Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Liver Transplantation
Ischemia/Reperfusion
Mitochondrial Activity and I/R
Mitochondrial Targeting...
Conclusions and Future...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(3):248-256. doi:10.7150/ijms.22891 This issue Cite
Review
Recent insights into mitochondrial targeting strategies in liver transplantation
Rui Miguel Martins1, ![]() , João Soeiro Teodoro2, Emanuel Furtado3, Anabela Pinto Rolo2, Carlos Marques Palmeira2, José Guilherme Tralhão4
, João Soeiro Teodoro2, Emanuel Furtado3, Anabela Pinto Rolo2, Carlos Marques Palmeira2, José Guilherme Tralhão4
1. Department of Surgery, Instituto Português de Oncologia de Coimbra, Coimbra, Portugal
2. Department of Life Sciences, Faculty of Sciences and Technology, University of Coimbra, Coimbra, Portugal; and Center of Neurosciences and Cell Biology, University of Coimbra, Coimbra, Portugal
3. Unidade de Transplantação Hepática de Crianças e Adultos, Hospitais da Universidade de Coimbra, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal
4. Department of Surgery A, Hospitais da Universidade de Coimbra, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal; Clínica Universitária de Cirurgia III, Faculty of Medicine, University of Coimbra, Coimbra, Portugal; and Center for Investigation on Environment, Genetics and Oncobiology (CIMAGO), Faculty of Medicine, University of Coimbra, Coimbra, Portugal
Received 2017-9-19; Accepted 2017-12-21; Published 2018-1-8
Abstract
Ischemia/reperfusion (I/R) injury in liver transplantation can disrupt the normal activity of mitochondria in the hepatic parenchyma. This potential dysfunction of mitochondria after I/R injury could be responsible for the initial poor graft function or primary nonfunction observed after liver transplantation. Thus, determining the mechanisms that lead to human hepatic mitochondrial dysfunction might contribute to improving the outcome of liver transplantation. Furthermore, early identification of novel prognostic factors involved in I/R injury could serve as a key endpoint to predict the outcome of liver grafts and also to promote the early adoption of novel strategies that protect against I/R injury. Here, we briefly review recent advances in the study of mitochondrial dysfunction and I/R injury, particularly in relation to liver transplantation. Next, we highlight various pharmacological therapeutic strategies that could be applied, and discuss their relationship to relevant mitochondrion-related processes and targets. Lastly, we note that although considerable progress has been made in our understanding of I/R injury and mitochondrial dysfunction, further investigation is required to elucidate the cellular and molecular mechanisms underlying these processes, thereby identifying biomarkers that can help in evaluating donor organs.
Keywords: Liver transplantation, Mitochondria, Ischemia/reperfusion injury, Liver preservation solution, Pharmacological conditioning
Introduction
Ischemia/reperfusion (I/R) injury is a multifactorial process by which cellular damage is initiated in organs during hypoxia, after which cells are then stressed by restoration of oxygen delivery and rebalancing of pH. This phenomenon is a major factor underlying the injury that occurs in liver surgery, mostly during liver transplantation (LT), and remains a source of major complications affecting perioperative morbidity and mortality. Consequently, it is critical to clarify the molecular mechanisms and regulatory processes involved in organ damage after I/R injury, a complex process that comprises a cascade of events that promote inflammation and tissue damage, including energy loss, generation of reactive oxygen species (ROS), release of cytokines and chemokines, and, finally, activation of immune cells [1-5].
In I/R injury, one of the most notable features is the deterioration of mitochondrial function coupled with subsequent adjustment of energy metabolism. During ischemia, the absence of oxygen leads to cessation of oxidative phosphorylation (which plays a crucial role in energy production), heightened generation of ROS, and initiation of apoptosis [6].
Currently, to treat patients with end-stage liver diseases or irreversible tumors of hepatic origin, LT is an established therapeutic regimen. However, an obstacle to LT is related to the lack of a donor pool; consequently, the mortality rate among LT waiting-list patients has been estimated to exceed 20%. This shortage has encouraged the adoption of extended criteria for selecting donor organs; however, these organs are particularly susceptible to I/R injury [7-9]. In LT, functional and structural damage caused to donor organs by the process of cold preservation/warm reperfusion are major problems, and these can result in a non-functional graft or primary graft dysfunction [10, 11].
To understand liver damage caused by I/R injury, characterizing mitochondrial activity after I/R is critical. The early identification of the cellular and molecular changes that occur might allow the adoption of new strategies that protect against I/R injury, and thus help maintain mitochondrial function and liver energy balance.
Liver Transplantation
LT has developed over the past six decades from an experimental procedure to the standard of care for patients with end-stage liver disease. In LT, the long‑term outcome has been improved as a result of advances in surgical techniques, the subsequent immunosuppressive regimens, in donor liver selection, and in postoperative care. However, during the past few decades, the number of patients awaiting an organ for transplantation has increased [7, 12, 13], and this has necessitated the extension of the criteria for organ donation and the use of marginal donors previously considered inadequate for LT (e.g. allowing for an increase in the age considered suitable for donors, the use of organs after prolonged cold ischemia, or donation after cardiac death or hepatic steatosis) [14]. Notably, the risk of primary graft nonfunction after the transplant of fatty donor organs is markedly higher than that after non-steatotic grafts (60% vs 5%).
Severe macrovesicular steatosis (> 60%) has been linked with > 60% risk of primary nonfunction after transplantation, and this has been calculated to be responsible for the rejection of 25% of donor livers [15, 16]. As a consequence of the shortage of donors, the MELD score (Model for End-Stage Liver Disease score) was adopted in 2002. The MELD score is used to predict the 3-month mortality from the patient's liver disease, and it was adopted worldwide to help select patients from the recipient waiting list that should receive specific donor organs [17]. Selection of the correct donor, particularly in living donor LT, is critical to increase the survival of the graft and the recipient [18].
