Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(1):10-15. doi:10.7150/ijms.22812 This issue Cite
Research Paper
A FCGR3A Polymorphism Predicts Anti-drug Antibodies in Chronic Inflammatory Bowel Disease Patients Treated With Anti-TNF
Patricia Romero-Cara1, Daniel Torres-Moreno2,3, José Pedregosa4, Juan Antonio Vílchez4, María Sergia García-Simón5,6, Guadalupe Ruiz-Merino3,7, Senador Morán-Sanchez1,6, Pablo Conesa-Zamora3,4,5 ![]()
1. Gastroenterology Department, Santa Lucía General University Hospital (HGUSL), C/ Mezquita sn, 30202 Cartagena, Spain;
2. Pathology Department, HGUSL, Cartagena, Spain;
3. Institute for Biohealth Research from Murcia (IMIB), Cartagena, Spain;
4. Clinical Analysis Department, HGUSL. Instituto Murciano de Investigaciones Biosanitarias (IMIB-Arrixaca), Murcia, Spain;
5. Pharmacy Department, HGUSL, Cartagena, Spain;
6. Faculty of Health Sciences. Catholic University from Murcia (UCAM), Murcia, Spain;
7. Statistical Unit, Fundación para la Formación e Investigación Sanitarias (FFIS), C/ Luis Fontes Pagán 9, 30003 Murcia, Spain.
Received 2017-9-13; Accepted 2017-10-30; Published 2018-1-1
Abstract
Background. The production of anti-drug antibodies (ADAs) against IgG monoclonal antibodies (mAbs) targeting tumour necrosis factor (TNF) is an important cause of loss of response to anti-TNF mAbs in patients with inflammatory bowel diseases (IBD) such as Crohn's disease (CD) and ulcerative colitis (UC). Since receptors for the Fc portion of IgG (FCGRs) are involved in the degradation of IgG complexes, we hypothesised that a polymorphism in FCGR3A (V158F; rs396991) gene could be involved in anti-TNF ADA generation and treatment resistance. Material and Methods. A cohort of 103 IBD patients (80 CD, 23 UC) were genotyped and serum level of both anti-TNFs (infliximab or adalimumab) and ADA against them were measured. Results. No significant differences were observed between ADA occurrence or V158F genotype and type of disease or the kind of anti-TNF administrated. Interestingly, VV genotype correlated with patients producing ADA (VV: 37.5% vs. FV: 10.6% or FF: 5%; p=0.004) and was an independent predictor of this event after multivariate analysis. Moreover, VV genotype also correlated with those patients receiving anti-TNF dose intensification (p=0.03). Conclusion. FCGR3A V158F polymorphism seems to be associated with ADA production against mAbs and it could be taken into account when considering the dose and type of anti-TNF in IBD patients.
Keywords: Crohn's Disease, ulcerative colitis, infliximab, adalimumab, anti-drug antibody, pharmacogenetics.
Introduction
It is generally assumed that chronic inflammatory bowel diseases (IBD) encompassing Crohn's disease (CD) and ulcerative colitis (UC) are, to a great extent, genetically determined although a series of environmental factors also influence the susceptibility and pathophysiology of these conditions [reviewed in 1, 2]. In IBD, an increased secretion of proinflammatory cytokines such as tumour necrosis factor (TNF) in the large bowel lamina propria, plays an essential role in the initiation and propagation of the disease [3]. Therefore, it is not surprising that infliximab (IFX) and adalimumab (ADM), two anti-TNF IgG monoclonal antibodies (mAbs), have shown an increased efficacy over conventional therapies in CD and UC [4]. Although, the response to anti-TNF mAbs shows inter-individual variability [5], the decrease in serum anti-TNF and the synthesis of anti-drug antibodies (ADAs) against these biological drugs are crucial causes of loss of response. That is why a dose adjustment or shift to another anti-TNF type of drug is necessary and several studies have been carried out in order to obtain useful markers in this setting. Although the mAb mechanism of action is not entirely known, the main clearance route for these drugs is through the reticulo-endothelial system (ERS) which, in turn, depends on two cell receptors with antagonistic functions. On the one hand, the Brambell's receptor (FcRn), expressed by endothelial ERS cells, protects IgG from catabolism and increases its half-life. On the other hand, the Fc-gamma receptors (FcγRs) expressed by macrophages, NK cells and neutrophils, induce the degradation of IgG-FcγR complexes in the endolysosomes of these innate immune cells [6]. Therefore, the presentation of IgG antigens on these cells through the class II major histocompatibility complex (MHC) increases the probability for anti-IgG ADA production by activated plasma cells. In fact, a functional polymorphism (V158F) in one of the FcγR genes (FCGR3A) which affects antibody binding affinity has been associated with IFX response in CD [7] and anti-CD20 mAb response in non-Hodgkin lymphoma patients[8], although other studies could not confirm such relationships [9, 10]. Recently, it was reported that FCGR3A 158V/V genotype was associated with increased IFX elimination and risk of relapse after IFX discontinuation in CD patients [11]. However, to the best of our knowledge, no previous works have analysed the possible influence of FCGRs polymorphisms on the anti-TNF levels and ADA synthesis in IBD patients. The aim of our work was to evaluate whether V158F in FCGR3A is associated with serum levels of TNF, anti-TNF IgG mAb (IFX, ADM), ADAs against IFX and ADM or with dose intensification.
