectively, we consider that they are previously-unannotated genes which produce RNAs that should be regarded as regular, but not as chimeric, RNAs, via a mechanism identical to that for the production of all regular RNAs. Note that in the NCBI database, green and blue colors indicate mRNA and noncoding RNA, respectively, while boxes and lines indicate exons and introns, respectively. The NCBI draws the lengths of exons and introns in proportion to their number of nucleotides. Arrows point to the 5'-to-3' direction.
Another situation in most developed countries is that many medical researchers are not medical graduates and completely lack clinical experience. Most of them formulate their research projects only based on the literature, but not on bedside knowledge and experience, although some have realized and tried to fix this fragility by collaborating with clinicians. Some of the irrational study designs, such as the abovementioned comparisons of cancer cell lines with their normal counterparts from the same tissue origin in the studies of chemo drugs' cancer-specificity, may be due to the lack of bedside experience. In the meantime, many medical doctors lack sufficient training, especially hands-on experience, in sophisticated biotechnologies, but their research teams still routinely use these technologies to scout out the mechanisms behind various medical observations. This will inevitably create technically derived artifacts, with biased or erroneous data interpretations as the sequel. In general, many of us lack a global knowledge of biology and medicine that are the outer tiers of, but important for, our research projects, since many disciplines of biology and medicine are interrelated and hence many pieces of data make sense only when they are looked from a more distant and more global viewpoint.
In most developed countries, funds for research have, for many years, been dwindling and will be unlikely to burgeon again in the near future. While more funds are positively correlated with more scientific findings or achievements, statistically, we opine that much of the research funding has actually been squandered, making biomedical research prodigal. This is largely because we are too rushed in going into new technology without assimilating enough technical detail and figuring out potential pitfalls and corrective measures. Or, reiterated in a positive or melodious tone, if researchers slow down their pace and put more effort onto digesting the technical detail of modern technology or vanquishing the weakness of wanting clinical knowledge and experience, more-meaningful data can be achieved with less funding.
Some tacks may be taken as corrective measures to reverse the unhealthy trends
Most science journals and research funding agencies already have panels of reviewers to scrutinize ethical aspect, interest conflict and plagiarism of manuscripts or research proposals. We propose that journals and funding agencies should also establish a panel of experts to scrutinize technical details in all manuscripts and research proposals submitted, because most scientists as reviewers do not have all lines of technical expertise described in each manuscript and grant proposal. Experts in this technical panel will only focus on the technical flaws and feasibilities of the methods used in the manuscript or to be used in the proposal, such as whether the RT-PCR primers can amplify all the mRNA variants or can just amplify one specific variant and whether the primary antibody will simultaneously detect several protein isoforms of the gene in question in immunohistochemical staining. Only after the manuscript or proposal has passed the scrutiny on the technical details, it can be assigned to reviewers or to a study section for further evaluation of its scientific merits. This tack may help minimize technical flaws and artifacts in published papers and improve the applicability of research proposals. Since it is time-consuming to assimilate a broad literature and technical details and to cogitate in more depth over the study designs, major research funds, such as the NIH's RO1 type, should require at least 50% efforts of the principal investigator (PI), which will also decrease the number of awards one PI can attain and will in turn make PIs more focusing on fewer research projects.
