International Journal of Medical Sciences 3.2
Impact Factor
ISSN: 1449-1907
Impact Factor
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(1):31-35. doi:10.7150/ijms.21075 This issue Cite
Research Paper
Next Generation Sequencing expression profiling of mitochondrial subunits in men with Klinefelter syndrome
Michele Salemi1 ![]() , Laura Cimino2, Marika Marino2, Rossella Cannarella2, Rosita A. Condorelli2, Corrado Romano1, Sandro La Vignera2, Aldo E. Calogero2
, Laura Cimino2, Marika Marino2, Rossella Cannarella2, Rosita A. Condorelli2, Corrado Romano1, Sandro La Vignera2, Aldo E. Calogero2
1. Oasi Institute for Research on Mental Retardation and Brain Aging (IRCCS), Troina (EN), Italy;
2. Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy.
Received 2017-5-18; Accepted 2017-10-11; Published 2018-1-1
Citation:
Salemi M, Cimino L, Marino M, Cannarella R, Condorelli RA, Romano C, La Vignera S, Calogero AE. Next Generation Sequencing expression profiling of mitochondrial subunits in men with Klinefelter syndrome. Int J Med Sci 2018; 15(1):31-35. doi:10.7150/ijms.21075. https://www.medsci.org/v15p0031.htm
Other stylesAbstract
Objectives: Klinefelter syndrome (KS) is one of the most common sex-chromosome disorders as it affects up to 1 in every 600-1000 newborn males. Men with KS carry one extra X chromosome and they usually present a 47,XXY karyotype, but less frequent variants have also been reported in literature. KS typical symptoms include tall stature, gynecomastia, broad hips, hypogonadism and absent spermatogenesis. The syndrome is also related to a wide range of cognitive deficits, among which language-based learning disabilities and verbal cognition impairment are frequently diagnosed. The present study was carried out to investigate the role of mitochondrial subunits in KS, since the molecular mechanisms underlying KS pathogenesis are not fully understood.
Methods: The study was performed by the next generation sequencing analysis and qRT-PCR assay.
Results: We were able to identify a significant down-expression of mitochondrial encoded NADH: ubiquinone oxidoreductase core subunit 6 (MT-ND6) in men with KS.
Conclusion: It is known that defects of the mtDNA encoding mitochondrial subunits are responsible for the malfunction of Complex I, which will eventually lead to the Complex I deficiency, the most common respiratory chain defect in human disorders. Since it has been shown that decreased Complex I protein levels could induce apoptosis, wehypothesizethat the above-mentioned MT-ND6 down-expression contributes to the wide range of phenotypes observed in men with KS.
Keywords: Klinefelter syndrome, MT-ND6, cognitive deficits, NGS analysis, qRT-PCR.
Introduction
Klinefelter syndrome (KS), with a prevalence of 1 in 600-1000 newborn males [1], is one of the most common sex-chromosome disorders. Men affected by KS carry one or more extra X-chromosome and the classic aneuploidy, observed in about the 80% of the cases, presents a 47,XXY karyotype. The above-mentioned karyotype arises as a consequence of a failure in disjunction of paired X-chromosomes during the first or second meiotic division, either during oogenesis or spermatogenesis. Other karyotype variations, although uncommon, have also been reported. Among them 48,XXXY, 48,XXYY, 49,XXXXY and mosaicisms (e.g. 47,XXY/46,XY) are mentioned in literature [2].
KS clinical symptoms typically include gynecomastia in late puberty, broad hips, sparse facial and pubic hair, tall stature, hypogonadism, low testosterone levels and absent spermatogenesis. Moreover, the disorder is associated with cognitive deficits, such as decreased verbal intelligence, language-based learning disabilities, verbal cognition impairment and auditory hallucinations. It has been suggested that, with the increase of supernumerary X-chromosomes, speech disabilities and cognitive impairment deepen progressively, leading to a loss of about 16 points of the intelligence quotient per extra X chromosome [3].Unfortunately, since most symptoms generally appear with a late onset and the related phenotype is prone to a considerable variability, the disorder is frequently disregarded and undiagnosed. Indeed, it has been estimated that the majority of men with KS are diagnosed after reaching adulthood and, most important, only about 26% of the affected men [4] receive a correct diagnosis. As we can infer from these data, a late diagnosis leads to severe health complications that often result into a burdensome clinical management. For these reasons, one main concern is to improve diagnostic tools in order to overcome the delay in identification of affected individuals and related issues.