Material and Methods
Study population
The present cohort study included 103 IBD patients (80 CD, 23 UC) from Santa Lucia General University Hospital, Cartagena, Spain who were recruited between February 2014 and May 2015. Patients under anti-TNF induction phase were excluded, all study patients were receiving anti-TNF maintenance dose (IFX: 5 mg/kg every eight weeks, ADM: 40 mg from the third dose onwards every two weeks) [12, 13]. Dose intensification (every 6 weeks) was applied in 23 patients out of 66 (34.8%) for the IFX group and in 10 out of 37 patients (27%) for the ADM group (every week) following clinician criteria, which did not take into account trough levels of TNF, anti-TNF or ADA. In the intensification group, sample collection was obtained once dosage was adjusted. Patients with prior anti-TNF treatments were excluded from the study. A written informed consent was obtained from all the participants and the study was approved by the Hospital Ethics Committee being carried out in accordance to the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Routine determinations of serum hemoglobin, C reactive protein (CRP) and albumin were carried out in all patients. Demographic and clinico-pathological features of the study cases are described in Table 1. Clinical management of the patients was carried out without any knowledge of genotype or TNF/anti-TNF/ADA serum concentration results.
Demographic and clinicopathological features of the study cases
| IFX group n=66 | ADM group n=37 | Total series n=103 | ||
|---|---|---|---|---|
| Gender, n (%) | Female | 30 (45.4) | 24 (64.8) | 54 (52.4) |
| Age, mean [±SD] | Years | 44.1 [14.6] | 43.2 [11.8] | 43.7 [13.6] |
| Weight, mean [±SD] | Kg | 70.4 [15.0] | 70.0 [15.7] | 70.2 [15.2] |
| IBD disease, n (%) | CD | 49 (74.2) | 31 (83.8) | 80 (77.7) |
| UC | 17 (25.8) | 6 (16.2) | 23 (22.3) | |
| mAb Naïve, n (%) | Yes | 54 (81.8) | 20 (54) | 74 (71.8) |
| Immunomodulator, n (%) | AZA | 23 (34.8) | 13 (35.1) | 36 (35.0) |
| MCP | 2 (3) | 1 (0.9) | 3 (2.9) | |
| MTX | 3 (4.5) | 2 (5.4) | 5 (4.9) | |
| None | 38 (57.5) | 21 (20.8) | 59 (57.3) | |
| Anti-TNF intensification | Positive | 23 (34.8) | 10 (27) | 33 (32.0) |
| Duration anti-TNF, mean [± SD] | Years | 4.2 [2.3] | 3.6 [1.6] | 4.0 [2.1] |
| Albumin, mean [± SD] | mg/dL | 3.9 [0.5] | 4.1 [0.4] | 4.0 [0.5] |
| C reactive protein, mean [± SD] | mg/dL | 0.9 [1.3] | 0.6 [0.6] | 0.8 [1.1] |
| TNF, mean [± SD] | pg/mL | 283.1 [516.6] | 442.6 [362.9] | 341.1 [471.0] |
| ADA production, n (%) | Positive | 11 (16.7) | 2 (5.4) | 13 (12.6) |
| FCGR3 V158F polymorphism, n (%) | FF | 21 (31.8) | 19 (51.4) | 40 (38.8) |
| FV | 33 (50.0) | 14 (37.8) | 47 (44.6) | |
| VV | 12 (18.2) | 4 (10.8) | 16 (15.5) | |
IFX: infliximab, ADA: adalimmab, SD: Standard deviation, CD: Crohn's disease, UC: Ulcerative colitis, AZA, Azacytidine, MCP: Mercaptopurine, MTX: Methotrextate, ADA: Anti-drug antibody
Biochemical determinations
A blood sample was extracted from the patients the same day and prior to IFX or ADM infusion during anti-TNF maintenance dose. The samples were centrifuged at 2.200 g for 10 min at 4°C, and the supernatants were stored in aliquots at -80°C until further use. Serum TNF was measured by a solid-phase chemiluminescent immunometric assay using the IMMULITE 1000 analyzer (Siemens, Germany) and the IMMUNLITE TNF-α kit (measure range 1.7-1000 pg/mL; sensitivity: 1.7pg/mL; intra-assay VC: 3.2%; inter-assay VC: 5.2%) and following the purveyor's instructions.