Conclusions
Mistakes, however non-deliberate, have been omnipresent in the biomedical research, with examples described herein. These errors are largely because modern technology consists of sophistical technical details while many scientists, under a high strain of “publish or perish” from their environments [17, 138], do not give adequate thought to the study design and do not spend sufficient time digesting the details. In part because of these advertent or inadvertent mistakes, discrepant data and paradoxical conclusions are omnipresent in the biomedical literature, making us used to saying “on one hand… but on the other hand…” when describing almost all biomedical issues. We opine that research reports, reviews or perspectives should emphasize more the possible spuriousness, pitfalls, technical difficulties, constraints and adverse repercussions, since these issues usually are not properly addressed [139]. For example, research on circular RNAs or chimeric RNAs should emphasize the possible artificial sequences, especially when RT-PCR is involved. Studies using small regulatory RNAs as tools should focus more on “how can we be sure that there is not any additional gene being mistakenly targeted”, but not on “how to improve the specificity or efficacy” (again, it becomes a philosophical issue). Although getting more research funds is better, scientists can always let money go much further during the funding gloom by spending more time on rummaging through a much broader or older literature for valuable information and learning more about technical detail. Scientists, especially those at the pinnacle, should continue writing manuscripts themselves, not only perspectives and reviews but also research reports, so as to more correctly interpret the lab data and avoid being technical illiterates and thus being purged from the real research. A woeful but unspoken fact is that there are many scientists just using scientific research as a means for making a good living and prestige with little interest in science per se, although this, advantageously, makes the competition in genuine scientific research not as tough as it seems to be. The unhealthy trends described herein gut theoretical research more but, luckily, may mar translational research relatively less, since the results of the latter can be evaluated more quickly and easily by the market.
Abbreviations
cDNA: complementary DNA; gDNA: genomic DNA; iPS: induced pluripotent stem (cells); LC-MS/MS: liquid chromatography and tandem mass spectrometry; NCBI: National Center for Bioinformation; NIH: National Institute of Health of the United States; miRNA: microRNA; PI: principal investigator; saRNA: small activating RNA; shRNA: short-hairpin RNA; siRNA: small interfering RNA; PCR: polymerase chain reactions; RT: reverse transcription; SOP: standard operation procedure.
Acknowledgements
We would like to thank Dr. Fred Bogott at Austin Medical Center of Mayo Clinic, Austin of Minnesota, and Mr. Lucas Zellmer at Masonic Cancer Center of Minnesota University, for their excellent English editing of this manuscript.
Funding
The work is supported by grants from Chinese National Science Foundation to Yan He (grant number 31560306) and DJ Liao (grant No. 81660501).
Authors' contributions
YH outlined and drafted the manuscript. LC and NX contributed many examples presented. YL and HZ participated in discussions and contributed comments. DJL participated in paper outline and finalized the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ioannidis JP. How to make more published research true. PLoS Med. 2014;11(10e):1001747
