ISSN: 1449-1907International Journal of Medical Sciences
- Current issue
- Volume 21; 2024
- Volume 20; 2023
- Volume 19; 2022
- Volume 18; 2021
- Volume 17; 2020
- Archive
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Dengue Virus (DENV)
Development of Peptides as...
Mode of Action for Therapeutic...
Conclusions
Acknowledgements
References

Int J Med Sci 2017; 14(13):1342-1359. doi:10.7150/ijms.21875 This issue Cite
Review
Peptides as Therapeutic Agents for Dengue Virus
Miaw-Fang Chew1, Keat-Seong Poh2, Chit-Laa Poh1 ![]()
1. Research Centre for Biomedical Sciences, Sunway University, Bandar Sunway, Selangor 47500, Malaysia;
2. Department of Surgery, Faculty of Medicine, University of Malaya, Jalan Universiti, Kuala Lumpur, 50603, Malaysia.
Abstract
Dengue is an important global threat caused by dengue virus (DENV) that records an estimated 390 million infections annually. Despite the availability of CYD-TDV as a commercial vaccine, its long-term efficacy against all four dengue virus serotypes remains unsatisfactory. There is therefore an urgent need for the development of antiviral drugs for the treatment of dengue. Peptide was once a neglected choice of medical treatment but it has lately regained interest from the pharmaceutical industry following pioneering advancements in technology. In this review, the design of peptide drugs, antiviral activities and mechanisms of peptides and peptidomimetics (modified peptides) action against dengue virus are discussed. The development of peptides as inhibitors for viral entry, replication and translation is also described, with a focus on the three main targets, namely, the host cell receptors, viral structural proteins and viral non-structural proteins. The antiviral peptides designed based on these approaches may lead to the discovery of novel anti-DENV therapeutics that can treat dengue patients.
Keywords: Dengue virus, Drug discovery, Peptides, Antiviral therapeutics.
Introduction
Dengue is a mosquito-borne disease caused by the infection of dengue virus (DENV). It has been estimated that 390 million dengue infections occur annually, of which 96 million manifest clinically [1]. Before 1970, only nine countries experienced dengue epidemics. Currently, dengue is endemic in more than 100 countries, primarily in tropical and sub-tropical countries [2]. There are four dengue virus serotypes, DENV-1-4, which are genetically and antigenically distinct, although each serotype elicits a similar range of disease manifestations during infection [3]. In humans, dengue infection causes a spectrum of illnesses ranging from asymptomatic, fever, rash, joint pain and other mild symptoms to life-threatening dengue haemorrhagic fever (DHF) and dengue shock syndrome (DSS) [4]. Infection with one DENV serotype induces lifelong immunity against the homologous serotype but not against the other three heterologous serotypes. In fact, studies have shown that secondary infection with a different DENV serotype is an important risk factor in causing more severe complications, such as DHF and DSS, due to a phenomenon designated as antibody dependent enhancement (ADE) or the original antigenic sin [5-7].
One of the strategies that has been undertaken to halt DENV infection is by vector control. Aedes aegypti and Aedes albopictus are the primary transmission vectors for DENV [8]. Strategies such as fogging and the release of genetically modified mosquitoes which could lead to the production of fewer progenies [9] have failed to lessen the mosquito population, as witnessed by the emergence of new dengue cases in places that were dengue-free or had less dengue cases in the past [10-12]. While active research on vaccine development for dengue has been ongoing for the past few decades, the development of vaccines has been held back by several challenges. The major constraints for dengue vaccine development include the lack of good animal models, the complexity of developing a vaccine against all four antigenically distinct DENV serotypes, as well as the need to achieve balanced tetravalent responses that could exhibit significant immunity against all four viruses without the adverse effects of ADE or original antigenic sin [13]. The first dengue vaccine, Dengvaxia®, (CYD-TDV, chimeric yellow fever virus-tetravalent dengue vaccine) developed by Sanofi Pasteur was licensed in December 2015 in Mexico. It is a live-attenuated tetravalent vaccine comprising structural proteins (pre-membrane and envelope proteins) of DENV based on the yellow fever 17D virus backbone [14, 15]. The approved regimen involves three doses, given at the 0th, 6th and 12th months, for individuals between 9-45 years of age. Outcomes from phase III clinical trials showed that the vaccine successfully reduced dengue hospitalizations by 80%. However, its average efficacy against DENV was low, especially against DENV-1 at approximately 50% and against DENV-2 at 39%. Its average efficacy against DENV-3 and DENV-4, meanwhile, was slightly higher at 75% and 77%, respectively [16, 17]. Furthermore, previous clinical trials revealed that CYD-TDV vaccination caused elevated risks of hospitalization for children less than nine years of age [18]. The World Health Organization has therefore recommended the use of CYD-TDV vaccine only in countries where epidemiological data indicated a high burden of dengue [19].