To this purpose, it is important to deepen our knowledge on the molecular and cellular mechanisms underlying KS pathogenesis and to unveil the genetic aspects responsible for the disease that are still undetermined. In fact, KS is a complex and multifaceted disorder and the genetic causes that lead to it still need to be partially understood. What we currently know about the genetic background of KS is that the incidence of paternal and maternal non-disjunction during meiosis seems to be equally shared (circa 50% each) in affected men [5]. Moreover, in recent studies, a 4-fold increase in the prevalence of KS cases was showed in advanced aged mothers [>40] rather that young ones [<24]. This result suggests that advanced maternal age could represent a risk factor [6].
Additionally, evidences [7-9] point to a contribution to the disorder from X-linked genes escaping X-chromosome inactivation in the early female embryo. In particular, it is known that in the somatic cell line of females, one of the two X-chromosomes is inactivated with the purpose to provide a dosage compensation of the genes located on it to that of male cells. The random X-inactivation is ensured by the untranslated product of X-inactive-specific transcript gene (Xist), located in the long arm of the inactive chromosome. Studies have shown that, in humans, around 15% of X-linked genes escape transcription inactivation. These genes, located on the short arm of the X chromosome, would be present in a double dosage (comparable to that of females) in males affected by KS and have thus been indicated as main contributors to the syndrome's phenotype [10-12] .
Furthermore, a potential cause for spermatogenic failure in KS has been identified in the gene Testis-expressed 11 (TEX11) [13], whose expression is increased in the germ line of affected individuals. In fact, the protein encoded by the gene TEX11, found in spermatogonia and early spermatocytes testes, is involved in the suppression of cell proliferation. Hence, its overexpression in KS men could represent one of the causes for germ cell death.
Another gene whose relation to the KS phenotype has been clearly proved is the short stature homeobox-containing gene (SHOX), located in the pseudo-autosomal region 1 (PAR1) in the terminal region of Xp [14]. As shown, the over-expression of a growth-related gene such as SHOX is responsible for tall stature and long extremities, typical features of KS patients [15].
It has also been established that KS men show more frequently than controls X-linked copy number variations. It is particularly relevant that the majority of the above-mentioned CNVs are duplications and half of them interest regions enclosing genes escaping X-inactivation [16].
Furthermore, a significant progress has been made in disclosing the relation between mitochondria and male infertility [17]. As known, mitochondria play a key role in the energy production and thus a correlation between sperm motility and mitochondrial enzymatic activities has been suggested. Specifically, recent studies have shown that mutations in mitochondrial genes like ATPase6 and 8, COX II and ND1 perturb the ATP production, affecting spermatogenesis and sperm motility, and hence could be responsible for the absent spermatogenesis in males affected by KS [18, 19]. Moreover, in a population study conducted in Japan, it has been identified a mutation pattern in the hypervariable segment 1 (HV1) of mtDNA of patients affected by the disorder [20]. Of notice, the polymorphisms found in the KS affected population were rare in the normal samples. These results support the hypothesis of the involvement of mitochondria DNA inKS pathogenesis.
In the present study, we used next generation sequencing technology to investigate on whole genome expression profile in peripheral blood leukocyte samples, collected from both KS men (karyotype 47,XXY) and normal age-matched controls. Subsequently, the results were validated by quantitative real-time PCR (qRT-PCR). Of particular interest to us was gene expression in mitochondrial sub-units, towards which we focused our attention.
Materials and Methods
Selection Criteria for Patients and Controls
The study was approved by the Ethical Committee of the University teaching Hospital “Policlinico-Vittorio Emanuele (Catania, Italy), trial registration number 49/2015/PO (Register of the Ethics Committee opinions). All the participants in the study signed an informed consent.
Transcriptome analysis was performed on blood mononuclear cells (PBMCs) of 10 non-mosaic KS patients as well as 10 healthy controls of similar age. All men with KS (mean age 32.5) had 47,XXY karyotype, as confirmed by cytogenetic investigation performed on at least 50 metaphases. Control group individuals (mean age 32) had negative history of genetic diseases, normal testicular volume and normal reproductive hormones (FSH, LH, total testosterone) levels, as showed in Table 1. All men enrolled in this study (KS patients and controls) were Italian.