Diagnosing pre-existing liver disease is a crucial part of donor organ evaluation, and histopathological examination plays an essential role in this analysis and in the assessment of the donor liver. However, despite efforts to improve the quality of the donor liver pool, some of the LT patients will develop initial poor function and primary nonfunction [19, 20].
Currently, the three most common indications for LT are hepatocellular carcinoma, hepatitis C virus infection, and alcoholic cirrhosis. In this regard, other indications are also used, such as those for acute fulminant liver failure (e.g. acute acetaminophen overdose, mushroom poisoning, fulminant hepatitis A or B infection, Wilson's disease, acute Budd-Chiari syndrome, or failed LT), cholestatic liver disease (e.g. primary biliary cirrhosis), and metabolic disorders (e.g. α-1-antitrypsin deficiency, non-alcoholic fatty liver disease) [21, 22].
In LT, the outcome is potently affected by liver preservation, which is one of the most critical component steps of LT [23]. The standard practice of liver preservation involves the use of preservation solutions at low temperatures (2-4 °C) under static, cool preservation conditions. In the 1980s, Belzer and Southard designed the University of Wisconsin (UW) solution, which is probably the most commonly used static preservation solution employed under hypothermic conditions, wherein the organ is perfused with cool preservation solution and held on ice; this has become the prevalent method for liver allograft preservation. The UW solution features an intracellular-type electrolyte composition, and to prevent tissue edema, the solution contains three inhibitory molecules: lactobionate, raffinose, and colloidal hydroxyethyl starch [24]. Conversely, in the histidine-tryptophan-ketoglutarate (HTK) solution, whose potassium concentration is slightly lower than that of the UW solution, the main impairment molecule is the amino acid histidine, and the HTK solution does not contain a colloid [25]. Another preservation solution, Celsior, which was developed in early 1990, contains histidine, a low concentration of glutathione, and incorporates lactobionate and mannitol as inhibitors. Celsior and the HTK solution are considerably less viscous than the UW solution [26].
Lastly, in clinical LT, the application of ex vivo machine preservation/perfusion is currently under investigation, and various temperatures (hypothermia or normothermia) and diverse preservation solutions are being tested. The development of new techniques will likely lead to an alteration in the manner in which organs are perfused, preserved, and transported [27-29].
Ischemia/Reperfusion
I/R injury induces damage to a hypoxic organ after oxygen delivery is restored, and this might occur in several clinical situations, such as trauma, hemorrhage, shock, thermal injury, transplantation, and certain types of liver surgery. In contrast to the ischemia under such clinical conditions, cold ischemia is exclusively related to the transplant setting.
The specific period of cold ischemia (used to reduce metabolic activities of the graft) begins when a donor graft is harvested using a cold perfusion solution and ends after the tissue reaches the physiological temperature during the implantation procedures. The cold ischemia process is followed by a period of warm ischemia, which ends with the completion of surgical anastomosis after blood-flow restoration [30, 31]. Inevitably, this step is responsible for the major part of the LT injury and the development of graft failure that is coupled with considerable morbidity and mortality in patients [32] (Figure 1).
Schematic timeline of the liver transplantation phases of I/R injury.
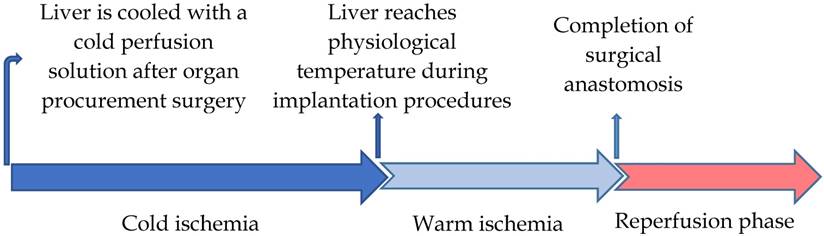
The cellular and molecular mechanisms of I/R injury are poorly understood; however, the injury is recognized to affect hepatocytes and biliary epithelial cells. The I/R injury caused by cold ischemia and warm ischemia can produce common and specific effects on various subsets of cells. For example, sinusoidal endothelial cells are more susceptible to the effects of cold preservation than are hepatic parenchymal cells. In the remaining viable endothelial cells, the expression of adhesion molecules is affected, and this accentuates the I/R injury (Figure 2).
I/R injury caused by cold ischemia and warm ischemia can produce common and specific effects on various subsets of cells. For example, sinusoidal endothelial cells are more susceptible than hepatic parenchymal cells to the effects of cold preservation, and the reperfusion phase amplifies the ischemic injury with the preferential involvement of the hepatic parenchymal cells; A- space of Disse; B- sinusoid; C- sinusoidal endothelial cells (with fenestrae); D- biliary canaliculus; E- Stellate cell; F- Kupffer cell; H- hepatocyte; N- nucleus.
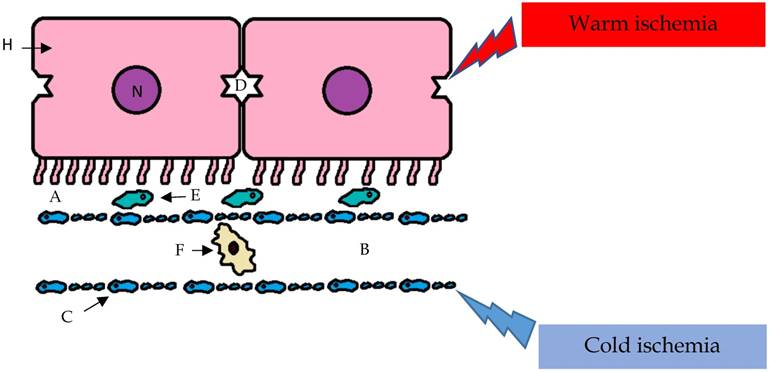
Conversely, in the reperfusion phase, ischemic injury can be amplified with the preferential involvement of hepatocytes. During this period, the generation of ROS causes nonspecific oxidative damage to lipids, proteins, and DNA [33].
