Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(1):46-58. doi:10.7150/ijms.20508 This issue Cite
Research Paper
Antioxydation And Cell Migration Genes Are Identified as Potential Therapeutic Targets in Basal-Like and BRCA1 Mutated Breast Cancer Cell Lines
Maud Privat1,2, Justine Rudewicz2, Nicolas Sonnier1,2,3, Christelle Tamisier2, Flora Ponelle-Chachuat1,2, Yves-Jean Bignon1,2,3 ![]()
1. Université Clermont Auvergne, Centre Jean Perrin, INSERM, U1240 Imagerie Moléculaire et Stratégies Théranostiques, F-63000 Clermont Ferrand, France
2. Département d'Oncogénétique, Centre Jean Perrin, F-63000 Clermont Ferrand, France
3. Biological Resources Center BB-0033-00075, Centre Jean Perrin, F-63000 Clermont Ferrand, France
Received 2017-9-19; Accepted 2017-10-11; Published 2018-1-1
Abstract
Basal-like breast cancers are among the most aggressive cancers and effective targeted therapies are still missing. In order to identify new therapeutic targets, we performed Methyl-Seq and RNA-Seq of 10 breast cancer cell lines with different phenotypes. We confirmed that breast cancer subtypes cluster the RNA-Seq data but not the Methyl-Seq data. Basal-like tumor hypermethylated phenotype was not confirmed in our study but RNA-Seq analysis allowed to identify 77 genes significantly overexpressed in basal-like breast cancer cell lines. Among them, 48 were overexpressed in triple negative breast cancers of TCGA data. Some molecular functions were overrepresented in this candidate gene list. Genes involved in antioxydation, such as SOD1, MGST3 and PRDX or cadherin-binding genes, such as PFN1, ITGB1 and ANXA1, could thus be considered as basal like breast cancer biomarkers. We then sought if these genes were linked to BRCA1, since this gene is often inactivated in basal-like breast cancers. Nine genes were identified overexpressed in both basal-like breast cancer cells and BRCA1 mutated cells. Amongst them, at least 3 genes code for proteins implicated in epithelial cell migration and epithelial to mesenchymal transition (VIM, ITGB1 and RhoA).
Our study provided several potential therapeutic targets for triple negative and BRCA1 mutated breast cancers. It seems that migration and mesenchymal properties acquisition of basal-like breast cancer cells is a key functional pathway in these tumors with a high metastatic potential.
Keywords: Basal-like breast cancer, BRCA1, RNA-Seq, cell migration, antioxydation
Introduction
Basal like breast cancers (BLBC) represent between 10 and 20% of breast cancers. They are associated with an aggressive phenotype, high histological grade, poor clinical behavior, and high rates of relapse (1). This cancer subgroup is characterized by lack of estrogen receptor (ER), progesterone receptor (PR), and HER2 amplification (TNBC: triple-negative breast cancers) with expression of basal cytokeratins 5/6, 14, 17, epidermal growth factor receptor (EGFR), and/or c-KIT. Currently, BLBC lack any specific targeted therapy, due to the fact that they do not express ER or HER2 and thus are typically refractory to endocrine therapy and to trastuzumab, a humanized monoclonal antibody that targets HER2.
The identification of new markers and therapeutic targets is thus necessary for this bad prognosis cancer type. First, BRCA1-associated BC are mostly BLBC (2) and sporadic BLBC (occurring in women without germline BRCA1 mutations) often show dysfunction of the BRCA1 pathway. The characteristics of hereditary BRCA1-associated BC found in sporadic BLBC cancers have thus been termed «BRCA-ness» with potential clinical implications (3). As BRCA1 pathway may be deficient in BLBC, these tumors may respond to specific therapeutic regimens, such as inhibitors of the poly (ADP-ribose) polymerase (PARP) enzyme (4). Cells deficient in BRCA1 have indeed a defect in the repair of DNA double strand breaks which could make them particularly sensitive to the chemotherapy drugs that generate such breaks, such as inhibitors of PARP enzyme. However, not all BLBC are associated with BRCA1 inactivation.
Then, EGFR could represent a therapeutic target as it is often overexpressed in BLBC. Recently, a phase II clinical trial showed good results (57% of pathological complete response) of panitumumab combined with an anthracycline/taxane-based chemotherapy in operable triple-negative breast cancer (5). Nevertheless, this study highlighted biological signatures correlated with treatment response. Heterogeneity of triple negative breast cancers requires subtyping in order to better identify molecular-based therapy. In 2006 already, Neve et al. separated BLBC cell lines in two subgroup (basal A and basal B) with different invasive properties (6). Lehmann et al. then identified 6 triple-negative breast cancer subtypes including 2 basal-like (BL1 and BL2), an immunomodulatory (IM), a mesenchymal (M), a mesenchymal stem-like (MSL) and a luminal androgen receptor (LAR) subtype (7).
All these subclassification of triple negative breast cancers were identified by studying transcriptomic profiles. Epigenetic modifications in breast cells could also allow identifying characteristics of these breast cancers. Roll et al. reported a hypermethylator phenotype in BLBC, characterized by methylation-dependent silencing of CEACAM6, CDH1, CST6, ESR1, GNA11, MUC1, MYB, SCNN1A, and TFF3 genes that are involved in a wide range of neoplastic processes relating to tumors with poor prognosis (8).
Our results of Methyl-Seq did not confirm this hypermethylator phenotype but we could not identify hypermethylated BLBC specific genes. On the other hand the RNA-Seq data allowed us to identify antioxidation and cell migration as specifically activated pathways in basal-like breast cancer cells.
Materials and methods
Biological material
The main characteristics of the cell lines used are presented in table 1. MDA-MB-231 and HCC1937 human breast cancer cell lines were purchased from the American Type Culture Collection (Rockville, MD, USA) and were grown in RPMI medium supplemented with 10% foetal calf serum, 2 mM L-glutamine and 20 μg/ml gentamicin. SUM149 and SUM1315 human breast cancer cell lines were obtained from Asterand (Hertfordshire, UK) and grown in Ham's F12 medium according to the manufacturer's instructions. SUM1315MO2 cells were transfected with a pLXSN plasmid containing the full-length BRCA1 cDNA using Fugene 6 transfection reagent (Roche Molecular Biochemicals). Control cells were transfected with the pLXSN empty vector. After selection in 721.5 µM G418 (Sigma Aldrich), clones were tested for BRCA1 expression by Western blotting (9). All cell lines were grown at 37˚C in a humidified atmosphere containing 5% CO2. All our cell lines are stored and managed by the CJP Biological Resources Center (BB-0033-00075).
Main characteristics of the cell lines.
| Cell line | Site of origin | Pathology | Molecular type (6) | Triple negative subtype (7) | BRCA1 status | TP53 status |
|---|---|---|---|---|---|---|
| MCF10A | Normal breast | Fibrocystic | Basal B | - | Wild type | Wild Type |
| MCF7 | Pleural effusion | Adenocarcinoma | Luminal | - | Wild type | Wild Type |
| T47D | Pleural effusion | Adenocarcinoma | Luminal | - | Wild type | Missense mutation |
| MDA231 | Pleural effusion | Adenocarcinoma | Basal B | MSL | Wild type | Missense mutation |
| MDA436 | Pleural effusion | Adenocarcinoma | Basal B | MSL | 5382insC | Nonsense mutation |
| HCC1937 | Primary tumor | Infiltrating ductal carcinoma | Basal A | BL1 | 5396 + 1G>A | Nonsense mutation |
| SUM149 | Primary tumor | Inflammatory breast carcinoma | Basal B | BL2 | 2288delT | Missense mutation |
| SUM1315 | Skin metastasis | Infiltarting ductal carcinoma | Basal B | - | 185delAG | Missense mutation |
| SUM1315-LXSN (SL) | Skin metastasis | Infiltarting ductal carcinoma | Basal B | - | 185delAG | Missense mutation |
| SUM1315-BRCA1 (SB) | Skin metastasis | Infiltarting ductal carcinoma | Basal B | - | 185delAG + sauvage | Missense mutation |
Cell immunohistochemistry
Cells were fixed in Preservcyt solution (Thinprep) and cytoblocks were prepared with Shandon Cytoblock kit (Thermo Scientific). Hormone receptors (ER and PR), HER2, EGFR and cytokeratin status were studied as already described (5). The immunostainings were scored semi-quantitatively by an expert pathologist under a light upright microscope.