RNA Sequencing and Next Generation Sequencing
Total RNA was extracted using the standard RNA extraction method with TRIzol (Invitrogen, Carlsbad, CA, USA). Before use, RNA concentration in each sample was assayed with a ND-1000 spectrophotometer (NanoDrop) and its quality assessed with the Agilent 2100 Bioanalyzer via Agilent RNA 6000 nano kit (Agilent Technologies, Santa Clara, CA, USA).
Next generation sequencing experiments were performed by Genomix4life S.R.L. (Baronissi, Salerno, Italy). Indexed libraries were prepared from 1 ug/ea purified RNA with TruSeq Stranded Total RNA (Illumina) Library Prep Kit according to the manufacturer's instructions. Libraries were quantified using the Agilent 2100 Bioanalyzer (Agilent Technologies) and pooled such that each index-tagged sample was present in equimolar amounts, with final concentration of the pooled samples of 2nM. The pooled samples were subject to cluster generation and sequencing using an Illumina HiSeq 2500 System (Illumina) in a 2x100 paired-end (RNA-seq) format. The raw sequence files generated (.fastq files) underwent quality control analysis using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Data Analysis of Next Generation Sequencing
Bioinformatics analysis were performed by Genomix4Life srl (Baronissi(SA), Italy).The raw sequence files generated (fastq files) underwent quality control analysis using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and the quality checked reads were trimmed with cutadapt [21] v.1.10 and then aligned to the human genome (hg19 assembly) using STAR v.2.5.2 [22], with standard parameters. Differentially expressed mRNAs were identified using DESeq2 v.1.12 [23].
Firstly, gene annotation was obtained for all known genes in the human genome, as provided by Ensembl (GRCh37). Using the reads mapped to the genome, we calculated the number of reads mapping to each transcript with HTSeq-count v.0.6.1 [24]. These raw read counts were then used as input to DESeq2 for calculation of normalized signal for each transcript in the samples, and differential expression was reported as Fold Change along with associated adjusted p-values (computed according to Benjamini-Hochberg). Differential expression data were further confirmed using Cuffdiff (25).
Table 1
Clinical and biochemical characteristics of men with Klinefelter syndrome (KS) and controls (C).
| Patient | Age (years) | BMI (Kg/m2) | LH (mUI/ml) | FSH (mUI/ml) | T (ng/ml) | Right testicular volume (ml) | Left testicular volume (ml) |
|---|---|---|---|---|---|---|---|
| C1 | 34 | 26.2 | 7.78 | 3.5 | 6.36 | 15 | 15 |
| KS1 | 35 | 28 | 24.3 | 34.9 | 3.2 | 1 | 1 |
| C2 | 32 | 27.1 | 4.57 | 3.69 | 6.13 | 22 | 25 |
| KS2 | 32 | 39.9 | 9.9 | 19 | 2.3 | 2 | 1.5 |
| C3 | 31 | 27.3 | 4.93 | 4.5 | 6 | 18 | 18 |
| KS3 | 33 | 20.1 | 31.1 | 55 | 3.3 | 1 | 1 |
| C4 | 31 | 25.1 | 2.1 | 3.5 | 4.7 | 16 | 16 |
| KS4 | 29 | 27.3 | 15 | 25 | 2.1 | 1.5 | 1.5 |
| C5 | 36 | 23.6 | 2.1 | 3.5 | 4.7 | 16 | 16 |
| KS5 | 38 | 22 | 26.5 | 33.3 | 5.17 | 18 | 18 |
| C6 | 24 | 23 | 7.78 | 3.5 | 6.36 | 20 | 20 |
| KS6 | 23 | 17.6 | 11.6 | 13.7 | 9.03 | 5 | 5 |
| C7 | 24 | 20 | 4 | 3.2 | 10.5 | 20 | 15 |
| KS7 | 21 | 29.3 | 22.2 | 25.6 | 1.03 | 2 | 1.5 |
| C8 | 50 | 24.7 | 5.2 | 2.08 | 2.7 | 21 | 19 |
| KS8 | 48 | 30.1 | 18 | 23 | 5 | 2 | 1.4 |
| C9 | 28 | 24.5 | 7.04 | 3.84 | 5.13 | 22 | 21 |
| KS9 | 26 | 24.9 | 29.1 | 64.6 | 2.8 | 2 | 1.5 |
| C10 | 41 | 29.6 | 4.6 | 4 | 4.3 | 15 | 15 |
| KS10 | 39 | 22.5 | 17.2 | 35.6 | 2.6 | 2 | 2 |
Quantitative Real-Time PCR Expression (qRT-PCR)
To validate the results obtained via the NGS analysis of KS subjects described above, we compared gene expression in 10 KS patients and 10 normal subjects via qRT-PCR. In this step, the subjects studied in the NGS analysis were included. Once again, KS and control individuals were recruited in the study only after obtaining family and/or personal informed consent.