IFX and ADM serum concentrations were measured, following the manufacturer's instructions, by two ELISA (Enzyme-linked Immunosorbent assay) immunoassays, both approved for in vitro diagnostic: Promonitor-IFX (IVD reference: 5060230000) and Promonitor-ADM (IVD ref: 5080230000) both provided by Progenika Biopharma (Bizkaia, Spain). The kit is a capture ELISA provided in microplate (96 wells) configuration with wells coated with mouse monoclonal anti-human TNFα antibodies bound to recombinant human TNF, the signal obtained being proportional to the amount of mAb in the patient sample. The cut-points selected for positivity were those provided by Promonitor® (IFX: 0.035 μg/mL, ADM: 0.024 μg/mL).
Serum concentration of ADAs against IFX and ADM were determined in a semi-quantitative manner by using the Promonitor®-anti-IFX kit and Promonitor®-anti-ADM (IVD references 5070230000 and 5090230000, respectively). These assays are binding ELISA tests in which the signal detected is proportional to the amount of antibodies directed against IFX or ADM in the patient sample. The cut-points considered were provided in the Promonitor® kit (anti-IFX: 2 RU/mL, anti- ADM: 3.5 RU/mL) and drug concentrations and ADAs samples were measured in the fully automated Triturus® system platform (Grifols, Barcelona, Spain).
FCGR3A V158F genotyping
DNA was extracted from patient buffy coat using the QiaAmp DNA Mini Kit (ref: 51306) and Qiacube automatic extractor by Qiagen (Hilden, Germany). FcGR3A V158F genotyping was performed by nested polymerase chain reaction (PCR) and restriction fragment length polymorphism (RFLP) as described previously [14] in order to selectively amplify the FCGR3A and not FCGR3B gene. Briefly, in the first PCR, 1 μl of DNA was amplified in reaction mixture of 10 μl volume, containing 0.4mM dNTPs, 3 pmol of each primer, 7.5 mM MgCl2, and 1 U Taq DNA polymerase (annealing temperature, 57ºC). The PCR product was amplified under same conditions as in previous PCR except for primers and annealing temperature (64ºC). All PCR reagents were provided by Promega (Madison, WI). Five microliters of the amplicon from the second PCR was digested with 0.5 l NlaIII (Fermentas, Vilnius, Lithuania) in 10-μl volume for 10 hours. Digestion products were 1:20 diluted visualized using the QiAxcell high-resolution DNA separation matrix (cat 929002, Qiagen). Genotype evaluation was performed without previous knowledge of clinico-pathological information.
Statistical analysis
Sample size estimation was calculated after assuming an alpha signification level <0.05 and a beta power of 0.8. Considering the results of a meta-analysis reporting a 18% proportion of positive ADA [15] we would need 88 patients for an accuracy of 0.08. Assuming a possible loss of 10% patients we finally recruited 103 consecutive patients in this study. Continuous variables were tested for normal distribution by the Kolmogorov-Smirnov test. Continuous variables are represented as means±SD or medians (interquartile range) and categorical variables as percentages. Logarithmic transformation (ln) was applied to TNF serum concentrations due to its wide range of values. Differences between groups were assessed by the unpaired t test for independent samples, the Mann-Whitney U test (as appropriate) for continuous variables and the ANOVA or Kruskal-Wallis test (as appropriate) for more than two groups. Correlation between two continuous variables was performed by Spearman test. To study the association between qualitative variables we used the Chi-square test. For those variables that were significant with a p-value <0.1 we did a logistic regression analysis. A p value of < 0.05 was accepted as statistically significant and the statistical analysis was performed using SPSS version 19.0 for Windows (IBM, Chicago, IL, USA).