Two distinct phases follow reperfusion: an early phase that lasts for the first 2 h after reperfusion; and a late phase that extends from 6 to 48 h after reperfusion.
The early phase is characterized by the activation of immune cells and oxidative stress. In the initial stages, the activation of Kupffer cells leads to ROS generation, which causes moderate hepatocellular injury. This oxidative damage is then increased as a consequence of the release of several proinflammatory chemokines and cytokines (e.g. tumor necrosis factor (TNF)‑α, and interleukin (IL)-12 and IL-1β), and this promotes and amplifies the later secondary inflammatory phase [34, 35].
The occurrence of the late phase is mediated by neutrophils, whose involvement depends on the chemokines released in the early stage. These neutrophils release proteases and other cytotoxic enzymes (e.g. collagenase, elastase, cathepsin G, and heparanase) that act within cellular membranes and on matrix components, thereby promoting cellular degradation [36, 37].
Mitochondrial Activity and I/R
Mitochondrial activity is involved in the I/R process, and the change in in this parameter might be critical for I/R injury. The most crucial change induced by I/R injury is related to the deterioration of mitochondrial function and the consequent alteration in energy metabolism.
In cold ischemia, oxygen deprivation and metabolite reduction lead to a reduction in the natural function of the mitochondrial respiratory chain and in ATP synthesis; this results in failure of ATP-dependent enzymes and a concomitant rise in ADP, AMP, and Pi concentrations, coupled with the consequent disturbances in membrane ion translocation and cytoskeletal disruption. During this period, any ATP that is produced is used to preserve the mitochondrial membrane potential, and the ATP yield from glycolysis is insufficient [36,37].
During ischemia, an increase in the intracellular concentrations of H+, Na+, and Ca2+ causes mitochondrial dysfunction. This increase in Na+ is associated with ATP depletion, which inhibits Na+/K+ ATPases. The increased Na+ concentration promotes Ca2+ influx through the Na+-Ca2+ exchanger, which is responsible for the irreversible cell injury that occurs. The intracellular Ca2+ increase associated with Ca2+-ATPase failure mainly affects sinusoidal endothelial cells [38].
The source of ROS generation during hepatic I/R remains unclear; however, it might involve complexes I and III of the electron transport chain or possibly xanthine/xanthine oxidase. ROS promote the peroxidation of the components of the phospholipids (unsaturated fatty acids) of the inner mitochondrial membrane, and this disrupts the electron flow through the electron transport chain. Moreover, during the reperfusion phase, the damage caused to mitochondrial lipids and proteins enhances ROS generation. If the tissue damage occurs for only a short time, mitochondria can repair themselves and continue to generate ATP; however, if a critical period is exceeded, mitochondrial recovery is not possible [39].
During mitochondrial damage, once mitochondrial permeability transition (MPT) has been permanently initiated, the mitochondrial inner membrane collapses, which enables solutes with a molecular mass of up to 1.5 kDa to cross the inner membrane. MPT promotes the release of certain apoptotic factors (such as cytochrome c) from the mitochondrial intermembrane space into the cytosol through channels formed by Bax (a proapoptotic Bcl-2 family member). After I/R, the predominant type of cell death is necrosis, but the onset of MPT can induce apoptosis in the ischemic liver [4, 40-42]. MPT is a common pathway leading to both types of cell death after I/R: necrosis and either apoptosis or necroptosis.
Damaged mitochondria are cleared through the selective autophagy process of mitophagy, a catabolic pathway that favors cell survival by preserving energy levels and preventing the accumulation of damaged mitochondria and cytotoxic mitochondrial subproducts [43, 44]. At least two types of mitophagy exist: the phosphatidylinositol-3-kinase-dependent and -independent types [45]. In normoxia or short ischemia, the demand for mitophagy is negligible because only a few mitochondria are damaged. By contrast, in prolonged ischemia and reperfusion, the increase in Ca2+ and ROS levels induce numerous damaged mitochondria, which must be rapidly removed via mitophagy to prevent autophagy failure caused by the increase in the number of injured mitochondria [46].
Mitochondrial Targeting Strategies against I/R Injury in Liver Transplantation
I/R is a multifactorial process and the animal models used to study it have limitations; thus, most of the animal studies on I/R have not translated to human trials [4]. In the literature, multiple therapeutic strategies against hepatic I/R injury have been reported. Furthermore, numerous experimental investigations have suggested that the use of various drugs (synthetic and natural derivatives) could prevent or reduce the injury related to I/R; however, despite these efforts, no 'optimal' drug has been identified to date. Nevertheless, the strong implication of mitochondrial involvement in I/R injury justifies a careful analysis of various available therapeutic options in relation to their effects on mitochondrial function. Diverse therapeutic approaches have been attempted thus far, including those involving the storage process (cold storage, machine perfusion), manual conditioning, and multiple pharmacological conditioning. These approaches can promote a reduction in I/R injury, which indicates the importance of the relationship between mitochondrial activity and the mitigation of I/R injury.