RNA extraction from peripheral blood leukocytes has been performed using RNeasy Mini Handbook (Qiagen Sciences, Germantown, USA), following the manufacturer's protocol.
2-ΔΔCTqRT-PCR was performed with a Light Cycler 480 software v1.5 (Roche Molecular System Inc., Pleasanton, CA, USA) on KS subjects to measure gene expression and the results were compared with controls, as previously reported by Salemi et al. (2015) [26].
Gene expression levels were then normalized to GAPDH level and target mean Cp (crossing point, cycle number at detection threshold) definition was used to indicate the mean normalized cycle threshold.
Distribution analysis of the expression values was performed using Shapiro - Wilk test and statistical analysis of results was carried out using the Wilcoxon rank-sum test. A p value <0.05 was been considered significant.
mRNA levels of the following mitochondrial subunits have been evaluated: ATP6, ATP8, CO1, CO2, CO3, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5 and MT-ND6.
Results
As described in Materials and Methods, blood mononuclear cells samples from patients with non-mosaic KS and controls were examined in an NGS transcriptome analysis. The results obtained showed that the transcription of MT-ND6 mitochondrial subunit (locus MT:14148-14673) was down-regulated in the pool of men with KS with a fold-change of 0.166 (p<0.00005).
With the purpose of validating these results, we performed a qRT-PCR, for the high sensitivity of this assay is optimal for ensuring accuracy in the evaluation of gene expression. We examined mRNA levels of mitochondrial subunits ATP6, ATP8, CO1, CO2, CO3, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5 and MT-ND6 in the 10 KS men and the 10 controls. Data acquired from this analysis confirmed the significant down-expression of MT-MD6 mitochondrial subunit in KS affected individuals compared to that of controls, as showed in Figure 1.
Discussion
The mechanisms underlying KS clinical symptoms have been extensively studied and many hypotheses have been proposed to date. Nevertheless, how the supernumerary X-chromosome affects the phenotype and determines the typical features of KS is poorly understood. As discussed in the Introduction, gene-dosage dysregulations have been largely implicated in the pathogenesis of KS [27-29], suggesting that epigenetic modifications play a pivotal role in the process. In the present study, we have shown that MT-ND6 (mitochondrially encoded NADH: ubiquinone oxido reductase core subunit 6), a mitochondrial subunit belonging to Complex I, encoded by mtDNA and thus thought to be involved in proton translocation and ubiquinone binding during the respiratory chain, is down-expressed in men with KS.
Figure 1
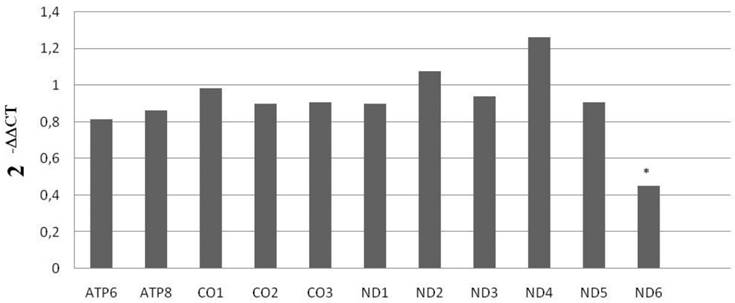
mRNA mitochondrial expression in KS subjects and normal controls. Data shown were obtained via qRT-PCR. *= p<0.00005
It has been proposed that mutations in mitochondrial subunits encoded by mtDNA, such as MT-ND6, are responsible for great defects in the assembling of Complex I [30, 31]. These malfunctions result in the Complex I deficiency, which is the most common respiratory chain defect in human disorders as it has been proved its involvement in many pathological conditions, such as adult onset neurodegenerative disorders, isolated mitochondrial myopathy and lethal infantile mitochondrial disease [32-34].
Using next generation sequencing analysis, we identified variants in the sequences of different mitochondrial subunits, but no statistically significant difference between KS and controls was observed. Therefore, the above-mentioned variants are not a factor influencing the phenotypes related to KS.