Results
Of total study cases, 74 (71.8%) were naïve for anti-TNF treatment (54 (81.8%) in the IFX group and 20 (54%) in the ADM group). A comparative study including those variables that could affect anti-TNF trough concentration (age, weight, years of anti-TNF treatment and serum TNF, albumin, CRP concentrations) revealed no significant differences between the IFX and ADM group except for slightly higher mean albumin concentration in the ADM group (4.1 g/dL±0.4 vs. 3.86 g/dL±0.5; p=0.03) (Table 1). V158F genotype distribution in the study cases (FF: 38.8%, FV: 44.6% VV: 15.5%) was consistent with Hardy-Weinberg equilibrium (p=0.722). No significant differences were obtained between the genotype distribution and the type of IBD (CD: 37.5% FF, 46.3% FV. 16.3% VV vs. UC: 43.5% FF, 45.5% FV, 13% VV; p=0.85).
Table 2 shows the association between the V158F polymorphism and serum levels of TNF, IFX, ADM and ADAs against anti-TNFs.
Serum TNF concentrations (pg/mL) (ln) were higher in FF carriers than in FV and the lowest were found in VV patients. The ANOVA test, although not reaching statistical significance, shows a tendency between groups (FF: 5.4±1.4, FV: 5.1±1.3, VV: 4.9±1.2, respectively; p=0.09) which was mainly due to the FF vs. VV comparison (p=0.055) and, to a lesser extent, from FF vs. FV (p=0.08). When only ADM group was considered, FF patients showed higher TNF concentration than FV carriers (p=0.035).
Similarly, FF patients frequently had higher IFX or ADM (µg/mL±SD) serum concentrations than VV patients (IFX: 2.4±1.9 vs. 1.8±1.7 and ADM: 6.3±3.8 vs. 3.4±3.5) but without statistical significance (p=0.20 and p=0.38, respectively).
Of the study IBD subjects, 13 (12.6%) (11 CD (13.8%), 2 UC (8.7%)) developed ADAs against the anti-TNFs (11 anti-IFX (16.7%) and 2 anti-ADM (5.4%)) without significant differences for the type of anti-TNF (p=0.086) or type of IBD (p=0.52). In the ADA-producing group of patients, 23.1% were taking an immunomodulator drug and 76.9% were not, although this tendency did not reach statistical significance (p=0.12). A tendency was observed for the association between TNF serum concentration and ADA (p=0.09). Intriguingly, amongst IFX patients, those with ADA production showed lower trough IFX concentration (0.61±1.65 vs. 3.24±3.15 μg/ml; p=0.009).
Of note, the duration of anti-TNF treatment did not correlate with ADA occurrence (Mean years ±SD for ADA negative (n=90): 3.93 ±2.1 and 4.52 ±1.8 for ADA positive cases (n=13); p=0.42).
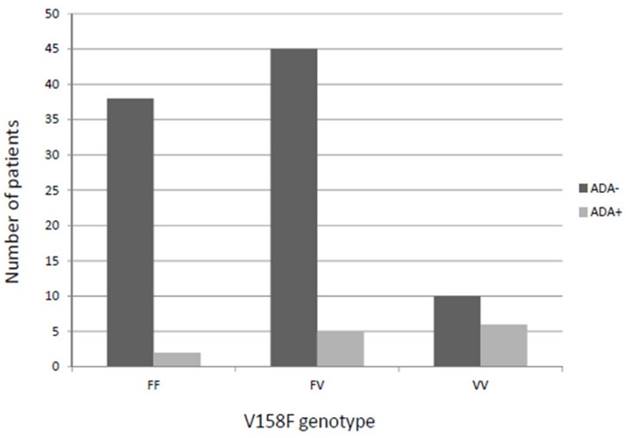
Intriguingly, the percentage of VV patients showing ADA production was higher than in FV or FF carriers (37.5% vs. 10.6% or 5%, respectively), this difference being statistically significant (p=0.004) and allele-dose dependent (Figure 1). Similar results were obtained when restricting the analysis to IFX patients (VV vs. FF+FV (41.7% vs. 11.1%; p=0.01) and a tendency when only ADM patients were considered (VV vs. FF+FV (25% vs. 3.0%; p=0.066).