Nucleic acid extraction
DNA extraction was performed using the QIAamp DNA mini kit (Qiagen) for cell lines and the QIAamp DNA micro kit (Qiagen) for tumors. RNA extraction was performed using RNeasy mini kit (Qiagen). The quantity and quality of the nucleic acids obtained were measured spectrophotometrically at 260nm and 280nm. RNA were also checked on 2100 Bioanalyzer (Agilent Technologies).
RNA sequencing and data processing
First, mRNA were purified using Oligotex mRNA mini kit (Qiagen). cDNA libraries were then generated following the GS-FLX Titanium cDNA Rapid Library Preparation Method Manual (Roche). Finally, emPCR amplification and 454 sequencing were performed according to the manufacturer's protocol (emPCR Amplification Manual- Lib-L LV and Sequencing Method Manual-GS FLX Titanium Series, Roche). RNA-Seq data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-5465.
Sequence reads were aligned on the human genome (hg19) with GS Reference Mapper software (Roche) and mapped on the human exome using a home-made software named AGSA. Data was then normalized by calculating the 'reads per kilo base per million mapped reads' (RPKM) for each gene. When the RPKM value was below the threshold of 0.3, then it was considered as background noise and replaced by zero.
Validation of gene regulation by q-RT-PCR
Total RNAs were extracted from cell lines using RNeasy mini kit (Qiagen) according to the manufacturer's protocol. Quality of RNAs was checked using the 2100 BioAnalyzer (Agilent Technologies). Five microgram RNA was then reverse-transcribed using First-strand cDNA synthesis kit (GE Healthcare). Multiplex quantitative RT-PCR was performed using a 7900HT Fast Real-Time PCR System (Applied Biosystems).
Predesigned and validated gene-specific probe-based Taq-Man Gene Expression Assays were used and relative gene expression was determined using the comparative threshold cycle method. Ribosomal 18S was chosen as the endogenous control gene.
Methyl-DNA sequencing and data processing
First, DNA was fragmented by nebulization during 1min30sec at 2.1 bar of nitrogen pressure. After DNA purification using Qiaquick PCR purification kit (Qiagen), methylated DNA was captured using MethylCap kit (Diagenode) following the supplier recommendations. Libraries were generated using GS FLX Titanium Rapid Library Preparation Kit (Roche). Finally, emPCR amplification and 454 sequencing were performed according to the manufacturer's protocol (emPCR Amplification Manual- Lib-L LV and Sequencing Method Manual-GS FLX Titanium Series, Roche). Methyl-Seq data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-5468.
Sequence reads were aligned on the human genome (hg19) with GS Reference Mapper software (Roche) and mapped on the human proximal promotors using a home-made software named AGSA. Data was then normalized by calculating the 'reads per million mapped reads' (RPM) for each gene. When the RPM value was below the threshold of 0.3, then it was considered as background noise and replaced by zero.
TCGA data analysis
Both clinical and RNA sequencing data (Illumina HiSeq RNAseq Version 2 data) of invasive breast cancers were downloaded from The Cancer Genome Atlas (TCGA) database. A total of 449 patients with information on ER, PR and HER2 status were selected to compare the expression profiles of genes in the respective tumors. 71 cases were found to have a negative ER, PR end HER2 phenotype (i.e., triple-negative), whereas 371 cases were positive for at least one of these receptors.
Statistical analysis
Statistical analysis of data was performed using R software. Significant differences between cell line groups were sought by Wilcoxon test. Statistical overrepresentation test were performed using PANTHER classification system (10). For TCGA data, Student's t-test was used to assess statistical differences in mean normalized expression between triple negative and non triple negative groups. A p-value ≤ 0.05 was considered statistically significant.
Results
Data normalization
Our transcriptome data consisted of 16,008 genes before and 13,168 genes after RPKM normalization. Methylome data consisted of 6,140 genes before and 6,109 genes after RPM normalization. Eliminated genes are those whose expression or methylation, for each cell line, did not exceed the background noise.
For the transcriptome, the average standard of each cell line data was much higher than the median and the third quartile revealing a very high concentration of data around zero and a very large number of reads for some genes as observed in the box plots (Fig 1 and Table 2). Regarding the methylome, median and third quartile were zero while the average was between 0.8 and 1.8 readings per gene revealing again a very high concentration of data around zero and a very large number of reads for some genes. In addition, for the transcriptome and methylome, all cell lines displayed a strong deviation above the average of the number of reads per gene. This revealed a very high dispersion of data.
Data standardization. The distribution of transcriptome and methylome data are represented in boxplot. For most of the cell lines, the distribution has many values close to zero and a minority of extreme values.
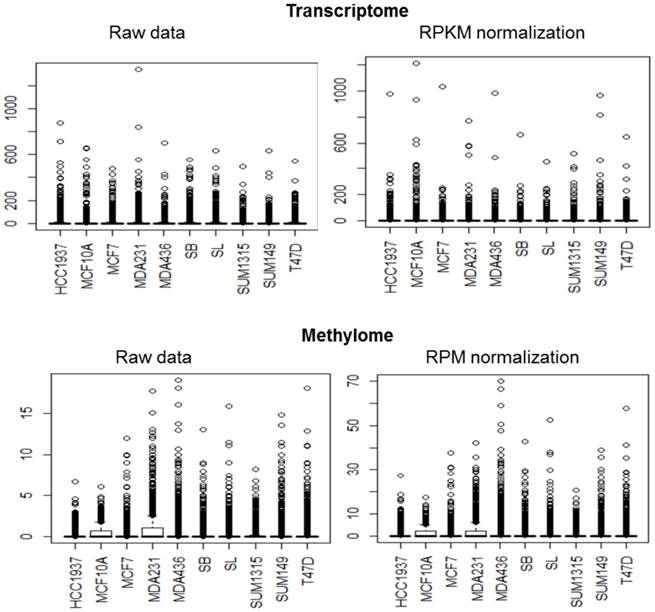
Summary of transcriptome and methylome data.
| MCF10A | MCF7 | T47D | MDA231 | MDA436 | HCC1937 | SUM149 | SUM1315 | SL | SB | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Transcriptome (RPKM) | Mean | 3.08 | 2.11 | 2.12 | 2.87 | 2.07 | 2.99 | 2.58 | 2.24 | 1.91 | 2.01 |
| Median | 0.55 | 0.53 | 0.58 | 0.57 | 0.53 | 0.60 | 0.58 | 0.54 | 0.48 | 0.49 | |
| 3rd quartile | 1.56 | 1.47 | 1.58 | 1.76 | 1.45 | 1.82 | 1.63 | 1.46 | 1.37 | 1.43 | |
| Maximum | 1213.98 | 1033.06 | 645.80 | 774.17 | 982.73 | 975.89 | 965.45 | 517.51 | 453.00 | 664.35 | |
| Standard deviation | 21.12 | 11.91 | 9.980 | 14.65 | 12.31 | 14.55 | 15.56 | 11.65 | 8.33 | 9.43 | |
| Methylome (RPM) | Mean | 0.95 | 0.89 | 0.95 | 1.53 | 1.34 | 0.88 | 0.83 | 0.75 | 0.92 | 0.76 |
| Median | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 3rd quartile | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Maximum | 17.56 | 37.71 | 57.87 | 42.33 | 70.12 | 27.30 | 39.14 | 20.69 | 52.60 | 42.95 | |
| Standard deviation | 1.77 | 2.08 | 2.70 | 3.06 | 3.90 | 1.99 | 2.33 | 1.61 | 2.33 | 2.18 |
Means, medians, 3rd quartile, maximum values and standard deviations of the normalized data RPKM (transcriptome) and RPM (methylome) are presented for each cell line.
From RPKM or RPM data, log normalization was performed to dilate the low values and strengthen high values. Standardization was also performed to obtain a standardized normal distribution. These two normalizations could be coupled to give reduced centered log normalized data.
Non supervised analysis
First we investigated how breast cancer cell lines clustered by RNA-Seq and Methyl-Seq.
For RNA-Seq, a hierarchical clustering on the RPKM normalized RNA-Seq data was generated from Euclidean distances according to Ward's method (Fig 2). This hierarchical clustering was performed for the base matrix (Fig 2A), the log normalized data (Fig 2B), the standardized data (Fig 2C) and the log-standardized data (Fig 2D). This showed that whatever the normalization, the SUM1315 lines, SB and SL are always classified together, like the two luminal T47D and MCF7 cell lines. As already observed with RNA microarrays, transcriptomic analyse by RNA-Seq thus allowed to separate luminal and basal-like breast cancer cells.
Ascending hierarchical classification of breast cancer cell line RNA-Seq data. Classification of cell lines according to the Euclidean distances of gene expression for the basic matrix (A), log normalized (B), centered reduced (C) and log normalized and centered reduced (D).