2. Begley CG, Ioannidis JP. Reproducibility in science: improving the standard for basic and preclinical research. Circ Res. 2015;116(1):116-126
3. Amrhein V, Korner-Nievergelt F, Roth T. The earth is flat (p > 0.05): significance thresholds and the crisis of unreplicable research. PeerJ. 2017;5:e3544
4. Freedman LP, Venugopalan G, Wisman R. Reproducibility2020: Progress and priorities. F1000Res. 2017;6:604
5. Baker M, Dolgin E. Cancer reproducibility project releases first results. Nature. 2017;541(7637):269-270
6. Mobley A, Linder SK, Braeuer R, Ellis LM, Zwelling L. A survey on data reproducibility in cancer research provides insights into our limited ability to translate findings from the laboratory to the clinic. PLoS One. 2013;8(5):e63221
7. Baker M. Reproducibility: Respect your cells!. Nature. 2016;537(7620):433-435
8. Baker M. 1,500 scientists lift the lid on reproducibility. Nature. 2016;533(7604):452-454
9. Reality check on reproducibility. Nature. 2016;533(7604):437. doi: 10.1038/533437a
10. Gall T, Ioannidis JPA, Maniadis Z. The credibility crisis in research: Can economics tools help? PLoS Biol. 2017;15(4e):2001846
11. Annesley T, Scott M, Bastian H, Fonseca V, Ioannidis JP, Keller MA. et al. Biomedical Journals and Preprint Services: Friends or Foes? Clin Chem. 2017;63(2):453-458
12. Chalmers I, Glasziou P. Avoidable waste in the production and reporting of research evidence. Lancet. 2009;374(9683):86-89
13. Kleinert S, Horton R. How should medical science change? Lancet. 2014;383(9913):197-198
14. Glasziou P, Altman DG, Bossuyt P, Boutron I, Clarke M, Julious S. et al. Reducing waste from incomplete or unusable reports of biomedical research. Lancet. 2014;383(9913):267-276
15. Bhopal RS. Increasing value and reducing waste in biomedical research. Lancet. 2016;388(10044):562-doi 10.1016/S0140-6736(16)31216-8
16. Furst T, Strojil J. A patient called Medical Research. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2017;161(1):54-57
17. Alberts B, Kirschner MW, Tilghman S, Varmus H. Rescuing US biomedical research from its systemic flaws. Proc Natl Acad Sci U S A. 2014;111(16):5773-5777
18. Hvistendahl M. China's publication bazaar. Science. 2013;342(6162):1035-1039
19. Sun Q, Xin Q, Wei L, Liu C, Gao G. "Science Citation Index Worship" in China. Iran J Public Health. 2013;42(8):921-922
20. Bustin SA, Huggett JF. Reproducibility of biomedical research - The importance of editorial vigilance. Biomol Detect Quantif. 2017;11:1-3
21. Vasilevsky NA, Brush MH, Paddock H, Ponting L, Tripathy SJ, Larocca GM. et al. On the reproducibility of science: unique identification of research resources in the biomedical literature. PeerJ. 2013;1(e):148
22. Dinis-Oliveira RJ, Magalhaes T. The Inherent Drawbacks of the Pressure to Publish in Health Sciences: Good or Bad Science. F1000Res. 2015;4:419-doi 10.12688/f1000research.6809.2
23. Stephan P. Research efficiency: Perverse incentives. Nature. 2012;484(7392):29-31
24. Collins FS, Tabak LA. Policy: NIH plans to enhance reproducibility. Nature. 2014;505(7485):612-613
25. Jarvis MF, Williams M. Irreproducibility in Preclinical Biomedical Research: Perceptions, Uncertainties, and Knowledge Gaps. Trends Pharmacol Sci. 2016;37(4):290-302
26. Errington TM, Iorns E, Gunn W, Tan FE, Lomax J, Nosek BA. An open investigation of the reproducibility of cancer biology research. Elife. 2014:3 -doi: 10.7554/eLife.04333
27. Mullane K, Williams M. Enhancing reproducibility: Failures from Reproducibility Initiatives underline core challenges. Biochem Pharmacol. 2017;138:7-18
28. Rosenblatt M. An incentive-based approach for improving data reproducibility. Sci Transl Med. 2016;8(336):336ed5-doi 10.1126/scitranslmed.aaf5003