On the other hand, our results suggest that the down-expression of MT-ND6 could contribute to the etiology of KS. In fact, it has been established that decreased complex I protein levels could induce apoptosis [35] and contribute to the phenotype of men with KS.
Moreover, we can speculate that, by severely interfering with the assembling of complex I, and thus with the process of mitochondrial oxidative phosphorylation (OXPHOS), the reduced expression of MT-ND6 can cooperate with the above-mentioned gene-dosage dysregulation and epigenetic factors to establish the physical as well as psychological features typically associated with KS. Other studies, currently under development, will be necessary to determine the modalities and the extension of mitochondrial subunits involvement in KS pathogenesis.
In conclusion, this study showed a significant down-expression of the mitochondrial subunit MT-ND6 in men with non-mosaic KS. The above-mentioned down-expression could play an important role in determining the typical pathological phenotypes associated with the syndrome.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Aksglaede L, Link K, Giwercman A, Jørgensen N, Skakkebaek NE, Juul A. 47,XXY Klinefelter syndrome: clinical characteristics and age-specific recommendations for medical management. Am J Med Genet C Semin Med Genet. 2013;163C:55-63
2. Maiburg M, Repping S, Giltay J. The genetic origin of Klinefelter syndrome and its effect on spermatogenesis. Fertil Steril. 2012;98:253-60
3. Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995;96(4 Pt 1):672-82
4. Bojesen A, Kristensen K, Birkebaek NH, Fedder J, Mosekilde L, Bennett P. et al. The metabolic syndrome is frequent in Klinefelter's syndrome and is associated with abdominal obesity and hypogonadism. Diabetes Care. 2006;29:1591-8
5. Bojesen A, Gravholt CH. Klinefelter syndrome in clinical practice. Nat Clin Pract Urol. 2007;4:192-204
6. Bojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. J Clin Endocrinol Metab. 2003;88:622-6
7. Wistuba J, Luetjens CM, Stukenborg JB, Poplinski A, Werler S, Dittmann M. et al. Male 41, XXY* mice as a model for Klinefelter syndrome: hyperactivation of leydig cells. Endocrinology. 2010;151:2898-910
8. Kleinheinz A, Schulze W. Klinefelter's syndrome: new and rapid diagnosis by PCR analysis of XIST gene expression. Andrologia. 1994;26:127-9
9. Tuttelmann F, Gromoll J. Novel genetic aspects of Klinefelter's syndrome. Mol Hum Reprod 16(6): 386-395. 2010
10. Yang F, Babak T, Shendure J, Disteche CM. Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res. 2010;20:614-22
11. Berletch JB, Yang F, Disteche CM. Escape from X inactivation in mice and humans. Genome Biol. 2010;11:213
12. Carrel L, Willard HF. Heterogeneous gene expression from the inactive X chromosome: An X-linked gene that escapes X inactivation in some human cell lines but is inactivated in others. Proc Natl Acad Sci USA. 1999;96:7364-9
13. Yu YH, Siao FP, Hsu LC, Yen PH. TEX11 modulates germ cell proliferation by competing with estrogen receptor beta for the binding to HPIP. Mol Endocrinol. 2012;26:630-42
14. Helena Mangs A, Morris BJ. The human pseudoautosomal region (PAR): origin, function and future. Curr Genomics. 2007;8:129-36
15. Ottesen AM, Aksglaede L, Garn I, Tartaglia N, Tassone F, Gravholt CH, et al.Increased number of sex chromosomes affects height in a nonlinear fashion. a study of 305 patients with sex chromosome aneuploidy. Am J Med Genet. 2010;152:1206-12
16. Rocca MS, Pecile V, Cleva L, Speltra E, Selice R, Di Mambro A. et al. The Klinefelter syndrome is associated with high recurrence of copy number variations on the X chromosome with a potential role in the clinical phenotype. Andrology. 2016;4:328-34
17. Kumar DP, Sangeetha N. Mitochondrial DNA mutations and male infertility. Indian J Hum Genet. 2009;15:93-7
18. Guney AI, Javadova D, Kırac D, Ulucan K, Koc G, Ergec D. et al. Detection of Y chromosome microdeletions and mitochondrial DNA mutations in male infertility patients. Genet Mol Res. 2012;11:1039-48