With regard to the aforementioned relationship, the most important mitochondrion-related processes and targets are the following: (1) MPT onset, (2) calcium channel inhibition, (3) autophagy, (4) antioxidants, (5) nitric oxide (NO), (6) TNF-α, (7) apoptosis, and (8) nucleic acids as drugs (Figure 3).
Mitochondrion-related processes and targets
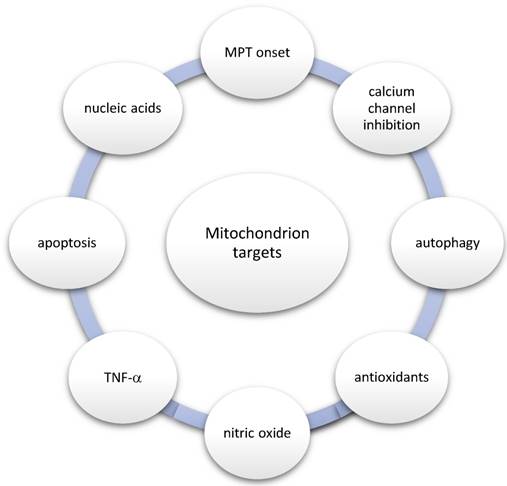
MPT is a phenomenon involved in calcium signaling and cell destruction. A previous study showed that MPT inhibition with cyclosporine A reduced mitochondrial ROS production in response to calcium [47]. Moreover, Mg2+, acidic pH, and trifluoperazine were shown to prevent the opening of permeability transition pores; whereas calcium, inorganic phosphate, alkaline pH, and ROS were shown to promote the onset of MPT [48].
Mitochondrial Ca2+ accumulation and overloading are responsible for the cell abnormality associated with I/R injury. In one study, pretreatment with the calcium-channel blocker amlodipine restored cellular normality and counteracted the alteration in mitochondrial enzymes induced by I/R injury [49]. In another study, the calcium-channel inhibitor, 2-aminoethoxydiphenyl borate, prevented Ca2+ overloading, cytochrome c release, and cell death during I/R [50].
Mitochondrial autophagy can play a protective role in liver I/R injury [51]. Heme oxygenase-1 can prevent liver I/R injury by suppressing inflammation and eliciting an antiapoptotic response, and inhibition of this enzyme reduced autophagy and upregulated apoptosis [52]. Furthermore, autophagy inhibition aggravated starvation-induced ROS accumulation, which contributed to hepatocyte necrosis [53].
The deleterious effects produced by ROS could potentially be reduced using antioxidants. For example, mangafodipir trisodium, a powerful antioxidant, exerts a protective effect when administrated to the donor before organ harvesting [54]. Furthermore, herbal antioxidants, such as green tea catechins, tetrandrine, quercetin, and trans-resveratrol can efficiently reduce I/R injury and could act directly as antioxidants and indirectly through the activation of Nrf2 [55-57]. Another example is glutathione, a crucial molecule in the cell's defense against oxidative stress, and N-acetylcysteine, a glutathione precursor, might help to maintain or replenish hepatic glutathione stores [58]. Pretreatment with N-acetylcysteine can improve glutathione homeostasis, enhance ATP regeneration, and increase survival [59].
Mitochondria reduce nitrite to NO, and this is usually sufficient to inactivate redox-active iron ions. NO is a signaling mediator involved in numerous cellular activities, such as the regulation of microcirculation and the inhibition of caspase activity in apoptosis pathways [60]. Nitrite protects against I/R injury and improves mitochondrial function by inhibiting the iron-mediated oxidative reactions that occur as a consequence of the release of iron ions during hypoxia [61]. During liver I/R injury, the protective effects of NO, including the potentiation of hepatic ATP levels, reduce oxidative damage and alleviate the adverse effects of endothelin. However, the safe therapeutic window of NO is limited because large amounts of NO can damage liver tissue [62, 63].
TNF-α is a proinflammatory cytokine that plays a major role in hepatocyte apoptosis and triggers apoptotic liver damage. In mitochondria, TNF-α induces the formation of MPT pores, the release of cytochrome c, and the activation of caspases [64-66]. In animal models, TNF-α induces apoptotic liver injury only when hepatocyte-specific transcription is inhibited, whereas in the absence of this inhibition, it protects against liver damage. Thus, TNF-α preconditioning with low doses of TNF-α or the blockade of TNF-α action (e.g. with anti-TNF-α antibodies) prevents hepatocellular apoptosis and liver injury [67].
As a consequence of I/R injury, the mitochondrial respiratory chain is disrupted, and this can lead to ATP loss and initiation of apoptosis through caspase activation and cytochrome c release. Cyclosporine A treatment could serve as a promising adjunct therapeutic approach, because cyclosporine A limits the activation of the apoptotic machinery by inhibiting MPT [68]. Moreover, supplementation with dibutyryl-cAMP could promote the inhibition of mitochondrial apoptosis by stimulating the cAMP second-messenger signaling pathway and subsequently reducing the release of cytochrome c into the cytosol [69].
Current data indicate that circulating microRNAs could serve as non-invasive biomarkers because of their association with liver diseases and liver injury. Farid et al. demonstrated that serum levels of microRNAs (e.g., miR-122) increased before an elevation of transaminase levels [70]. This could represent a critical finding because the currently used biochemical blood parameters related to liver disease/injury and dysfunction are nonspecific [71].
Some studies relate the role of mitochondria with the genetic regulation by RNA interference[72-75](Figure 4).