29. Wang G, Chen L, Yu B, Zellmer L, Xu N, Liao DJ. Learning about the Importance of Mutation Prevention from Curable Cancers and Benign Tumors. J Cancer. 2016;7(4):436-445
30. Jia Y, Chen L, Ma Y, Zhang J, Xu N, Liao DJ. To Know How a Gene Works, We Need to Redefine It First but then, More Importantly, to Let the Cell Itself Decide How to Transcribe and Process Its RNAs. Int J Biol Sci. 2015;11(12):1413-1423
31. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353-358
32. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033-1038
33. An Y, Furber KL, Ji S. Pseudogenes regulate parental gene expression via ceRNA network. J Cell Mol Med. 2017;21(1):185-192
34. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344-352
35. Maddox J. Where next with peer-review? Nature. 1989;339(6219):11-doi 10.1038/339011a0
36. Conover E. Einstein's milestones. Science. 2015;347(6226):1085 1087-1088, 1092
37. Kennefick D, Blume M. Reviewing Einstein. Science. 2015;349(6244):149-doi 10.1126/science.349.6244.149-a
38. Kennefick.D. Einstein Versus the Physical Review. Physics Today. 2005;58(9):43-doi 10.1063/1.2117822
39. Liu B, Ezeogu L, Zellmer L, Yu B, Xu N, Liao DJ. Protecting the normal in order to better kill the cancer. Cancer Med. 2015;4(9):1394-1403
40. Zhang J, Lou XM, Jin LY, Zhou RJ, Liu SQ, Xu NZ. et al. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: clearance of a few misconceptions. Oncoscience. 2014;1(6):407-422
41. Ma Y, Jia Y, Chen L, Ezeogu L, Yu B, Xu N. et al. Weaknesses and Pitfalls of Using Mice and Rats in Cancer Chemoprevention Studies. J Cancer. 2015;6(10):1058-1065
42. Zhang J, Lou X, Zellmer L, Liu S, Xu N, Liao DJ. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience. 2014;1(9):580-590
43. Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7(3):143-164
44. Liao DJ, Natarajan G, Deming SL, Jamerson MH, Johnson M, Chepko G. et al. Cell cycle basis for the onset and progression of c-Myc-induced, TGFalpha-enhanced mouse mammary gland carcinogenesis. Oncogene. 2000;19(10):1307-1317
45. Liao DJ, Wang Y, Wu J, Adsay NV, Grignon D, Khanani F. et al. Characterization of pancreatic lesions from MT-tgfalpha, Ela-myc and MT-tgfalpha/Ela-myc single and double transgenic mice. J Carcinog. 2006;5:DOI 10.1186/1477-3163-5-19
46. Liao JD, Adsay NV, Khannani F, Grignon D, Thakur A, Sarkar FH. Histological complexities of pancreatic lesions from transgenic mouse models are consistent with biological and morphological heterogeneity of human pancreatic cancer. Histol Histopathol. 2007;22(6):661-676
47. Boyle EA, Andreasson JOL, Chircus LM, Sternberg SH, Wu MJ, Guegler CK. et al. High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding. Proc Natl Acad Sci U S A. 2017;114(21):5461-5466
48. Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB. et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550(7676):407-410
49. Hannus M, Beitzinger M, Engelmann JC, Weickert MT, Spang R, Hannus S. et al. siPools: highly complex but accurately defined siRNA pools eliminate off-target effects. Nucleic Acids Res. 2014;42(12):8049-8061
50. Jamal M, Ullah A, Ahsan M, Tyagi R, Habib Z, Rehman K. Improving CRISPR-Cas9 On-Target Specificity. Curr Issues Mol Biol. 2017;26:65-80
51. Kim D, Lim K, Kim ST, Yoon SH, Kim K, Ryu SM. et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol. 2017;35(5):475-480
52. Koo T, Lee J, Kim JS. Measuring and Reducing Off-Target Activities of Programmable Nucleases Including CRISPR-Cas9. Mol Cells. 2015;38(6):475-481
53. Mendoza BJ, Trinh CT. Enhanced guide-RNA Design and Targeting Analysis for Precise CRISPR Genome Editing of Single and Consortia of Industrially Relevant and Non-Model Organisms. Bioinformatics. 2017 -doi: 10.1093/bioinformatics/btx564