19. Holyoake AJ, Sin IL, Benny PS, Sin FY. Association of a novel human mtDNA ATPase6 mutation with immature sperm cells. Andrologia. 1999;31:339-45
20. Oikawa H, Tun Z, Young DR, Ozawa H, Yamazaki K, Tanaka E. et al. The specific mitochondrial DNA polymorphism found in Klinefelter's syndrome. Biochemic Biophys Res Commun. 2002;297:341-5
21. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal. 2011;17:1
22. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S. et al. Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15-21
23. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
24. Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166-9
25. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;5:511-5
26. Salemi M, Barone C, Morale MC, Caniglia S, Romano C, Salluzzo MG. et al. Killer-specific secretory (Ksp37) gene expression in subjects with Down's syndrome. Neurol Sci. 2016;37:793-5
27. Iitsuka Y, Bock A, Nguyen DD, Samango-Sprouse CA, Simpson JL, Bischoff FZ. Evidence of skewed X-chromosome inactivation in 47,XXY and 48,XXYY Klinefelter patients. Am J Med Genet. 2001;98:25-31
28. Bruining H, van Rijn S, Swaab H, Giltay J, Kates W, Kas MJ. et al. The parent-of-origin of the extra X chromosome may differentially affect psychopathology in Klinefelter syndrome. Biol Psychiatry. 2010;68:1156-62
29. Mehta A, Malek-Jones M, Bolyakov A, Mielnik A, Schlegel PN, Paduch DA. Methylation-specific PCR allows for fast diagnosis of X chromosome disomy and reveals skewed inactivation of the X chromosome in men with Klinefelter syndrome. J Androl. 2012;33:955-62
30. Ugalde C, Triepels RH, Coenen MJ, van den Heuvel LP, Smeets R, Uusimaa J. et al. Impaired complex I assembly in a Leigh syndrome patient with a novel missense mutation in the ND6 gene. Ann. Neurol. 2003;54:665-9
31. Kirby DM, McFarland R, Ohtake A, Dunning C, Ryan MT, Wilson C. et al. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J Med Genet. 2004;41:784-9
32. Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology. 1999;52:1255-64
33. Triepels RH, Van Den Heuvel LP, Trijbels JM, Smeitink JA. Respiratory chain complex I deficiency. Am J Med Genet. 2001;106:37-45
34. Distelmaier F, Koopman WJ, van den Heuvel LP, Rodenburg RJ, Mayatepek E, Willems PH. et al. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833-42
35. Wolvetang EJ, Johnson KL, Krauer K, Ralph SJ, Linnane AW. Mitochondrial respiratory chain inhibitors induce apoptosis. FEBS Letters. 1994;339:40-44
Author contact
![]() Corresponding author: Dr. Michele Salemi, Oasi Institute for Research on Mental Retardation and Brain Aging (IRCCS), Troina, Italy. Tel: +39 935 936440; Fax: +39 935 936593; e-mail: msalemien.it
Corresponding author: Dr. Michele Salemi, Oasi Institute for Research on Mental Retardation and Brain Aging (IRCCS), Troina, Italy. Tel: +39 935 936440; Fax: +39 935 936593; e-mail: msalemien.it
Citation styles
APA
Salemi, M., Cimino, L., Marino, M., Cannarella, R., Condorelli, R.A., Romano, C., La Vignera, S., Calogero, A.E. (2018). Next Generation Sequencing expression profiling of mitochondrial subunits in men with Klinefelter syndrome. International Journal of Medical Sciences, 15(1), 31-35. https://doi.org/10.7150/ijms.21075.
ACS
Salemi, M.; Cimino, L.; Marino, M.; Cannarella, R.; Condorelli, R.A.; Romano, C.; La Vignera, S.; Calogero, A.E. Next Generation Sequencing expression profiling of mitochondrial subunits in men with Klinefelter syndrome. Int. J. Med. Sci. 2018, 15 (1), 31-35. DOI: 10.7150/ijms.21075.
NLM
Salemi M, Cimino L, Marino M, Cannarella R, Condorelli RA, Romano C, La Vignera S, Calogero AE. Next Generation Sequencing expression profiling of mitochondrial subunits in men with Klinefelter syndrome. Int J Med Sci 2018; 15(1):31-35. doi:10.7150/ijms.21075. https://www.medsci.org/v15p0031.htm
CSE
Salemi M, Cimino L, Marino M, Cannarella R, Condorelli RA, Romano C, La Vignera S, Calogero AE. 2018. Next Generation Sequencing expression profiling of mitochondrial subunits in men with Klinefelter syndrome. Int J Med Sci. 15(1):31-35.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.