Interaction between the regulation of gene expression by RNA interference due to the presence of pre-mature (pre-miRNA) and mature (miRNA) microRNAs and the mitochondria system
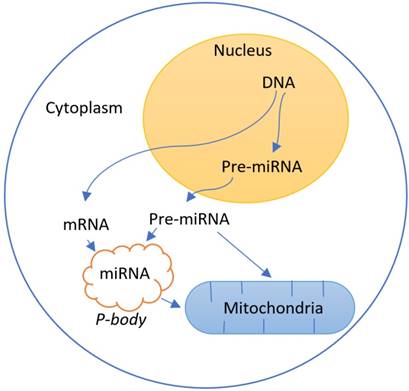
The use of nucleic acids as drugs represents the ultimate therapy [76]. RNA interference (RNAi) is a biological process in which RNA molecules neutralize targeted mRNA molecules by inhibiting gene expression or translation. Several options are available for synthetic and expressed RNAi. The most commonly used form of synthetic RNAi involves the use of small interfering RNAs (siRNAs), which occur naturally in the cytoplasm or are synthesized outside and then introduced into the cell. Intraportal administration of siRNAs targeting caspase-8 and caspase-3 promoted a reduction in lesions induced in the liver by warm I/R via RNAi-mediated inhibition of the expression of caspase-8 and caspase-3, which are both components of the apoptotic process [77]. Other RNAi therapies that have been applied to prevent I/R injury targeted IL-1β/nuclear factor kappa B (NF- κB) (transcription-related factors), Fas cell surface death receptor (Fas) and acid sphingomyelinase (ASMase) (apoptosis), and adiponectin (oxidative stress)[78-80]. Recent advances in nanomedicine have led to progress in the design of RNA/DNA drug-delivery systems, such as the development of a multifunctional envelope-type nano device that can control intracellular trafficking in specific cells in vivo and enables drug targeting to the mitochondrial system [81, 82]. MITO-Porter is a specific delivery system to mitochondria that allows the introduction of macromolecules cargoes into mitochondria. To date, this system was used to delivery antisense oligo-RNA with functional effect on mitochondria[83, 84].
One of the therapeutic approaches that has attracted the most attention recently is the use of machine perfusion. The first randomized controlled trial comparing normothermic machine perfusion with cold storage revealed that machine perfusion is safe and can preserve liver function outside the body for 24 h. Moreover, using this technique, liver function can be assessed, including bile production and clearance of lactic acidosis [85]. In the future, it is believed that it should be possible to assess specific miRNAs during organ preservation to evaluate the potential liver injury related to the I/R process and the RNAi that might be active during normothermic preservation or the reperfusion phase.
Conclusions and Future Perspectives
Although considerable effort has been devoted to studying I/R injury, the molecular and cellular mechanisms involved in this process remain incompletely determined and require further investigation.
In evaluating the quality of donor organs for LT, a critical aspect could be the identification of biomarkers. For example, microRNAs have been established as key posttranscriptional regulators in the liver, and could be used in LT as valuable biomarkers and potential therapeutic targets.
To improve the outcome of the LT, pharmacological agents could be added to the preservation solutions used for the donor liver. Although this has been extensively investigated using animal models, few clinical trials have been conducted, because most of these studies were conducted in unrealistic conditions without the potential to be translated for clinical use [86].
The development of new mitochondrial drug delivery systems could be helpful to use some of these mitochondrial targets directly into the mitochondria [87].
Nowadays, the most promisor's mitochondrion-related targets are the antioxidant agents or caspase inhibitors, which are being studied in Phase II trials [88].
Lastly, because I/R injury is a multifactorial process, it will probably be necessary to perform studies to assess the results of treatment with emergent pharmacological drugs that act on multiple therapeutic targets. Translational research could represent a solution to increase the donor liver pool and improve the outcome of LT [89, 90].
Acknowledgements
This work was supported by Sociedade Portuguesa de Transplantação (SPT), Astellas Pharma and Centro de Investigação do Meio Ambiente, Genética e Oncobiologia (CIMAGO). JST is a recipient of a Portuguese Fundação para a Ciência e a Tecnologia (FCT) post-doctoral Grant (SFRH/BPD/94036/2013).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jassem W, Fuggle SV, Rela M, Koo DD, Heaton ND. The role of mitochondria in ischemia/reperfusion injury. Transplantation. 2002;73:493-9
2. Jassem W, Battino M, Cinti C, Norton SJ, Saba V, Principato G. Biochemical changes in transplanted rat liver stored in University of Wisconsin and Euro-Collins solutions. The Journal of surgical research. 2000;94:68-73
3. Jaeschke H, Woolbright BL. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplantation reviews. 2012;26:103-14
4. Go KL, Lee S, Zendejas I, Behrns KE, Kim JS. Mitochondrial Dysfunction and Autophagy in Hepatic Ischemia/Reperfusion Injury. BioMed research international. 2015;2015:183469
5. Varela AT, Rolo AP, Palmeira CM. Fatty liver and ischemia/reperfusion: are there drugs able to mitigate injury? Current medicinal chemistry. 2011;18:4987-5002
6. Jassem W, Heaton ND. The role of mitochondria in ischemia/reperfusion injury in organ transplantation. Kidney international. 2004;66:514-7