Moreover, the percentage of VV patients with anti-TNF dose intensification was higher than those VV carriers receiving the standard dose (27.3% vs. 10%) whereas the opposite was observed for FF (27.3% vs. 45.7%). FV carriers, again, showed an intermediate phenotype (45.5% vs. 44.6%) (p=0.03).
Multivariate analysis revealed that only FCGR3A polymorphism (FF+FV vs. VV) was an independent predictor of ADA production (p=0.032; OR=6.084; CI (95%)=1.16-31.84).
Serum levels of TNF, IFX/ADM and ADA according to the FCGR3 V158F polymorphism
| TNF | n | IFX | n | ADM | ADAs | anti-TNF intensification | ||
|---|---|---|---|---|---|---|---|---|
| (pg/mL±SD) (ln) | (µg/mL±SD) | (µg/mL±SD) | No. (%) | No. % | ||||
| V158F FCGR3A | FF (n=40) | 5.42±1.37 | 21 | 2.37±1.92 | 19 | 6.28±3.79 | 2 (5) | 9 (22.5) |
| FV (n=47) | 5.09±1.28 | 33 | 3.45±3.91 | 14 | 5.01±3.81 | 5 (10.6) | 15 (31.9) | |
| VV (n=16) | 4.93±1.17 | 12 | 1.76±1.67 | 4 | 3.37± 3.52 | 6 (37.5) | 9 (56.3) | |
| p value | 0.098 | 0.2 | 0.58 | 0.004 | 0.03 |
IFX: infliximab, ADA: adalimmab, SD: Standard deviation, ADA: Anti-drug antibody
Patients who developed anti-drug antibodies against anti-TNF (pooled analysis) according to the V158F genotype. ADA: Anti-drug antibody
Discussion
The production of antibodies against anti-TNF biological therapy constitutes an important cause of loss of response in patients with chronic inflammatory diseases such as IBD [16, 17]. Therefore, there is a need to understand the molecular basis of this resistance and, eventually, identify biomarkers which could allow us to discern which patients are more likely to develop ADAs; as well as under which circumstances dose adjustment or mAb shift would be interesting clinical options. Besides, this therapy is both expensive and not exempt of considerable adverse reactions. As human immunoglobulins, anti-TNF mAbs, such as IFX and ADM, are IgG with an Fc fraction that, regardless of its target antigen, enable them to exert their effector functions by means of antibody-dependent cellular cytotoxicity (ADCC) in which Fc binds to Fc gamma receptors (FCGRs) expressed on the surface of innate immune cells. In fact, several works have demonstrated associations of a functional polymorphism (V158F) in FCGR3A, a FCGRs family member, with response to mAbs in different neoplastic and chronic inflammatory diseases, including IBD [7, 8, 18, 19]. However, others did not find such associations[9,20] and one explanation for this controversy could be the fact that clinical response depends, not only on pharmacodynamic or pharmacokinetic features, but also on therapy adherence, concurrent medication, disease presentation and patient life style factors. For these reasons, the use of clinical disease activity indexes is not always suitable for pharmacogenetics studies.
Retrospective studies have demonstrated that anti-TNF trough concentrations within therapeutic range relate to better response to therapy in IBD [21, 22], although there is no common consensus as to which anti-TNF concentrations are considered as therapeutic. This can explain the high frequency of responders with IFX levels below the therapeutic range found in our study. Nevertheless, ADA production is associated with worse treatment results, not only for high risk of hipersensitivity reactions, but also for a loss of response due to a lower bioavailability caused by higher anti-TNF clearance [23, 24]. In the present study, we have analysed the relationship of V158F polymorphism with serum levels of the antigen (TNF), the therapy (anti-TNF) and the ADAs (anti-anti-TNF) being, to the best of our knowledge, the first study of this kind in inflammatory diseases. Of note, no statistical associations were found between ADA occurrence and anti-TNF type or treatment duration, thus suggesting that this anti-TNF resistance might be genetically determined. In fact, it was found here that patients with VV genotype have more risk of developing ADAs and of being subjects to anti-TNF intensification. The V allelic version of FCGR3A has higher affinity for IgG binding than the F allele. Therefore, it is not surprising that Ternant et al. demonstrated that patients harbouring the VV genotype showed a higher clearance rate of IFX [11]. Our study shows that ADM FF carriers have increased TNF serum concentration and a tendency to higher anti-TNF levels associated with this genotype. Furthermore, patients with ADAs have lower IFX trough concentrations thus suggesting that probably the increased elimination of this anti-TNF may be induced by the higher V-allele-related immunogenicity. Therefore, the generation of ADAs would ultimately facilitate, the opsonization and fagocytosis of anti-TNF agents. The lower TNF concentration associated with VV patients, although not significant, could be due to the increased anti-TNF dose which may diminish the serum levels of this cytokine.