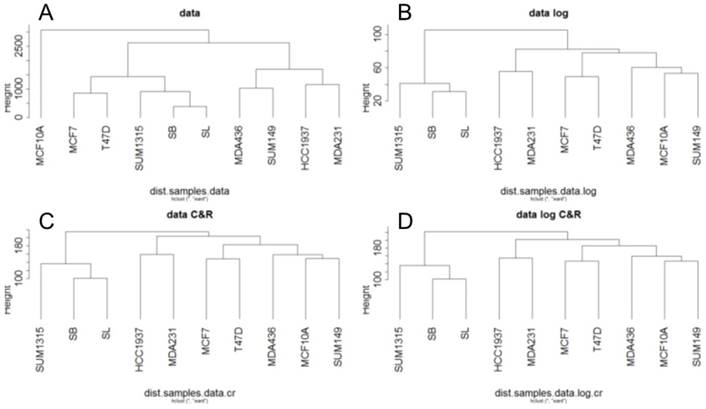
In contrast, the benign MCF10A line appears to be different from the tumor lines only for the base matrix (Fig 2A). This is in agreement with the fact that this cell line was classified as basal-like in several studies (6,11).
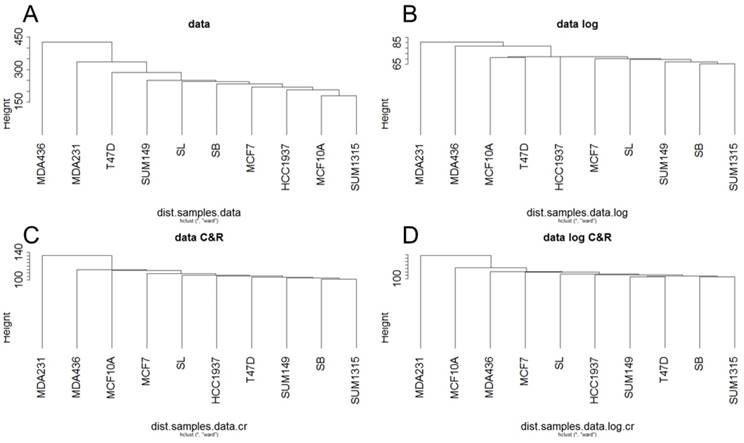
For Metyl-seq, a hierarchical clustering on the RPM normalized Methyl-Seq data was generated from Euclidean distances according to Ward's method (Fig 3). This hierarchical clustering was performed for the base matrix (Fig 3A), the log normalized data (Fig 3B), the standardized data (Fig 3C) and the log-standardized data (Fig 3D). Metyl-seq data were neither massively influenced by breast cancer subtype nor by BRCA1 mutation. It seems like MDA231 and MDA436 present a hypermetylated phenotype. These two cell lines have thus a higher mean of methylation (1.535 and 1.343 versus 0.7 to 0.95 for all the other cell lines).
Ascending hierarchical classification of breast cancer cell line Methyl-Seq data. Classification of cell lines according to the Euclidean distances of gene methylation for the basic matrix (A), log normalized (B), centered reduced (C) and log normalized and centered reduced (D).
Search for genes most significantly regulated
To determine whether the data are normally distributed, a Kolmogorov-Smirnov test was performed by taking as reference the normal distribution on basic matrix (data), normalized log (data log), centered reduced (data C & R) and centered reduced normalized log (log data C & R). For transcriptome and methylome data, the p-value was less than 2.10-16 for the four matrices. Thus, the p-value is very significantly lower the first degree risk α, set at 0.01. It is recognized that the data does not follow a normal distribution. Wilcoxon non-parametric statistical test was thus performed for each gene to select genes significantly regulated between two groups of cell lines.
Subtype: 1205 genes with p<0.05
First of all, we compared the luminal to the basal cell lines. 1205 genes were found significantly different for expression in luminal versus basal-like cell lines (p<0.05). Among them, we found well-known basal-specific genes, such as EGFR, VIM, CAV1 and CAV2 (1). Conversely, ESR1, coding for the estrogen receptor alpha, is only expressed in the two luminal cell lines. Luminal keratins (KRT8, KRT18 and KRT19) are also significantly more expressed in luminal cell lines.
For the genes used for clinical classification of basal-like breast cancers, we could compare our RNA-Seq data to immunohistochemistry results (Table 3). A very good correlation was observed between mRNA expression analysed by RNA-Seq and protein expression studied by immunocytochemistry.
Comparison of RNA-Seq and immunohistochemistry data.
| Gene | Protein | MCF10A | MCF7 | T47D | MDA231 | MDA436 | HCC1937 | SUM149 | SUM1315 | SL | SB | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RNA-Seq | ESR1 | ERalpha | 0 | 1.05 | 0.88 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| IHC | ER | - | + (90%) | + (50%) | - | - | - | - | - | - | - | |
| RNA-Seq | PGR | PR | 0 | 0 | 0.45 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| IHC | PR | - | + (40%) | + (80%) | - | - | - | - | - | - | - | |
| RNA-Seq | ERBB2 | HER2 | 0.40 | 1.31 | 0.90 | 0.46 | 0 | 0.43 | 0.36 | 0 | 0 | 0 |
| IHC | HER2 | - | - | - | - | - | - | - | - | - | - | |
| RNA-Seq | KRT5 | CK5 | 148.0 | 0 | 0 | 0 | 0 | 11.7 | 6.3 | 0 | 0 | 0 |
| KRT6A | CK6 | 102.9 | 0 | 0 | 0 | 0 | 0.8 | 8.2 | 0 | 0 | 0 | |
| KRT6B | CK6 | 6.8 | 0 | 0 | 0 | 0 | 0 | 5.1 | 0 | 0 | 0 | |
| KRT6C | CK6 | 3.7 | 0 | 0 | 0 | 0 | 0.3 | 2.4 | 0 | 0 | 0 | |
| IHC | CK5/6 | + (90%) | - | - | - | - | + (25%) | + (25%) | - | - | - | |
| RNA-Seq | KRT14 | CK14 | 76.1 | 0 | 0 | 0.3 | 0 | 5.5 | 0.4 | 0 | 0 | 0 |
| IHC | CK14 | + (80%) | - | - | - | - | + (20%) | + (<1%) | - | - | - | |
| RNA-Seq | EGFR | EGFR | 0.90 | 0 | 0 | 0.88 | 0.82 | 0.50 | 1.22 | 0.44 | 0.51 | 0.62 |
| IHC | EGFR | + (100%) | - | + (40%) | + (100%) | + (90%) | + (100%) | + (100%) | + (90%) | + (60%) | + (80%) |
RNA-Seq results are presented as RPKM values. Results of immunohistochemistry are presented as negative (-) or positive (+), specifying the percentage of labeled cells.
IHC: immunohistochemistry; ER: estrogen receptor; PR: progesteron receptor; CK: cytokeratin.
With the objective of identifying new therapeutic targets for basal-like breast cancers, we selected genes that were highly expressed (>10 RPKM) and significantly up-regulated in basal cell lines. This reduced the list to 77 candidate genes (Table 4). Thanks to the expression data extracted from the TCGA project "Breast Invasive Carcinoma", we confirmed significant overexpression for 48 genes of this list in triple negative breast tumors compared to non triple-negative breast tumors. Among them, some have already been described as basal-like markers, such as Annexin A1 (12,13) and Vimentin (14,15).