54. Morgens DW, Wainberg M, Boyle EA, Ursu O, Araya CL, Tsui CK. et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat Commun. 2017;8:15178-doi 10.1038/ncomms15178
55. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281-2308
56. Riba A, Emmenlauer M, Chen A, Sigoillot F, Cong F, Dehio C. et al. Explicit Modeling of siRNA-Dependent On- and Off-Target Repression Improves the Interpretation of Screening Results. Cell Syst. 2017;4(2):182-193
57. Rosenbluh J, Xu H, Harrington W, Gill S, Wang X, Vazquez F. et al. Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat Commun. 2017;8:15403-doi 10.1038/ncomms15403
58. Schmich F, Szczurek E, Kreibich S, Dilling S, Andritschke D, Casanova A. et al. gespeR: a statistical model for deconvoluting off-target-confounded RNA interference screens. Genome Biol. 2015;16:220-doi 10.1186/s13059-015-0783-1
59. Seok H, Lee H, Jang ES, Chi SW. Evaluation and control of miRNA-like off-target repression for RNA interference. Cell Mol Life Sci. 2017 -doi: 10.1007/s00018-017-2656-0
60. Suter SR, Sheu-Gruttadauria J, Schirle NT, Valenzuela R, Ball-Jones AA, Onizuka K. et al. Structure-Guided Control of siRNA Off-Target Effects. J Am Chem Soc. 2016;138(28):8667-8669
61. Tasan I, Zhao H. Targeting Specificity of the CRISPR/Cas9 System. ACS Synth Biol. 2017;6(9):1609-1613
62. Ui-Tei K. Is the Efficiency of RNA Silencing Evolutionarily Regulated?. Int J Mol Sci. 2016:17 (5):-pii: E719. doi: 10.3390/ijms17050719
63. Wang Q, Ui-Tei K. Computational Prediction of CRISPR/Cas9 Target Sites Reveals Potential Off-Target Risks in Human and Mouse. Methods Mol Biol. 2017;1630:43-53
64. Wolt JD. Safety, Security, and Policy Considerations for Plant Genome Editing. Prog Mol Biol Transl Sci. 2017;149:215-241
65. Yilmazel B, Hu Y, Sigoillot F, Smith JA, Shamu CE, Perrimon N. et al. Online GESS: prediction of miRNA-like off-target effects in large-scale RNAi screen data by seed region analysis. BMC Bioinformatics. 2014;15:192-doi 10.1186/1471-2105-15-192
66. Zhong R, Kim J, Kim HS, Kim M, Lum L, Levine B. et al. Computational detection and suppression of sequence-specific off-target phenotypes from whole genome RNAi screens. Nucleic Acids Res. 2014;42(13):8214-8222
67. Peng Z, Yuan C, Zellmer L, Liu S, Xu N, Liao DJ. Hypothesis: Artifacts, Including Spurious Chimeric RNAs with a Short Homologous Sequence, Caused by Consecutive Reverse Transcriptions and Endogenous Random Primers. J Cancer. 2015;6(6):555-567
68. Yuan C, Han Y, Zellmer L, Yang W, Guan Z, Yu W. et al. It Is Imperative to Establish a Pellucid Definition of Chimeric RNA and to Clear Up a Lot of Confusion in the Relevant Research. Int J Mol Sci. 2017;18(4):pii E714. doi: 10.3390/ijms18040714
69. Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009;27(5):1050-1056
70. Riggs JW, Barrilleaux BL, Varlakhanova N, Bush KM, Chan V, Knoepfler PS. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev. 2013;22(1):37-50
71. Laplane L, Beke A, Vainchenker W, Solary E. Concise Review: Induced Pluripotent Stem Cells as New Model Systems in Oncology. Stem Cells. 2015;33(10):2887-2892
72. Yamada Y, Haga H, Yamada Y. Concise review: dedifferentiation meets cancer development: proof of concept for epigenetic cancer. Stem Cells Transl Med. 2014;3(10):1182-1187
73. Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D. et al. Function of alternative splicing. Gene. 2005;344:1-20
74. Bollig-Fischer A, Thakur A, Sun Y, Wu J-S, Liao DJ. The predominant proteins that react to the MC-20 estrogen receptor alpha antibody differ in molecular weight between the mammary gland and uterus in the mouse and rat. Int J Biomed Sci. 2012;8(1):51-63
75. Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB. et al. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol. 1995;9(11):1441-1454
76. Denger S, Reid G, Kos M, Flouriot G, Parsch D, Brand H. et al. ERalpha gene expression in human primary osteoblasts: evidence for the expression of two receptor proteins. Mol Endocrinol. 2001;15(12):2064-2077
77. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993;90(23):11162-11166
78. Sun Y, Lou X, Yang M, Yuan C, Ma L, Xie BK. et al. Cyclin-dependent kinase 4 may be expressed as multiple proteins and have functions that are independent of binding to CCND and RB and occur at the S and G 2/M phases of the cell cycle. Cell Cycle. 2013;12(22):3512-3525
79. Zamore PD, Haley B. Ribo-gnome: the big world of small RNAs. Science. 2005;309(5740):1519-1524
80. Ui-Tei K. Is the Efficiency of RNA Silencing Evolutionarily Regulated? Int J Mol Sci. 2016;17(5):pii E719. doi: 10.3390/ijms17050719
81. Sarett SM, Nelson CE, Duvall CL. Technologies for controlled, local delivery of siRNA. J Control Release. 2015;218:94-113
82. Sun Y, Wu J, Wu SH, Thakur A, Bollig A, Huang Y. et al. Expression profile of microRNAs in c-Myc induced mouse mammary tumors. Breast Cancer Res Treat. 2009;118(1):185-196
83. Sun Y, Cao S, Yang M, Wu S, Wang Z, Lin X. et al. Basic anatomy and tumor biology of the RPS6KA6 gene that encodes the p90 ribosomal S6 kinase-4. Oncogene. 2013;32(14):1794-1810
84. Lin A, Giuliano CJ, Sayles NM, Sheltzer JM. CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. Elife. 2017:6 -pii: e24179. doi: 10.7554/eLife.24179
85. Zhang J, Lou X, Shen H, Zellmer L, Sun Y, Liu S. et al. Isoforms of wild type proteins often appear as low molecular weight bands on SDS-PAGE. Biotechnol J. 2014;9(8):1044-1054
86. Liu X, Wang Y, Yang W, Guan Z, Yu W, Liao DJ. Protein multiplicity can lead to misconduct in western blotting and misinterpretation of immunohistochemical staining results, creating much conflicting data. Prog Histochem Cytochem. 2016;51(3-4):51-58
87. Voskuil JL. The challenges with the validation of research antibodies. F1000Res. 2017;6:161-doi 10.12688/f1000research.10851.1
88. Weber J, Peng H, Rader C. From rabbit antibody repertoires to rabbit monoclonal antibodies. Exp Mol Med. 2017;49(3):e305-doi 10.1038/emm.2017.23
89. Zhao A, Tohidkia MR, Siegel DL, Coukos G, Omidi Y. Phage antibody display libraries: a powerful antibody discovery platform for immunotherapy. Crit Rev Biotechnol. 2016;36(2):276-289
90. Shukra AM, Sridevi NV, Dev C, Kapil M. Production of recombinant antibodies using bacteriophages. Eur J Microbiol Immunol (Bp). 2014;4(2):91-98
91. Ioannidis JP, Khoury MJ. Improving validation practices in "omics" research. Science. 2011;334(6060):1230-1232
92. Dijkstra JR, van Kempen LC, Nagtegaal ID, Bustin SA. Critical appraisal of quantitative PCR results in colorectal cancer research: can we rely on published qPCR results? Mol Oncol. 2014;8(4):813-818
93. Yuan C, Liu Y, Yang M, Liao DJ. New methods as alternative or corrective measures for the pitfalls and artifacts of reverse transcription and polymerase chain reactions (RT-PCR) in cloning chimeric or antisense-accompanied RNA. RNA Biol. 2013;10(6):958-967
94. Labaj PP, Kreil DP. Sensitivity, specificity, and reproducibility of RNA-Seq differential expression calls. Biol Direct. 2016;11(1):66-doi 10.1186/s13062-016-0169-7
95. Sun Y, Li Y, Luo D, Liao DJ. Pseudogenes as Weaknesses of ACTB (Actb) and GAPDH (Gapdh) Used as Reference Genes in Reverse Transcription and Polymerase Chain Reactions. PLoS One. 2012;7(8):e41659-doi 10.1371/journal.pone.0041659
96. Bustin S, Nolan T. Talking the talk, but not walking the walk: RT-qPCR as a paradigm for the lack of reproducibility in molecular research. Eur J Clin Invest. 2017 -doi: 10.1111/eci.12801
97. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105-111
98. Yang W, Wu JM, Bi AD, Ou-Yang YC, Shen HH, Chirn GW. et al. Possible Formation of Mitochondrial-RNA Containing Chimeric or Trimeric RNA Implies a Post-Transcriptional and Post-Splicing Mechanism for RNA Fusion. PLoS One. 2013;8(10):e77016-doi 10.1371/journal.pone.0077016
99. Felsher DW. Tumor dormancy: death and resurrection of cancer as seen through transgenic mouse models. Cell Cycle. 2006;5(16):1808-1811
100. Felsher DW. Tumor dormancy and oncogene addiction. APMIS. 2008;116(7-8):629-637
101. Shachaf CM, Felsher DW. Tumor dormancy and MYC inactivation: pushing cancer to the brink of normalcy. Cancer Res. 2005;65(11):4471-4474
102. Shachaf CM, Felsher DW. Rehabilitation of cancer through oncogene inactivation. Trends Mol Med. 2005;11(7):316-321
103. Davis JS. III. The Effect of Scarlet Red, in Various Combinations, upon the Epitheliation of Granulating Surfaces. Ann Surg. 1910;51(1):40-51
104. VASILIEV JM, CHEUNG AB. Evolution of epithelial proliferation induced by scarlet red in the skin of normal and carcinogen-treated rabbits. Br J Cancer. 1962;16:238-245
105. Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16(4):313-317
106. Cabot S, Shear N, Shear MJ, Perrault A. Studies in carcinogenesis: XI. Development of skin tumors in mice painted with 3:4-benzpyrene and creosote oil fractions. Am J Pathol. 1940;16(3):301-312
107. D'Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE. et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7(2):235-239
108. Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199-207
109. Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ. et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15(24):3249-3262
110. Fujiki H. Gist of Dr. Katsusaburo Yamagiwa's papers entitled "Experimental study on the pathogenesis of epithelial tumors" (I to VI reports). Cancer Sci. 2014;105(2):143-149
111. Haslam SZ, Bern HA. Histopathogenesis of 7,12-diemthylbenz(a)anthracene-induced rat mammary tumors. Proc Natl Acad Sci U S A. 1977;74(9):4020-4024
112. Huggins C, BRIZIARELLI G, SUTTON H Jr. Rapid induction of mammary carcinoma in the rat and the influence of hormones on the tumors. J Exp Med. 1959;109(1):25-42
113. Huggins C, GRAND LC, BRILLANTES FP. Mammary cancer induced by a single feeding of polymucular hydrocarbons, and its suppression. Nature. 1961;189:204-207
114. Li Z, Huang X, Zhan H, Zeng Z, Li C, Spitsbergen JM. et al. Inducible and repressable oncogene-addicted hepatocellular carcinoma in Tet-on xmrk transgenic zebrafish. J Hepatol. 2012;56(2):419-425
115. McGuire WL, Chamness GC, Costlow ME, Shepherd RE. Hormone dependence in breast cancer. Metabolism. 1974;23(1):75-100
116. Mider GB, Morton JJ. Skin tumors following a single application of methylcholanthrene in C57 brown mice. Am J Pathol. 1939;15(3):299-302
117. Mobbs BG. Uptake of (3H)oestradiol by dimethylbenzanthracene-induced rat mammary tumours regressing spontaneously or after ovariectomy. J Endocrinol. 1969;44(3):463-464
118. Nguyen AT, Emelyanov A, Koh CH, Spitsbergen JM, Parinov S, Gong Z. An inducible kras(V12) transgenic zebrafish model for liver tumorigenesis and chemical drug screening. Dis Model Mech. 2012;5(1):63-72
119. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S. et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431(7012):1112-1117
120. Sun L, Nguyen AT, Spitsbergen JM, Gong Z. Myc-induced liver tumors in transgenic zebrafish can regress in tp53 null mutation. PLoS One. 2015;10(1):e0117249