7. Plin C, Tillement JP, Berdeaux A, Morin D. Resveratrol protects against cold ischemia-warm reoxygenation-induced damages to mitochondria and cells in rat liver. European journal of pharmacology. 2005;528:162-8
8. Wertheim JA, Petrowsky H, Saab S, Kupiec-Weglinski JW, Busuttil RW. Major challenges limiting liver transplantation in the United States. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11:1773-84
9. Singal AK, Guturu P, Hmoud B, Kuo YF, Salameh H, Wiesner RH. Evolving frequency and outcomes of liver transplantation based on etiology of liver disease. Transplantation. 2013;95:755-60
10. Clavien PA, Harvey PR, Strasberg SM. Preservation and reperfusion injuries in liver allografts. An overview and synthesis of current studies. Transplantation. 1992;53:957-78
11. Strasberg SM, Howard TK, Molmenti EP, Hertl M. Selecting the donor liver: risk factors for poor function after orthotopic liver transplantation. Hepatology. 1994;20:829-38
12. Castaldo ET, Chari RS. Liver transplantation for acute hepatic failure. HPB: the official journal of the International Hepato Pancreato Biliary Association. 2006;8:29-34
13. Jia JJ, Li JH, Jiang L, Lin BY, Wang L, Su R. et al. Liver protection strategies in liver transplantation. Hepatobiliary & pancreatic diseases international: HBPD INT. 2015;14:34-42
14. Elwood D, Pomposelli JJ. Hepatobiliary surgery: lessons learned from live donor hepatectomy. The Surgical clinics of North America. 2006;86(vii):1207-17
15. He S, Atkinson C, Evans Z, Ellett JD, Southwood M, Elvington A. et al. A role for complement in the enhanced susceptibility of steatotic livers to ischemia and reperfusion injury. J Immunol. 2009;183:4764-72
16. Limanond P, Raman SS, Lassman C, Sayre J, Ghobrial RM, Busuttil RW. et al. Macrovesicular hepatic steatosis in living related liver donors: correlation between CT and histologic findings. Radiology. 2004;230:276-80
17. Bernardi M, Gitto S, Biselli M. The MELD score in patients awaiting liver transplant: strengths and weaknesses. Journal of hepatology. 2011;54:1297-306
18. Dirican A, Baskiran A, Dogan M, Ates M, Soyer V, Sarici B. et al. Evaluation of Potential Donors in Living Donor Liver Transplantation. Transplantation proceedings. 2015;47:1315-8
19. Drebber U, Dienes HP. [Histopathology in liver transplantation]. Der Pathologe. 2008;29:84-92
20. Markin RS, Wisecarver JL, Radio SJ, Stratta RJ, Langnas AN, Hirst K. et al. Frozen section evaluation of donor livers before transplantation. Transplantation. 1993;56:1403-9
21. O'Leary JG, Lepe R, Davis GL. Indications for liver transplantation. Gastroenterology. 2008;134:1764-76
22. Mukherjee S, Mahmoudi TM, Mukherjee U. Liver transplant for viral hepatitis and fulminant hepatic failure. Minerva gastroenterologica e dietologica. 2009;55:83-100
23. Briceno J, Marchal T, Padillo J, Solorzano G, Pera C. Influence of marginal donors on liver preservation injury. Transplantation. 2002;74:522-6
24. Jamieson NV, Sundberg R, Lindell S, Claesson K, Moen J, Vreugdenhil PK. et al. Preservation of the canine liver for 24-48 hours using simple cold storage with UW solution. Transplantation. 1988;46:517-22
25. Steininger R, Roth E, Holzmuller P, Reckendorfer H, Grunberger T, Sperlich M. et al. Comparison of HTK- and UW-solution for liver preservation tested in an orthotopic liver transplantation model in the pig. Transplant international: official journal of the European Society for Organ Transplantation. 1992;5(Suppl 1):S403-7
26. Tolba RH, Akbar S, Muller A, Glatzel U, Minor T. Experimental liver preservation with Celsior: a novel alternative to University of Wisconsin and histidine-tryptophan-alpha-ketoglutarate solutions? European surgical research Europaische chirurgische Forschung Recherches chirurgicales europeennes. 2000;32:142-7
27. Perera T, Mergental H, Stephenson B, Roll GR, Cilliers H, Liang R. et al. First human liver transplantation using a marginal allograft resuscitated by normothermic machine perfusion. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2016;22:120-4
28. Schlegel A, Dutkowski P. Role of hypothermic machine perfusion in liver transplantation. Transplant international: official journal of the European Society for Organ Transplantation. 2015;28:677-89
29. Fontes P, Lopez R, van der Plaats A, Vodovotz Y, Minervini M, Scott V. et al. Liver preservation with machine perfusion and a newly developed cell-free oxygen carrier solution under subnormothermic conditions. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015;15:381-94
30. McLaren AJ, Friend PJ. Trends in organ preservation. Transplant international: official journal of the European Society for Organ Transplantation. 2003;16:701-8
31. Bahde R, Spiegel HU. Hepatic ischaemia-reperfusion injury from bench to bedside. The British journal of surgery. 2010;97:1461-75
32. Duval M, Plin C, Elimadi A, Vallerand D, Tillement JP, Morin D. et al. Implication of mitochondrial dysfunction and cell death in cold preservation-warm reperfusion-induced hepatocyte injury. Canadian journal of physiology and pharmacology. 2006;84:547-54
33. Ligeret H, Brault A, Vallerand D, Haddad Y, Haddad PS. Antioxidant and mitochondrial protective effects of silibinin in cold preservation-warm reperfusion liver injury. Journal of ethnopharmacology. 2008;115:507-14
34. Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection. Journal of gastroenterology and hepatology. 2003;18:891-902
35. Jaeschke H, Mitchell JR. Use of isolated perfused organs in hypoxia and ischemia/reperfusion oxidant stress. Methods in enzymology. 1990;186:752-9
36. Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and Kupffer cells. Hepatology. 1998;27:507-12
37. Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32:169-73
38. Inserte J, Garcia-Dorado D, Hernando V, Barba I, Soler-Soler J. Ischemic preconditioning prevents calpain-mediated impairment of Na+/K+-ATPase activity during early reperfusion. Cardiovascular research. 2006;70:364-73
39. Li J, Wang F, Xia Y, Dai W, Chen K, Li S. et al. Astaxanthin Pretreatment Attenuates Hepatic Ischemia Reperfusion-Induced Apoptosis and Autophagy via the ROS/MAPK Pathway in Mice. Marine drugs. 2015;13:3368-87
40. Ghribi O, DeWitt DA, Forbes MS, Herman MM, Savory J. Co-involvement of mitochondria and endoplasmic reticulum in regulation of apoptosis: changes in cytochrome c, Bcl-2 and Bax in the hippocampus of aluminum-treated rabbits. Brain research. 2001;903:66-73
41. Grunenfelder J, Miniati DN, Murata S, Falk V, Hoyt EG, Kown M. et al. Upregulation of Bcl-2 through caspase-3 inhibition ameliorates ischemia/reperfusion injury in rat cardiac allografts. Circulation. 2001;104:I202-6
42. Borutaite V, Jekabsone A, Morkuniene R, Brown GC. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. Journal of molecular and cellular cardiology. 2003;35:357-66
43. Lee S, Kim JS. Mitophagy: therapeutic potentials for liver disease and beyond. Toxicological research. 2014;30:243-50
44. Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends in cell biology. 2007;17:422-7
45. Czaja MJ, Ding WX, Donohue TM Jr, Friedman SL, Kim JS, Komatsu M. et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131-58
46. Kim JS, Nitta T, Mohuczy D, O'Malley KA, Moldawer LL, Dunn WA Jr. et al. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008;47:1725-36
47. Kim JS, Ohshima S, Pediaditakis P, Lemasters JJ. Nitric oxide: a signaling molecule against mitochondrial permeability transition- and pH-dependent cell death after reperfusion. Free radical biology & medicine. 2004;37:1943-50
48. Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y. et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochimica et biophysica acta. 1998;1366:177-96
49. Pronobesh C, Dagagi AV, Pallab C, Kumar WA. Protective role of the calcium channel blocker amlodipine against mitochondrial injury in ischemia and reperfusion injury of rat liver. Acta pharmaceutica. 2008;58:421-8
50. Nicoud IB, Knox CD, Jones CM, Anderson CD, Pierce JM, Belous AE. et al. 2-APB protects against liver ischemia-reperfusion injury by reducing cellular and mitochondrial calcium uptake. American journal of physiology Gastrointestinal and liver physiology. 2007;293:G623-30
51. Wang JH, Ahn IS, Fischer TD, Byeon JI, Dunn WA Jr, Behrns KE. et al. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology. 2011;141:2188-99 e6
52. Wang Y, Xiong X, Guo H, Wu M, Li X, Hu Y. et al. ZnPP reduces autophagy and induces apoptosis, thus aggravating liver ischemia/reperfusion injury in vitro. International journal of molecular medicine. 2014;34:1555-64
53. Sun K, Xie X, Liu Y, Han Z, Zhao X, Cai N. et al. Autophagy lessens ischemic liver injury by reducing oxidative damage. Cell & bioscience. 2013;3:26
54. Ben Mosbah I, Mouchel Y, Pajaud J, Ribault C, Lucas C, Laurent A. et al. Pretreatment with mangafodipir improves liver graft tolerance to ischemia/reperfusion injury in rat. PloS one. 2012;7:e50235
55. Hassan-Khabbar S, Cottart CH, Wendum D, Vibert F, Clot JP, Savouret JF. et al. Postischemic treatment by trans-resveratrol in rat liver ischemia-reperfusion: a possible strategy in liver surgery. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2008;14:451-9
56. Liang R, Nickkholgh A, Kern M, Schneider H, Benzing S, Zorn M. et al. Green tea extract ameliorates reperfusion injury to rat livers after warm ischemia in a dose-dependent manner. Molecular nutrition & food research. 2011;55:855-63
57. Firuzi O, Miri R, Tavakkoli M, Saso L. Antioxidant therapy: current status and future prospects. Current medicinal chemistry. 2011;18:3871-88
58. Nakano H, Boudjema K, Alexandre E, Imbs P, Chenard MP, Wolf P. et al. Protective effects of N-acetylcysteine on hypothermic ischemia-reperfusion injury of rat liver. Hepatology. 1995;22:539-45
59. Fukuzawa K, Emre S, Senyuz O, Acarli K, Schwartz ME, Miller CM. N-acetylcysteine ameliorates reperfusion injury after warm hepatic ischemia. Transplantation. 1995;59:6-9
60. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological reviews. 2007;87:315-424
61. Dungel P, Perlinger M, Weidinger A, Redl H, Kozlov AV. The cytoprotective effect of nitrite is based on the formation of dinitrosyl iron complexes. Free radical biology & medicine. 2015;89:300-10
62. Miyake T, Yokoyama Y, Kokuryo T, Mizutani T, Imamura A, Nagino M. Endothelial nitric oxide synthase plays a main role in producing nitric oxide in the superacute phase of hepatic ischemia prior to the upregulation of inducible nitric oxide synthase. The Journal of surgical research. 2013;183:742-51
63. Liu P, Xu B, Spokas E, Lai PS, Wong PY. Role of endogenous nitric oxide in TNF-alpha and IL-1beta generation in hepatic ischemia-repefusion. Shock. 2000;13:217-23
64. Neuman MG. Apoptosis in diseases of the liver. Critical reviews in clinical laboratory sciences. 2001;38:109-66
65. Kirillova I, Chaisson M, Fausto N. Tumor necrosis factor induces DNA replication in hepatic cells through nuclear factor kappaB activation. Cell growth & differentiation: the molecular biology journal of the American Association for Cancer Research. 1999;10:819-28