Additionally, it is worthy of mention that our study has important limitations regarding the limited sample size and the heterogeneity in the intestinal bowel diseases and anti-TNFs included. Nevertheless, although CD and UC display important pathological and molecular differences, they are both IBD and no differences were observed in terms of ADA production occurrence or FCGR3A genotype distribution. Secondly, although IFX and ADM display differences in the human component of the variable part of the mAb sequence -ADM being humanized and IFX chimeric, both are IgG monoclonal antibodies targeting TNF with similar Fc portions and therefore, downstream functions.
Another issue worth considering is that a more severe disease at drug initiation could promote drug clearance and potentially result in lower anti-TNF level and antibody formation. In order to minimize the indicated bias, this study excluded patients at the induction-phase and so albumin and CRP levels suggested disease stability in the study subjects.
For these reasons, our study makes necessary the validation on independent and larger cohorts in order to know the extent of our findings, although the association between genotype and ADAs synthesis would presumably be more evident in CD patients treated with IFX as, although not reaching statistical significance, ADA occurrence is higher in this set of patients.
In conclusion, our results provide an explanation for controversies in the relationships between FCGR3A V158F polymorphism and mAbs response, as well as an interesting starting point for further studies dealing with the involvement of the V158F polymorphism in the resistance and ADA production against anti-TNF treatment with the view of tailoring the dose and type of mAb in IBD patients.
Abbreviations
ADA: Anti-drug antibody; ADM: Adalimumab; CD: Crohn's disease; CRP: C reactive protein; FCGR: receptor for the Fc portion of IgG; IBC: Inflammatory bowel diseases; IgG: Immunoglobulin G; IVD: In vitro diagnostics; IFX: Infliximab; mAbs: Monoclonal Antibody; PCR: Polymerase chain reaction; SD: Standard deviation; TNF: Tumour necrosis factor alpha; UC: Ulcerative colitis.
Acknowledgements
We are grateful to Dr. Samantha Wasniewski for reviewing the English version of this manuscript. The work was supported by Fundación CajaMurcia (grant number: CM08/15-I).
Contributions
PRM and SMS participated in clinical follow-up of patients and data collection and analysis; DTM, JP, JAV and MSGS were responsible for setting up biochemical and molecular assays and for sample collection and analysis; GRM participated in the statistical analysis and PCZ in the study design and manuscript writing. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mirkov MU, Verstockt B, Cleynen I. Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol Hepatol. 2017;2:224-234
2. Tsianos EV, Katsanos KH, Tsianos VE. Role of genetics in the diagnosis and prognosis of Crohn's disease. World J Gastroenterol. 2012;18:105-18
3. Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP. et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993;94:174-81
4. Reenaers C, Louis E, Belaiche J. Current directions of biologic therapies in inflammatory bowel disease. Therap Adv Gastroenterol. 2010;3:99-106
5. Lacruz-Guzmán D, Torres-Moreno D, Pedrero F, Romero-Cara P, García-Tercero I, Trujillo-Santos J. et al. Influence of polymorphisms and TNF and IL1β serum concentration on the infliximab response in Crohn's disease and ulcerative colitis. Eur J Clin Pharmacol. 2013;69:431-8
6. Mould DR, Sweeney KR. The pharmacokinetics and pharmacodynamics of monoclonal antibodies-mechanistic modeling applied to drug development. Curr Opin Drug Discov Devel. 2007;10:86-96
7. Louis E, El Ghoul Z, Vermeire S, Dall'Ozzo S, Rutgeerts P, Paintaud G. et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn's disease. Aliment Pharmacol Ther. 2004;19:511-519
8. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P. et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754-758
9. Louis EJ, Watier HE, Schreiber S, Hampe J, Taillard F, Olson A. et al. Polymorphism in IgG Fc receptor gene FCGR3A and response to infliximab in Crohn's disease: a subanalysis of the ACCENT I study. Pharmacogenet Genomics. 2006;16:911-4
10. Kenkre VP, Hong F, Cerhan JR, Lewis M, Sullivan L, Williams ME. et al. Fc Gamma Receptor 3A and 2A Polymorphisms Do Not Predic Response to Rituximab in Follicular Lymphoma. Clin Cancer Res. 2016;22(4):821-6
11. Ternant D, Berkane Z, Picon L, Gouilleux-Gruart V, Colombel JF, Allez M. et al. Assessment of the Influence of Inflammation and FCGR3A Genotype on Infliximab Pharmacokinetics and Time to Relapse in Patients with Crohn's Disease. Clin Pharmacokinet. 2015;54:551-62
12. Present DH, Rutgeerts P, Targan S, Hanauer SB, Mayer L, van Hogezand RA. et al. Infliximab for the treatment of fistulas in patients with crohn's disease. N Engl J Med. 1999;340:1398-405
13. Billiet T, Rutgeerts P, Ferrante M, Van Assche G, Vermeire S. Targeting TNF-alfa for the treatment of inflammatory bowel disease. Expert Opin Biol Ther. 2014;14:75-101
14. Conesa-Zamora P, Santaclara V, Gadea-Niñoles E, Ortiz-Reina S, Perez-Guillermo M. Association of polymorphism in FcGR3A gene and progression of low-grade precursor lesions of cervical carcinoma. Hum Immunol. 2010;71:314-7
15. Pecoraro V, De Santis E, Melegari A, Trenti T. The impact of immunogenicity of TNFα inhibitors in autoimmune inflammatory disease. A systematic review and meta-analysis. Autoimmun Rev. 2017;16:564-75
16. Ordás I, Mould DR, Feagan BG, Sandborn WJ. Anti-TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics-based dosing paradigms. Clin Pharmacol Ther. 2012;91:635-646
17. Vermeire S, Gils A. Value of drug level testing and antibody assays in optimising biological therapy. Frontline Gastroenterol. 2013;4:41-43
18. Morales-Lara MJ, Conesa-Zamora P, García-Simón MS, Pedrero F, Santaclara V, Perez-Guillermo M. et al. Association between the FCGR3A V158Fpolymorphism and the clinical response to infliximab in rheumatoid arthritis and spondyloarthritis patients. Scand J Rheumatol. 2010;39:518-520
19. Mendrinou E, Patsatsi A, Zafiriou E, Papadopoulou D, Aggelou L, Sarri C. et al. FCGR3A-V158F polymorphism is a disease-specific pharmacogenetic marker for the treatment of psoriasis with Fc-containing TNFα inhibitors. Pharmacogenomics J. 2017;17:237-241
20. Montes A, Pérez-Pampín E, Joven B, Carreira P, Fernández-Nebro A, Del Carmen Ordoñez M. et al. FCGR polymorphisms in the treatment of rheumatoid arthritis with Fccontaining TNF inhibitors. Pharmacogenomics. 2015;16:333-345
21. Afif W, Loftus EV, Faubion WA, Kane SV, Bruining DH, Hanson KA. et al. Clinical utility of measuring infliximab and human anti-chimeric antibody concentrations in patients with inflammatory bowel disease. Am J Gastroenterol. 2010;105:1133-1139
22. Vande Casteele N, Ferrante M, Van Assche G, Ballet V, Compernolle G, Van Steen K. et al. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology. 2015;148:1320-9
23. Chaparro M, Guerra I, Muñoz-Linares P, Gisbert JP. Systematic review: antibodies and anti-TNF-α levels in inflammatory bowel disease. Aliment Pharmacol Ther. 2012;35:971-986
24. Vande Casteele N, Gils A, Singh S, Ohrmund L, Hauenstein S, Rutgeerts P. et al. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol. 2013;108:962-971
Author contact
![]() Corresponding author: Pablo Conesa-Zamora, Clinical Analysis Department, Molecular Diagnostic Lab. Santa Lucía University Hospital. Calle Mezquita s/n. 30202 Cartagena, Spain. Telephone: +34 968 325008. Fax: +34 968 326389. E-mail address: pablo.conesaes
Corresponding author: Pablo Conesa-Zamora, Clinical Analysis Department, Molecular Diagnostic Lab. Santa Lucía University Hospital. Calle Mezquita s/n. 30202 Cartagena, Spain. Telephone: +34 968 325008. Fax: +34 968 326389. E-mail address: pablo.conesaes