List of the 77 genes highly expressed and significantly up-regulated in basal-like breast cancer cell lines.
| MCF7 | T47D | MCF10A | MDA231 | HCC1937 | MDA436 | SUM149 | SUM1315 | SL | SB | |
|---|---|---|---|---|---|---|---|---|---|---|
| MIR198 | 0 | 14,19 | 93,95 | 774,17 | 975,89 | 982,74 | 814,74 | 517,52 | 82,52 | 70,38 |
| UQCRHL # | 58,16 | 77,51 | 430,89 | 182,15 | 184,14 | 222,29 | 352,09 | 412,39 | 223,07 | 181,27 |
| TMSB10 # | 25,42 | 9,40 | 150,63 | 508,74 | 111,48 | 85,40 | 215,62 | 244,01 | 142,27 | 192,18 |
| PFN1 # | 77,62 | 79,56 | 152,67 | 276,20 | 206,92 | 233,86 | 257,91 | 111,56 | 137,20 | 122,80 |
| S100A6 # | 22,98 | 19,45 | 416,84 | 181,76 | 87,12 | 197,18 | 213,62 | 134,53 | 77,28 | 78,92 |
| MIR638 | 24,28 | 25,53 | 56,63 | 163,59 | 310,95 | 27,49 | 26,78 | 37,29 | 45,69 | 43,01 |
| S100A2 *# | 0 | 0 | 396,33 | 1,83 | 6,93 | 1,03 | 58,95 | 13,39 | 12,77 | 4,03 |
| LGALS1 # | 4,33 | 8,97 | 94,73 | 31,84 | 40,83 | 66,74 | 88,92 | 76,64 | 45,56 | 35,35 |
| MRPL51 # | 9,48 | 6,10 | 33,88 | 40,25 | 83,61 | 53,47 | 79,59 | 53,90 | 43,90 | 33,53 |
| GADD45G IP1 # | 20,21 | 13,10 | 56,25 | 46,81 | 46,27 | 52,13 | 54,98 | 61,42 | 49,41 | 44,08 |
| VIM *# | 0,50 | 0 | 4,16 | 66,68 | 0,77 | 45,39 | 5,55 | 92,39 | 111,79 | 57,37 |
| GBA3 # | 23,01 | 13,33 | 38,60 | 63,11 | 41,76 | 26,47 | 47,50 | 44,67 | 42,65 | 43,55 |
| SF3B5 # | 21,57 | 11,73 | 60,91 | 26,86 | 30,75 | 65,28 | 22,73 | 44,05 | 40,86 | 47,65 |
| C12orf57 | 15,12 | 3,36 | 19,90 | 44,05 | 39,11 | 16,08 | 70,09 | 62,27 | 34,24 | 25,15 |
| SOD1 | 12,50 | 18,37 | 28,37 | 32,57 | 32,45 | 43,47 | 32,28 | 40,60 | 32,88 | 41,36 |
| SEP15 | 4,65 | 4,20 | 18,88 | 68,50 | 88,43 | 12,53 | 22,00 | 23,38 | 15,92 | 20,07 |
| RHOA * | 18,08 | 15,94 | 31,02 | 23,71 | 34,34 | 31,11 | 30,49 | 39,17 | 42,33 | 26,96 |
| ANXA1 # | 0 | 0,74 | 28,25 | 48,48 | 68,29 | 32,67 | 23,70 | 21,05 | 15,59 | 19,83 |
| CTSL1 | 2,72 | 1,46 | 8,66 | 5,45 | 11,97 | 42,06 | 7,19 | 97,48 | 35,75 | 48,36 |
| SEC61G # | 6,17 | 9,31 | 31,31 | 15,74 | 25,86 | 25,40 | 28,09 | 55,55 | 18,64 | 22,24 |
| PRKCDBP # | 0 | 0 | 12,95 | 110,28 | 58,21 | 1,48 | 16,03 | 5,36 | 5,97 | 11,87 |
| GPX1 | 0 | 0 | 45,87 | 21,07 | 56,00 | 26,42 | 32,33 | 6,82 | 13,15 | 10,05 |
| CAV1 | 0 | 0 | 19,90 | 9,06 | 11,40 | 12,18 | 8,70 | 62,73 | 35,85 | 44,37 |
| MIR548G | 7,94 | 0 | 17,20 | 54,05 | 17,83 | 23,58 | 26,84 | 20,86 | 19,93 | 19,37 |
| IGFBP3 # | 0 | 0 | 0,52 | 3,06 | 35,62 | 5,03 | 0,42 | 7,23 | 71,32 | 72,54 |
| MRPL18 # | 11,32 | 10,28 | 20,14 | 25,41 | 31,24 | 22,67 | 22,04 | 25,26 | 23,32 | 21,50 |
| TOMM5 # | 5,41 | 8,71 | 22,67 | 37,90 | 32,04 | 23,64 | 10,35 | 24,73 | 13,88 | 17,39 |
| NOL7 # | 5,33 | 8,45 | 10,25 | 27,78 | 22,53 | 8,94 | 11,42 | 42,43 | 28,90 | 29,18 |
| NCL # | 12,21 | 12,21 | 18,56 | 20,20 | 12,60 | 40,10 | 20,97 | 24,80 | 20,65 | 20,23 |
| LOC100134713 # | 1,05 | 0,40 | 45,26 | 32,04 | 38,63 | 13,57 | 5,78 | 18,77 | 12,09 | 9,76 |
| MIR221 | 0 | 0 | 22,90 | 18,12 | 16,71 | 20,65 | 22,83 | 31,35 | 12,88 | 30,41 |
| DRAP1 | 4,31 | 5,40 | 8,48 | 50,74 | 53,09 | 9,56 | 12,60 | 10,83 | 10,24 | 10,17 |
| PRNP # | 3,60 | 4,94 | 26,82 | 31,78 | 39,99 | 6,25 | 19,82 | 15,22 | 6,41 | 15,54 |
| SNX3 *# | 7,77 | 4,94 | 20,69 | 25,26 | 40,09 | 23,57 | 10,62 | 17,08 | 13,86 | 9,34 |
| GADD45A * | 0,78 | 1,59 | 8,91 | 31,59 | 32,62 | 17,73 | 2,67 | 20,19 | 27,45 | 12,92 |
| TM4SF1 # | 0,51 | 0 | 3,27 | 37,44 | 25,03 | 6,81 | 9,97 | 26,03 | 18,67 | 26,40 |
| MRPL34 # | 12,20 | 11,19 | 29,64 | 19,45 | 19,31 | 18,26 | 15,31 | 18,10 | 13,49 | 18,97 |
| MANF *# | 3,78 | 4,36 | 8,94 | 52,26 | 30,04 | 8,20 | 13,00 | 13,69 | 16,19 | 8,36 |
| PRDX1 # | 7,42 | 6,62 | 15,43 | 8,62 | 20,45 | 16,24 | 21,76 | 21,97 | 19,50 | 26,71 |
| MLF2 # | 9,78 | 7,32 | 13,41 | 13,15 | 26,36 | 20,47 | 23,37 | 16,44 | 19,19 | 16,94 |
| C17orf89 # | 4,00 | 5,19 | 13,21 | 32,82 | 51,74 | 7,74 | 12,94 | 8,25 | 8,05 | 11,30 |
| PARK7 | 4,73 | 9,46 | 17,09 | 28,64 | 29,03 | 11,81 | 23,99 | 12,18 | 10,79 | 12,07 |
| NDUFA1 | 3,53 | 3,62 | 27,60 | 31,00 | 16,17 | 15,70 | 17,73 | 22,27 | 5,95 | 7,62 |
| WBP5 # | 0 | 1,76 | 9,72 | 42,26 | 11,18 | 31,02 | 4,59 | 20,00 | 15,79 | 9,17 |
| MRPS33 | 9,14 | 9,83 | 34,59 | 11,75 | 17,39 | 17,43 | 16,19 | 13,81 | 15,79 | 10,16 |
| EEF1E1 # | 4,18 | 5,29 | 15,98 | 41,85 | 24,68 | 12,12 | 20,58 | 7,80 | 5,37 | 8,01 |
| EBNA1BP2 # | 3,20 | 7,18 | 9,58 | 23,10 | 18,68 | 10,83 | 9,71 | 17,32 | 26,00 | 16,38 |
| TGFBI # | 0 | 0 | 14,00 | 0,36 | 1,69 | 0,98 | 7,18 | 28,45 | 25,45 | 53,07 |
| TIMMDC1 | 4,62 | 4,31 | 7,56 | 9,96 | 45,53 | 14,17 | 12,67 | 16,41 | 6,67 | 8,83 |