121. Tilli MT, Furth PA. Conditional mouse models demonstrate oncogene-dependent differences in tumor maintenance and recurrence. Breast Cancer Res. 2003;5(4):202-205
122. Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J. et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS One. 2008;3(5):e2125-doi 10.1371/journal.pone.0002125
123. Uchiyama K, Watanabe D, Hayasaka M, Hanaoka K. A novel imprinted transgene located near a repetitive element that exhibits allelic imbalance in DNA methylation during early development. Dev Growth Differ. 2014;56(9):653-668
124. Yamagiwa K, Ichikawa K. Experimental study of the pathogenesis of carcinoma. CA Cancer J Clin. 1977;27(3):174-181
125. YOUNG S, COWAN DM. Spontaneous regression of induced mammary tumours in rats. Br J Cancer. 1963;17:85-89
126. Zheng W, Li Z, Nguyen AT, Li C, Emelyanov A, Gong Z. Xmrk, kras and myc transgenic zebrafish liver cancer models share molecular signatures with subsets of human hepatocellular carcinoma. PLoS One. 2014;9(3):e91179
127. Li Y, Li H, Spitsbergen JM, Gong Z. Males develop faster and more severe hepatocellular carcinoma than females in krasV12 transgenic zebrafish. Sci Rep. 2017;7:41280. doi: 10.1038/srep41280
128. Burrows H. Carcinoma mammae occurring in a male mouse under continued treatment with oestrin. Am J Cancer. 1935;24(3):613-616
129. Cutts JH, Froude GC. Regression of estrone-induced mammary tumors in the rat. Cancer Res. 1968;28(12):2413-2418
130. Cutts JH. Enzyme activities in regressing estrone-induced mammary tumors of the rat. Cancer Res. 1973;33(6):1235-1237
131. Geschickter CF, Lewis D, Hartman CG. Tumors of the breast related to the oestrin hormone. Am J Cancer. 1934;21:828-859
132. Huggins C. Endocrine-induced regression of cancers. Cancer Res. 1967;27(11):1925-1930
133. Huggins C. Endocrine-induced regression of cancers. Am J Surg. 1978;136(2):233-238
134. Mceuen CS. Occurrence of cancer in rats treated with oestrone. Am J Cancer. 1938;34:184-195
135. Noble RL, Cutts JH. Mammary tumors of the rat: a review. Cancer Res. 1959;19:1125-1139
136. Liao DJ, Dickson RB. Roles of androgens in the development, growth, and carcinogenesis of the mammary gland. J Steroid Biochem Mol Biol. 2002;80(2):175-189
137. Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10(1):57-67
138. Gualberto Buela-Casal. Pathological publishing: A new psychological disorder with legal consequences? Eur J Psychol, Appl Legal Context. 2014;6:91-97
139. Ioannidis JP. Limitations are not properly acknowledged in the scientific literature. J Clin Epidemiol. 2007;60(4):324-329
Author contact
![]() Corresponding authors: Dr. Yan He, Key Lab of Endemic and Ethnic Diseases of the Ministry of Education of China in Guizhou Medical University, Guiyang, Guizhou Province 550004, P. R. China, Email: annieheyanedu.cn; Dr. Chengfu Yuan, Department of Biochemistry, China Three Gorges University, Yichang City, Hubei Province 443002, P.R. China, Email: yuancf46edu.cn; Dr. Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, China, Email: xuningzhiac.cn; Dr. D. Joshua Liao, Department of Pathology, Guizhou Medical University Hospital, Guiyang, Guizhou 550004, China, Email: djliaoedu.cn
Corresponding authors: Dr. Yan He, Key Lab of Endemic and Ethnic Diseases of the Ministry of Education of China in Guizhou Medical University, Guiyang, Guizhou Province 550004, P. R. China, Email: annieheyanedu.cn; Dr. Chengfu Yuan, Department of Biochemistry, China Three Gorges University, Yichang City, Hubei Province 443002, P.R. China, Email: yuancf46edu.cn; Dr. Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, China, Email: xuningzhiac.cn; Dr. D. Joshua Liao, Department of Pathology, Guizhou Medical University Hospital, Guiyang, Guizhou 550004, China, Email: djliaoedu.cn