66. Bradham CA, Qian T, Streetz K, Trautwein C, Brenner DA, Lemasters JJ. The mitochondrial permeability transition is required for tumor necrosis factor alpha-mediated apoptosis and cytochrome c release. Molecular and cellular biology. 1998;18:6353-64
67. Sass G, Shembade ND, Haimerl F, Lamoureux N, Hashemolhosseini S, Tannapfel A. et al. TNF pretreatment interferes with mitochondrial apoptosis in the mouse liver by A20-mediated down-regulation of Bax. Journal of immunology. 2007;179:7042-9
68. Plin C, Haddad PS, Tillement JP, Elimadi A, Morin D. Protection by cyclosporin A of mitochondrial and cellular functions during a cold preservation-warm reperfusion of rat liver. European journal of pharmacology. 2004;495:111-8
69. Akbar S, Minor T. Significance and molecular targets of protein kinase A during cAMP-mediated protection of cold stored liver grafts. Cellular and molecular life sciences: CMLS. 2001;58:1708-14
70. Farid WR, Pan Q, van der Meer AJ, de Ruiter PE, Ramakrishnaiah V, de Jonge J. et al. Hepatocyte-derived microRNAs as serum biomarkers of hepatic injury and rejection after liver transplantation. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2012;18:290-7
71. Dirksen K, Verzijl T, Grinwis GC, Favier RP, Penning LC, Burgener IA. et al. Use of Serum MicroRNAs as Biomarker for Hepatobiliary Diseases in Dogs. Journal of veterinary internal medicine. 2016;30:1816-23
72. Barrey E, Saint-Auret G, Bonnamy B, Damas D, Boyer O, Gidrol X. Pre-microRNA and mature microRNA in human mitochondria. PloS one. 2011;6:e20220
73. Huang L, Mollet S, Souquere S, Le Roy F, Ernoult-Lange M, Pierron G. et al. Mitochondria associate with P-bodies and modulate microRNA-mediated RNA interference. The Journal of biological chemistry. 2011;286:24219-30
74. Das S, Bedja D, Campbell N, Dunkerly B, Chenna V, Maitra A. et al. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PloS one. 2014;9:e96820
75. Jagannathan R, Thapa D, Nichols CE, Shepherd DL, Stricker JC, Croston TL. et al. Translational Regulation of the Mitochondrial Genome Following Redistribution of Mitochondrial MicroRNA in the Diabetic Heart. Circulation Cardiovascular genetics. 2015;8:785-802
76. Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Advanced drug delivery reviews. 2011;63:152-60
77. Contreras JL, Vilatoba M, Eckstein C, Bilbao G, Anthony Thompson J, Eckhoff DE. Caspase-8 and caspase-3 small interfering RNA decreases ischemia/reperfusion injury to the liver in mice. Surgery. 2004;136:390-400
78. Li X, Zhang JF, Lu MQ, Yang Y, Xu C, Li H. et al. Alleviation of ischemia-reperfusion injury in rat liver transplantation by induction of small interference RNA targeting Fas. Langenbeck's archives of surgery. 2007;392:345-51
79. Llacuna L, Mari M, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology. 2006;44:561-72
80. Massip-Salcedo M, Zaouali MA, Padrissa-Altes S, Casillas-Ramirez A, Rodes J, Rosello-Catafau J. et al. Activation of peroxisome proliferator-activated receptor-alpha inhibits the injurious effects of adiponectin in rat steatotic liver undergoing ischemia-reperfusion. Hepatology. 2008;47:461-72
81. Zhang ZX, Min WP, Jevnikar AM. Use of RNA interference to minimize ischemia reperfusion injury. Transplantation reviews. 2012;26:140-55
82. Kajimoto K, Sato Y, Nakamura T, Yamada Y, Harashima H. Multifunctional envelope-type nano device for controlled intracellular trafficking and selective targeting in vivo. Journal of controlled release: official journal of the Controlled Release Society. 2014;190:593-606
83. Yamada Y, Harashima H. MITO-Porter for Mitochondrial Delivery and Mitochondrial Functional Analysis. Handbook of experimental pharmacology. 2017;240:457-72
84. Sato Y, Nakamura T, Yamada Y, Harashima H. Development of a multifunctional envelope-type nano device and its application to nanomedicine. Journal of controlled release: official journal of the Controlled Release Society. 2016;244:194-204
85. Laing RW, Mergental H, Mirza DF. Normothermic ex-situ liver preservation: the new gold standard. Current opinion in organ transplantation. 2017;22:274-80
86. Hu C, Li L. Pre-conditions for eliminating mitochondrial dysfunction and maintaining liver function after hepatic ischaemia reperfusion. Journal of cellular and molecular medicine. 2017;21:1719-31
87. Yamada Y, Harashima H. Targeting the mitochondrial genome via a dual function MITO-Porter: evaluation of mtDNA levels and mitochondrial function. Methods in molecular biology. 2015;1265:123-33
88. Pantazi E, Bejaoui M, Folch-Puy E, Adam R, Rosello-Catafau J. Advances in treatment strategies for ischemia reperfusion injury. Expert opinion on pharmacotherapy. 2016;17:169-79
89. Klune JR, Tsung A. Molecular biology of liver ischemia/reperfusion injury: established mechanisms and recent advancements. The Surgical clinics of North America. 2010;90:665-77
90. Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11:1563-9
Author contact
![]() Corresponding author: Rui Miguel Martins, MD, Department of Surgery, Instituto Português de Oncologia de Coimbra, Av. Bissaya Barreto 98, 3000-075 Coimbra, Portugal; r23martinscom Telephone: +351-239400200
Corresponding author: Rui Miguel Martins, MD, Department of Surgery, Instituto Português de Oncologia de Coimbra, Av. Bissaya Barreto 98, 3000-075 Coimbra, Portugal; r23martinscom Telephone: +351-239400200