| SSBP1 # | 4,59 | 4,44 | 22,03 | 17,02 | 17,64 | 12,73 | 7,92 | 17,30 | 12,90 | 11,92 |
| NDUFA6 | 4,88 | 6,14 | 22,30 | 13,72 | 20,13 | 15,85 | 15,91 | 16,97 | 7,08 | 7,03 |
| DEGS1 | 3,71 | 2,40 | 15,70 | 17,47 | 14,80 | 9,62 | 24,47 | 13,43 | 7,90 | 14,16 |
| POMP | 4,66 | 4,53 | 14,31 | 15,16 | 18,07 | 6,27 | 14,01 | 18,71 | 14,00 | 15,72 |
| FAM96B # | 2,86 | 2,49 | 7,98 | 20,27 | 37,20 | 3,58 | 15,76 | 9,45 | 14,00 | 7,22 |
| CNIH4 | 2,38 | 2,70 | 25,54 | 24,52 | 15,68 | 5,42 | 25,05 | 5,20 | 5,07 | 4,73 |
| TUBB6*# | 3,22 | 3,14 | 11,60 | 31,30 | 6,52 | 4,95 | 6,54 | 14,02 | 20,88 | 14,37 |
| MGST3 # | 2,93 | 3,59 | 28,31 | 6,61 | 13,63 | 12,62 | 10,69 | 14,69 | 12,31 | 10,71 |
| MRPL50 | 5,92 | 4,35 | 15,89 | 7,48 | 19,59 | 9,21 | 13,52 | 18,73 | 13,84 | 10,01 |
| EMP3 | 1,18 | 0,71 | 2,53 | 23,75 | 12,14 | 8,89 | 5,89 | 20,73 | 17,77 | 14,68 |
| ITGB1 * | 1,71 | 3,11 | 3,24 | 12,42 | 13,20 | 5,08 | 6,81 | 27,43 | 21,63 | 12,56 |
| CAPG # | 0,44 | 2,72 | 16,12 | 24,80 | 14,30 | 8,38 | 10,00 | 12,28 | 9,43 | 6,45 |
| ARGLU1 | 5,76 | 4,22 | 12,54 | 14,83 | 23,04 | 11,06 | 13,22 | 9,79 | 6,34 | 10,58 |
| NOP16 # | 7,04 | 3,55 | 17,47 | 11,58 | 11,93 | 11,64 | 11,67 | 17,28 | 9,52 | 10,16 |
| HIST1H2BC | 0 | 0 | 1,29 | 0,73 | 3,72 | 1,72 | 4,59 | 43,46 | 21,81 | 22,94 |
| UQCR11 | 5,03 | 2,05 | 43,52 | 7,93 | 6,62 | 6,19 | 8,99 | 10,77 | 6,97 | 8,51 |
| VKORC1L1 | 3,51 | 4,51 | 6,73 | 20,57 | 35,75 | 10,50 | 7,37 | 6,23 | 4,62 | 6,97 |
| MRPS15 # | 3,55 | 4,53 | 15,28 | 8,90 | 16,47 | 8,13 | 11,86 | 11,24 | 12,77 | 12,19 |
| UBA52 # | 4,68 | 3,17 | 18,72 | 8,21 | 8,33 | 11,36 | 14,47 | 17,47 | 8,43 | 8,56 |
| MRPS6 # | 1,55 | 3,25 | 10,22 | 6,78 | 4,26 | 8,21 | 16,38 | 26,15 | 9,38 | 12,48 |
| TRAPPC3 # | 5,10 | 4,38 | 11,16 | 13,86 | 11,94 | 7,53 | 9,19 | 14,14 | 13,21 | 12,60 |
| SSU72 # | 7,68 | 7,17 | 12,52 | 13,80 | 15,57 | 7,80 | 8,45 | 11,33 | 10,31 | 11,90 |
| RBX1 # | 1,51 | 1,99 | 8,76 | 22,44 | 22,06 | 7,59 | 9,59 | 10,66 | 2,05 | 3,22 |
| COTL1 # | 2,13 | 2,15 | 4,19 | 5,45 | 20,06 | 13,86 | 6,97 | 9,80 | 10,02 | 13,43 |
| DDT # | 3,88 | 4,63 | 29,74 | 5,75 | 6,53 | 5,77 | 9,57 | 9,91 | 7,70 | 8,61 |
| RPS12 # | 4,11 | 3,59 | 17,47 | 7,04 | 8,50 | 9,25 | 9,52 | 14,96 | 8,63 | 7,79 |
| NOP56 # | 3,23 | 4,49 | 7,52 | 20,87 | 19,58 | 7,55 | 8,79 | 4,96 | 5,14 | 7,90 |
| PRPS1L1* | 1,96 | 1,96 | 4,26 | 19,40 | 9,97 | 16,52 | 4,43 | 4,97 | 14,80 | 6,06 |
RNA-Seq results are presented as RPKM values.
* : genes that are also significantly overexpressed in BRCA1 mutated (SL) compared to BRCA1 restored (SB) cell lines.
# : genes that are significantly overexpressed in triple negative breast cancers in the TCGA RNAseq data.
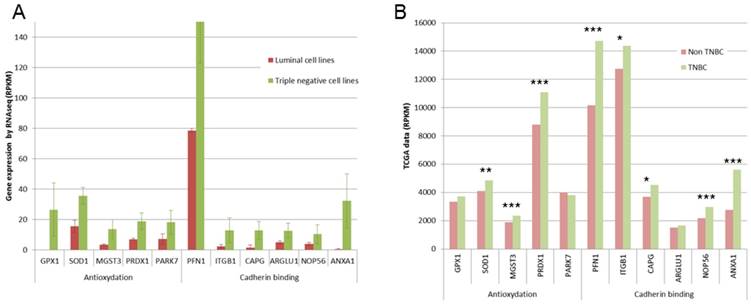
This 77 basal-like specific genes list was also submitted to Panther statistical overrepresentation test. Two molecular functions were given as overrepresented in this list (Fig 4A): antioxydant activity (5 genes, p=0.0318), and cadherin involved in cell-cell adhesions (6 genes, p=0.0231). For these 11 genes, TCGA data were compared for triple negative and non triple negative breast cancers (Fig 4B). Significant overexpression in triple negative breast cancers was found for SOD1, MGST3 and PRDX1 antioxydation genes and for PFN1, ITGB1, ARGLU1 and ANXA1 cadherin binding genes.
Expression of some genes involved in antioxydation and in cadherin binding. A. Gene expression found in our RNA-Seq study are presented as RPKM values. For all these genes, significant overexpression was observed in triple negative cell lines. B. TCGA data are presented as RPKM values. **: p <0.01; ***: p<0.001
Influence of BRCA1: specific study SL / SB: 979 genes with p<0.05
We studied our BRCA1 transfected cell lines: SL and SB come from the BRCA1 mutated SUM1315 cell line that was stably transfected with empty LXSN plasmid (SL: SUM1315-LXSN) or with a BRCA1 coding plasmid (SB: SUM1315-BRCA1). As already demonstrated (9), we could check that SB cell line expressed around 5 times more BRCA1 transcripts than SL cell line. Comparing these 2 cell line RNA sequencing, we found 979 genes significantly differently expressed. Among them, 304 genes were expressed at least twice more in SL comparing SB (Table 5). This gene list was submitted to Panther statistical overrepresentation test. The molecular functions the most overrepresented included semaphorin receptor activity (5 genes, p=0.0071), growth factor binding (12 genes, p=0.00327) and cell adhesion molecule binding (21 genes, p=0.0177).
List of the 304 genes significantly up-regulated in BRCA1 mutated cell line (SL) compared to BRCA1 wild-type cell line (SB).
| SL | SB | SL | SB | |||
|---|---|---|---|---|---|---|
| PPP1R13B | 0.480 | 0.000 | PROCR | 6.688 | 1.963 | |
| F2RL1 | 2.620 | 0.000 | UPP1 | 1.905 | 0.561 | |
| PAX8 | 0.830 | 0.000 | TADA3 | 3.076 | 0.906 | |
| FBXL16 | 0.910 | 0.000 | FURIN | 3.987 | 1.184 | |
| F2R | 0.750 | 0.000 | ZNF337 | 1.604 | 0.479 | |
| UBE2D1 | 0.660 | 0.000 | TDP1 | 1.103 | 0.331 | |
| UBASH3B | 0.900 | 0.000 | SAC3D1 | 2.500 | 0.750 | |
| SMYD4 | 0.550 | 0.000 | ZBED4 | 2.813 | 0.854 | |
| DBNDD2 | 1.540 | 0.000 | RFWD3 | 3.386 | 1.030 | |
| S1PR1 | 2.750 | 0.000 | ACTR8 | 1.420 | 0.442 | |
| PLXNB1 | 0.240 | 0.000 | ZNF467 | 1.333 | 0.417 | |
| CDC42EP3 | 4.000 | 0.000 | ATG7 | 1.053 | 0.331 | |
| DLX5 | 1.200 | 0.000 | RAB11A | 7.800 | 2.500 | |
| PLAGL2 | 2.620 | 0.000 | LEO1 | 2.505 | 0.806 | |
| TMEM164 | 0.360 | 0.000 | PTPN23 | 1.141 | 0.369 | |
| SLC25A20 | 0.610 | 0.000 | CPT1A | 1.714 | 0.560 | |
| MRE11A | 0.260 | 0.000 | TRIP11 | 1.221 | 0.399 | |
| FLRT2 | 2.000 | 0.000 | CRTC3 | 0.659 | 0.217 | |
| LOC101448202 | 0.980 | 0.000 | ENO2 | 2.669 | 0.882 | |
| NR1I3 | 0.910 | 0.000 | RPLP1 | 14.833 | 4.938 | |
| SOX9 | 1.750 | 0.000 | ZNF346 | 0.875 | 0.292 | |
| PSTK | 0.680 | 0.000 | FAM72D | 1.000 | 0.333 | |
| HOXB9 | 1.120 | 0.000 | GSE1 | 1.125 | 0.377 | |
| SPP1 | 1.000 | 0.000 | EFTUD1 | 1.548 | 0.522 | |
| NOV | 1.140 | 0.000 | PTPN1 | 1.518 | 0.513 | |
| FAM173A | 0.970 | 0.000 | TCF7 | 1.475 | 0.500 | |
| DACT1 | 1.060 | 0.000 | CSTB | 21.750 | 7.417 | |
| SQRDL | 1.040 | 0.000 | PRSS23 | 10.250 | 3.500 | |
| PRSS12 | 0.440 | 0.000 | SEC22C | 1.535 | 0.526 | |
| SERPINE2 | 1.020 | 0.000 | SNAPC1 | 2.508 | 0.863 | |
| CIB2 | 0.970 | 0.000 | TGFB2 | 1.280 | 0.442 | |
| NHLRC2 | 0.310 | 0.000 | ZDHHC9 | 1.146 | 0.396 | |
| LMBRD1 | 0.570 | 0.000 | PPP1R13L | 1.224 | 0.424 | |
| SORT1 | 0.710 | 0.000 | PINX1 | 1.670 | 0.583 | |
| AKAP12 | 1.650 | 0.000 | TOR1A | 4.850 | 1.700 | |
| RGS4 | 1.880 | 0.000 | FEM1B | 6.750 | 2.375 | |
| CACNG7 | 1.140 | 0.000 | DDAH1 | 1.058 | 0.375 | |
| ANKRD50 | 1.480 | 0.000 | SORBS3 | 3.436 | 1.222 | |
| THBS1 | 0.980 | 0.000 | TGFB1 | 3.345 | 1.190 | |
| KRTAP2-3 | 3.250 | 0.000 | LASP1 | 1.970 | 0.702 | |
| FGF5 | 7.750 | 0.000 | C11orf71 | 3.500 | 1.250 | |
| PCCA | 0.288 | 0.000 | PALM2-AKAP2 | 1.069 | 0.383 | |
| LAMB3 | 0.318 | 0.000 | VAT1 | 4.042 | 1.450 | |
| RAD51D | 1.058 | 0.000 | MBOAT2 | 0.972 | 0.350 | |
| MMD | 0.300 | 0.000 | PDZD8 | 1.621 | 0.588 | |
| EVA1A | 2.000 | 0.000 | GPC1 | 2.712 | 0.994 | |
| PLA2G16 | 1.125 | 0.000 | ZFC3H1 | 0.919 | 0.337 | |
| GTDC2 | 2.750 | 0.000 | GTF2A2 | 5.250 | 1.938 | |
| IQCC | 0.838 | 0.000 | ZNF410 | 2.091 | 0.772 | |
| TCAIM | 0.546 | 0.000 | TMEM126B | 3.375 | 1.250 | |
| DCLRE1C | 0.854 | 0.000 | TMEM5 | 2.675 | 0.992 | |
| NUP210 | 0.394 | 0.000 | TRAPPC6B | 1.225 | 0.458 | |
| SLC38A6 | 0.482 | 0.000 | ZFR | 1.473 | 0.552 | |
| EEF1A2 | 1.529 | 0.000 | MDM2 | 1.375 | 0.517 | |
| PODXL | 6.777 | 0.167 | GOLPH3 | 2.542 | 0.958 | |
| SIX1 | 3.000 | 0.125 | NINJ1 | 1.875 | 0.708 | |
| RAD18 | 0.879 | 0.050 | TNNT1 | 2.757 | 1.048 | |
| BAK1 | 1.458 | 0.083 | USP4 | 1.222 | 0.464 | |
| ATP5O | 1.167 | 0.083 | TRIP13 | 1.326 | 0.504 | |
| SPOCK1 | 1.079 | 0.083 | LRIG1 | 0.539 | 0.205 | |
| FXYD5 | 1.217 | 0.100 | ALDH5A1 | 2.469 | 0.948 | |
| ZNF12 | 1.500 | 0.125 | CSTF2T | 3.250 | 1.250 | |
| SLC19A2 | 1.700 | 0.150 | CHCHD3 | 1.263 | 0.488 | |
| HOXC10 | 1.375 | 0.125 | SIN3A | 3.334 | 1.293 | |
| TAGLN | 3.167 | 0.292 | FIBP | 2.121 | 0.826 | |
| TSR2 | 1.333 | 0.125 | TSC22D4 | 1.708 | 0.667 | |
| GEMIN2 | 0.521 | 0.050 | GINS1 | 2.704 | 1.063 | |
| FNDC1 | 1.734 | 0.167 | CD9 | 12.719 | 5.000 | |
| TAF3 | 0.646 | 0.063 | HYAL2 | 2.000 | 0.792 | |
| FAN1 | 0.831 | 0.083 | GNA13 | 3.938 | 1.563 | |
| ADCK1 | 1.417 | 0.143 | FLNB | 1.526 | 0.606 | |
| TMOD3 | 2.295 | 0.250 | ABHD6 | 0.942 | 0.375 | |
| RYBP | 1.708 | 0.188 | EZR-AS1 | 6.250 | 2.500 | |
| CTSH | 0.567 | 0.063 | NBEAL2 | 0.646 | 0.258 | |
| RAB27A | 0.750 | 0.083 | KIF7 | 1.077 | 0.431 | |
| FAM174A | 1.125 | 0.125 | YME1L1 | 5.181 | 2.073 | |
| ZBTB47 | 0.742 | 0.083 | NAP1L1 | 2.651 | 1.063 | |
| ALOX12B | 1.104 | 0.125 | SRP54 | 2.849 | 1.149 | |
| ITPKC | 0.521 | 0.063 | SEL1L | 1.385 | 0.559 | |
| CLP1 | 1.000 | 0.125 | SEC23A | 3.282 | 1.325 | |
| GCSH | 1.292 | 0.167 | EAPP | 2.067 | 0.838 | |
| ZDHHC3 | 1.746 | 0.229 | TTC8 | 1.029 | 0.418 | |
| MST4 | 0.750 | 0.100 | TAF1D | 1.963 | 0.800 | |
| FAM102A | 1.369 | 0.188 | IKBIP | 5.313 | 2.167 | |
| MORC4 | 0.876 | 0.121 | IDH2 | 2.602 | 1.064 | |
| HSBP1 | 2.958 | 0.417 | NUDT5 | 0.958 | 0.400 | |
| STC1 | 10.625 | 1.500 | RALY | 6.750 | 2.821 | |
| ATE1 | 1.598 | 0.226 | VIM | 118.611 | 49.583 | |
| ZNF672 | 1.750 | 0.250 | PHF7 | 0.893 | 0.375 | |
| UFD1L | 0.575 | 0.083 | SVIL | 1.001 | 0.421 | |
| CTNNBIP1 | 5.083 | 0.750 | MET | 2.561 | 1.084 | |
| PMEPA1 | 0.833 | 0.125 | TGFBR2 | 2.798 | 1.185 | |
| RAB8B | 0.883 | 0.133 | SMARCC1 | 1.714 | 0.733 | |
| TDRKH | 0.960 | 0.146 | ROCK1 | 0.875 | 0.375 | |
| TK2 | 1.092 | 0.167 | NFATC3 | 1.302 | 0.558 | |
| FHL1 | 0.792 | 0.125 | RRP1B | 3.012 | 1.303 | |
| PTPN2 | 1.467 | 0.244 | NUDT21 | 6.107 | 2.643 | |
| RAB13 | 2.646 | 0.458 | SEC13 | 4.444 | 1.929 | |
| HDAC9 | 0.713 | 0.125 | LARP6 | 7.625 | 3.313 | |
| ATP1A3 | 0.225 | 0.042 | ANP32B | 2.036 | 0.887 | |
| ZNF529 | 0.875 | 0.167 | TPX2 | 8.578 | 3.813 | |
| OCIAD2 | 2.833 | 0.542 | KIF5B | 5.363 | 2.392 | |
| RAP1GAP2 | 1.389 | 0.266 | DHCR24 | 2.361 | 1.054 | |
| RAB5A | 3.104 | 0.604 | ZC3H14 | 2.023 | 0.903 | |
| TTLL5 | 0.736 | 0.144 | NPC2 | 7.950 | 3.550 | |
| S100A2 | 5.625 | 1.125 | UNC45A | 4.306 | 1.924 | |
| DEDD | 3.063 | 0.625 | NHLRC3 | 0.838 | 0.375 | |
| SIMC1 | 0.700 | 0.146 | TRPC4AP | 3.223 | 1.450 | |
| RPSA | 4.771 | 1.000 | ATP6V0A2 | 0.702 | 0.317 | |
| COMMD7 | 2.185 | 0.467 | APEX2 | 2.196 | 0.992 | |
| SH3KBP1 | 1.694 | 0.371 | FBXO34 | 4.417 | 2.000 | |
| SPTLC2 | 1.413 | 0.313 | TERF2IP | 8.083 | 3.667 | |
| TMEM251 | 3.375 | 0.750 | RINT1 | 1.524 | 0.692 | |
| SRPX | 2.377 | 0.533 | SUPV3L1 | 1.802 | 0.821 | |
| EIF1AD | 1.479 | 0.333 | UBP1 | 2.618 | 1.196 | |
| ASNA1 | 5.226 | 1.192 | DARS2 | 1.533 | 0.708 | |
| THAP10 | 2.375 | 0.542 | HSD17B11 | 1.730 | 0.800 | |
| PPP3CC | 0.770 | 0.179 | MEX3B | 8.375 | 3.875 | |
| ZKSCAN1 | 1.063 | 0.250 | KIFC3 | 1.531 | 0.710 | |
| PLXDC1 | 1.908 | 0.458 | SMC2 | 1.902 | 0.883 | |
| TSPAN31 | 1.342 | 0.325 | DDX18 | 2.071 | 0.962 | |
| PTPRF | 3.353 | 0.813 | ZBTB5 | 1.792 | 0.833 | |
| TMED10 | 4.875 | 1.188 | POLG | 2.531 | 1.177 | |
| UXT | 1.188 | 0.292 | SNUPN | 3.223 | 1.500 | |
| DYNLRB1 | 4.292 | 1.063 | MFGE8 | 1.527 | 0.714 | |
| WSB2 | 1.963 | 0.492 | CNOT10 | 1.595 | 0.746 | |
| UBXN2A | 0.729 | 0.183 | ATG4B | 0.851 | 0.400 | |
| VTI1B | 2.292 | 0.583 | SLC30A6 | 1.250 | 0.590 | |
| ITPR3 | 2.743 | 0.707 | UBR7 | 1.366 | 0.650 | |
| RAB39B | 3.375 | 0.875 | RAB1A | 5.250 | 2.500 | |
| SS18 | 0.864 | 0.225 | REEP4 | 2.141 | 1.021 | |
| SLC2A1 | 2.184 | 0.579 | PGM3 | 1.866 | 0.890 | |
| CTGF | 14.850 | 3.950 | STYX | 1.004 | 0.479 | |
| PRKACA | 3.103 | 0.825 | PKM | 52.114 | 24.886 | |
| PRPS1L1 | 7.500 | 2.000 | AGPAT2 | 2.267 | 1.083 | |
| PIP4K2A | 1.003 | 0.271 | QRICH1 | 3.250 | 1.558 | |
| ITPK1 | 1.982 | 0.536 | CENPN | 2.008 | 0.964 | |
| ELK1 | 2.125 | 0.575 | BRD3 | 1.119 | 0.539 | |
| PWWP2A | 1.375 | 0.375 | SLC38A1 | 1.973 | 0.955 | |
| RAC1 | 4.458 | 1.217 | RARS | 2.777 | 1.344 | |
| SMC5 | 0.835 | 0.228 | MYBL2 | 1.482 | 0.718 | |
| RAB18 | 3.279 | 0.905 | PPM1G | 7.431 | 3.600 | |
| BAHD1 | 1.075 | 0.300 | RPL27A | 3.508 | 1.700 | |
| ZBTB1 | 6.208 | 1.750 | HDGFRP3 | 5.250 | 2.550 | |
| EPC1 | 0.990 | 0.281 | AQR | 0.596 | 0.292 | |
| VPS18 | 0.950 | 0.271 | GADD45A | 15.313 | 7.500 | |
| CHMP2B | 1.833 | 0.525 | QARS | 2.197 | 1.089 | |
| RANBP10 | 1.012 | 0.292 | NRP1 | 1.489 | 0.738 | |
| KIF3B | 2.313 | 0.667 | KIF23 | 4.444 | 2.208 | |
| PUSL1 | 1.292 | 0.375 | HK1 | 3.444 | 1.713 | |
| ADAM19 | 1.526 | 0.444 | SNX3 | 9.000 | 4.500 | |
| GORAB | 1.708 | 0.500 | TRAK1 | 2.001 | 1.001 |
RNA-Seq results are presented as RPKM values.
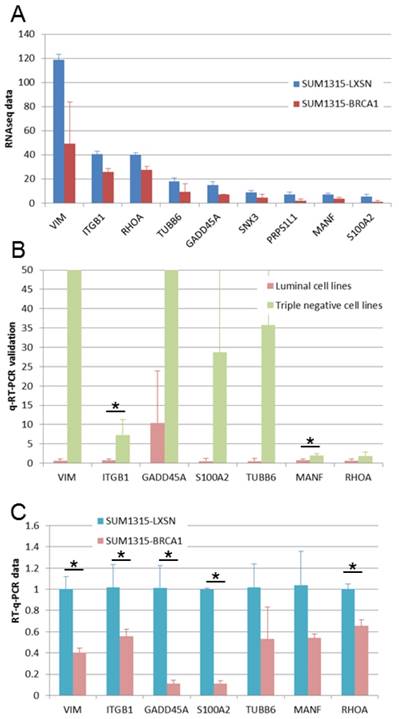
We found 9 genes that were overexpressed both in basal-like cell lines compared to luminal cell lines and in BRCA1 mutated (SL) compared to BRCA1 restored (SB) cell lines (Fig 5A). We performed q-RT-PCR experiments in order to validate these overexpressed genes. For 7 of these genes, we could validate higher expression in basal-like cell lines (Figure 5B) and in BRCA1 mutated cell line (Figure 5C). These 7 genes can be considered as basal-like biomarkers and potential therapeutic targets, particularly in BRCA1-mutated cancers. Above these genes, four are linked to cytoskeleton and could thus be implicated in epithelial cell migration: VIM, ITGB1, RHOA and TUBB6.
Expression of genes overexpressed in triple negative and in BRCA1-mutated cell lines. A. Gene expressions found in our RNA-Seq study are presented as RPKM values. For all these genes, significant overexpression was observed both in triple negative versus luminal cell lines and in BRCA1 mutated (SL) versus BRCA1 (SB) restored cell lines. B. Gene overexpression in basal-like breast cancer cell lines was confirmed by q-RT-PCR. *: p <0.05 C. Gene overexpression in BRCA1 mutated (SL) compared to BRCA1 restored (SB) cell lines was confirmed by q-RT-PCR. *: p <0.05
Discussion
Our study tested RNA-Seq and Methyl-Seq as sensitive methods to categorize breast cancer tumor cells. We found that Methyl-Seq is not an appropriate method to differentiate breast cancer subtypes. The weak quantity of data generated with our technology is a limit that could probably be overcome with the latest generations of sequencers. Nevertheless it seems that the subtypes of breast cancers do not broadly influence genome methylation. Among the genes published by Roll et al.(8), only ESR1 was found to be methylated in the MDA-MB-231 and MCF10A cell lines. We also observed a specific profile of the MDA-MB-231 and MDA-MB-436 cell lines, which appear to be globally hypermethylated. Deeper study will be needed to understand why these cell lines are hypermethylated.
By contrast, RNA-Seq was proven as a reliable method for breast cancer classification. We showed that different subtypes could be separated by global gene clustering. Moreover transcript expression analyzed by RNA-Seq was shown to correlate with protein expression evaluated by immunohistochemistry. With the steady decrease in sequencing prices, RNA-Seq could become the standard method for the molecular characterization of breast tumors. This could make it possible to propose a personalized treatment according to the therapeutic targets overexpressed in the tumor.
In our study we chose to focus on basal-like breast tumors. We thus identified some potentially new targets in this breast cancer subtype. PANTHER analysis (10) revealed that genes involved in cadherin binding were overrepresented in the basal-like overexpressed genes. For most of them, we could confirm overexpression in triple negative breast cancers thanks to the Cancer Genome Atlas data. Among them PFN1 codes for profilin1, a regulator of actin polymerization but it is known to be downregulated in breast cancers (16). In contrast, our study confirms overexpression of two genes involved in breast cancer progression, the ANXA1 and ITGB1 genes. ANXA1 was already shown to be associated with triple negative breast cancers (13,17). A recent study already identified ITGB1, that codes for integrin beta 1, as a potential prognosis biomarker in triple negative breast cancers (18).
Furthermore, genes involved in cell oxidation regulation were also found to be overexpressed in basal-like breast cancers. GPX1 codes for glutathione peroxidase 1 that protects cells against oxidative stress. A polymorphism of this gene has been associated to breast cancer risk (19). The PARK7 gene (also known as DJ-1) is involved in neuron protection against oxidative stress and cell death. It was recently shown to interact with HER3 receptor (20). For these two genes, our results showed at least a 3 fold overexpression in basal-like breast cancer cell lines but in TCGA triple negative breast tumors this overexpression was not confirmed. For SOD1, PRDX1 and MGST3, triple negative cell overexpression was observed both in our cell lines and in TCGA tumors. MGST3 is another gene belonging to antioxidant system shown to be overexpressed in some melanomas (21). Superoxyde dismutase 1 is an enzyme coded by the gene SOD1. It has already been suggested as a potential anti-cancer drug (22,23). At last, PRDX1 has a controversial role in oxidization-reduction balance and its expression seems to be up-regulated in breast cancer tissues (24). Oxydative stress could thus be targeted in basal-like breast cancers. This therapeutic strategy has already been proposed in other types of cancer (25,26).
BRCA1 is a double strand break repair gene known to be frequently inactivated in basal-like breast cancers. In our results, BRCA1 mutation does not appear to influence massively the transcriptome of breast cancer cells. Nevertheless, we looked for genes overexpressed both in basal-like and in BRCA1 mutated breast cancer cells. ITGB1 is one of these genes, BRCA1 could thus be proposed as a regulator of integrin beta 1. It could be particularly interesting to inhibit ITGB1 in BRCA1 mutated basal-like breast cancers. VIM gene codes for vimentin protein, a mesenchymal marker, and we also found it overexpressed in BLBC cell lines and in BRCA1 mutated cell line. It is also an interesting biomarker of triple negative and BRCA1 mutated breast cancers. MicroRNA-138, which targets vimentin, has thus been proposed as a therapeutic agent for breast cancer (28). RhoA and TUBB6 are also overexpressed in BRCA1 mutated and basal-like breast cancers. TUBB6 gene codes for a tubulin protein, the major constituent of microtubule cytoskeleton. RhoA is a small GTPase involved in actin cytoskeleton organization and it thus regulates cell shape and motility (27). Physical reorganization of the cytoskeleton appears to be important in BRCA1 mutated breast cancers. This ability to remodel the cellular form could explain the high metastatic capacities of these cancers. Targeting the proteins involved in this function could thus be an effective therapeutic strategy for basal type breast cancers.
Acknowledgements
The results published here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We used the PANTHER classification system to analyze RNA-Seq data (10).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Valentin MD, da Silva SD, Privat M, Alaoui-Jamali M, Bignon Y-J. Molecular insights on basal-like breast cancer. Breast Cancer Res Treat. 2012Jul;134(1):21-30
2. Waddell N, Arnold J, Cocciardi S, da Silva L, Marsh A, Riley J. et al. Subtypes of familial breast tumours revealed by expression and copy number profiling. Breast Cancer Res Treat. 2010Oct;123(3):661-77
3. Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FBL, Imholz ALT. et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer. 2013May28;108(10):2172-7
4. Lee J, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014Jan;25(1):32-40
5. Nabholtz JM, Abrial C, Mouret-Reynier MA, Dauplat MM, Weber B, Gligorov J. et al. Multicentric neoadjuvant phase II study of panitumumab combined with an anthracycline/taxane-based chemotherapy in operable triple-negative breast cancer: identification of biologically defined signatures predicting treatment impact. Ann Oncol. 2014Aug1;25(8):1570-7
6. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T. et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006Dec;10(6):515-27
7. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011Jul1;121(7):2750-67
8. Roll JD, Rivenbark AG, Sandhu R, Parker JS, Jones WD, Carey LA. et al. Dysregulation of the epigenome in triple-negative breast cancers: Basal-like and claudin-low breast cancers express aberrant DNA hypermethylation. Exp Mol Pathol. 2013;95(3):276-87
9. Privat M, Aubel C, Arnould S, Communal Y, Ferrara M, Bignon Y-J. Breast cancer cell response to genistein is conditioned by BRCA1 mutations. Biochem Biophys Res Commun. 2009Feb13;379(3):785-9
10. Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D. et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2016 gkw1138
11. Subik K, Lee J-F, Baxter L, Strzepek T, Costello D, Crowley P. et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer Basic Clin Res. 2010May20;4:35-41
12. Sobral-Leite M, Wesseling J, Smit VTHBM, Nevanlinna H, van Miltenburg MH, Sanders J. et al. Annexin A1 expression in a pooled breast cancer series: association with tumor subtypes and prognosis. BMC Med. 2015:13
13. Bhardwaj A, Ganesan N, Tachibana K, Rajapakshe K, Albarracin CT, Gunaratne PH. et al. Annexin A1 Preferentially Predicts Poor Prognosis of Basal-Like Breast Cancer Patients by Activating mTOR-S6 Signaling. PLoS ONE. 2015:10 (5)
14. Zelenko Z, Gallagher EJ, Tobin-Hess A, Belardi V, Rostoker R, Blank J. et al. Silencing vimentin expression decreases pulmonary metastases in a pre-diabetic mouse model of mammary tumor progression. Oncogene. 2016 Aug 29
15. Tanaka K, Tokunaga E, Inoue Y, Yamashita N, Saeki H, Okano S. et al. Impact of Expression of Vimentin and Axl in Breast Cancer. Clin Breast Cancer.
16. Valenzuela-Iglesias A, Sharma VP, Beaty BT, Ding Z, Gutierrez-Millan LE, Roy P. et al. Profilin1 regulates invadopodium maturation in human breast cancer cells. Eur J Cell Biol. 2015Feb;94(2):78-89
17. Sobral-Leite M, Wesseling J, Smit VTHBM, Nevanlinna H, van Miltenburg MH, Sanders J. et al. Annexin A1 expression in a pooled breast cancer series: association with tumor subtypes and prognosis. BMC Med. 2015 Jul 2;13
18. Klahan S, Huang W-C, Chang C-M, Wong HS-C, Huang C-C, Wu M-S. et al. Gene expression profiling combined with functional analysis identify integrin beta1 (ITGB1) as a potential prognosis biomarker in triple negative breast cancer. Pharmacol Res. 2016;104:31-7
19. Hu J, Zhou G-W, Wang N, Wang Y-J. GPX1 Pro198Leu polymorphism and breast cancer risk: a meta-analysis. Breast Cancer Res Treat. 2010Nov1;124(2):425-31
20. Zhang S, Mukherjee S, Fan X, Salameh A, Mujoo K, Huang Z. et al. Novel association of DJ-1 with HER3 potentiates HER3 activation and signaling in cancer. Oncotarget. 2016Aug25;7(40):65758-69
21. Bracalente C, Ibañez IL, Berenstein A, Notcovich C, Cerda MB, Klamt F. et al. Reprogramming human A375 amelanotic melanoma cells by catalase overexpression: Upregulation of antioxidant genes correlates with regression of melanoma malignancy and with malignant progression when downregulated. Oncotarget. 2016May10;7(27):41154-71
22. Che M, Wang R, Li X, Wang H-Y, Zheng XFS. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov Today. 2016Jan;21(1):143-9
23. Papa L, Manfredi G, Germain D. SOD1, an unexpected novel target for cancer therapy. Genes Cancer. 2014Jan;5(1-2):15-21
24. Ding C, Fan X, Wu G. Peroxiredoxin 1 - an antioxidant enzyme in cancer. J Cell Mol Med. 2016 Sep 1
25. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009Jul1;8(7):579-91
26. Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61(4):290-302
27. O'Connor K, Chen M. Dynamic functions of RhoA in tumor cell migration and invasion. Small GTPases. 2013Jul1;4(3):141-7
28. Zhang J, Liu D, Feng Z, Mao J, Zhang C, Lu Y. et al. MicroRNA-138 modulates metastasis and EMT in breast cancer cells by targeting vimentin. Biomed Pharmacother. 2016;77:135-41
Author contact
![]() Corresponding author: Yves-Jean.BIGNONunicancer.fr
Corresponding author: Yves-Jean.BIGNONunicancer.fr