The lack of efficient vector control strategies and the uncertainty of long-term protective efficacy of CYD-TDV vaccine against all four DENV serotypes call for an urgent need for dengue therapeutics, especially in endemic countries with poor resource setting. There are no antiviral drugs available and at present, supportive treatment with emphasis on fluid therapy and close clinical monitoring during the critical phase of illness are the only course of action for dengue disease. Many antiviral candidates have failed to reach clinical trials due to their poor selectivity and physiochemical or pharmacokinetic properties [20]. Although nucleoside analogs, such as NITD-008 and balapiravir, have entered preclinical animal safety study and clinical trials, they were terminated due to lack of potency [21]. Balapiravir, for instance, did not improve the clinical and virological parameters in patients in the phase II clinical trial, although it was shown to have good in vitro antiviral activities with EC50 values of 1.3-3.2 µM in DENV infection assays using primary human macrophages [21]. Treatment of DENV-infected mice with another nucleoside analog NITD-008, on the other hand, completely prevented mice death, but severe adverse events were observed in rats and dogs after two weeks of oral dosing [20, 22]. Likewise, other anti-DENV candidates, including chloroquine, prednisolone, celgosivir and lovastatin, have gone through clinical trials but failed to meet the defined primary end points, whereby neither significant viremia nor evidence of beneficial effects on clinical manifestations was observed [23-26]. At present, two candidates, namely ivermectin and ketotifen, are undergoing clinical trials (trial number NCT02045069 and NCT02673840, respectively). However, their long-term clinical efficacies remain to be determined. In contrast to small molecules, peptides are generally known to have high selectivity and possess relatively safe characteristics which make them attractive pharmacological candidates [27]. Due to their attractive pharmacological profiles, this review will highlight the current status and the rational drug design of antiviral peptides and peptidomimetics as therapeutics for dengue.
Dengue Virus (DENV)
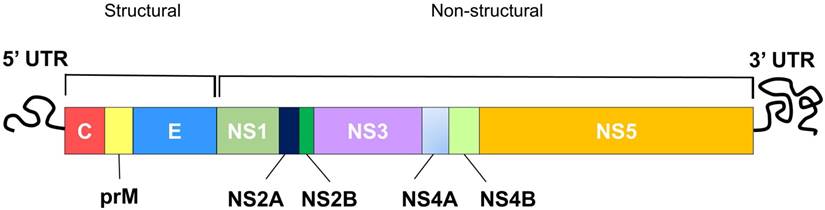
DENV is an enveloped, positive, single-stranded (ss) RNA virus classified under the genus Flavivirus of the Flaviridae family [28]. Other closely related viruses classified under the Flavivirus include yellow fever virus (YFV), west nile virus (WNV), japanese encephalitis virus (JEV) and zika virus. The dengue virion is a spherical particle, approximately 50 nm in diameter with envelope (E) and precursor-membrane (prM) / membrane (M) proteins located on its surface, while the capsid (C) proteins sit underneath the lipid bilayer, encapsulating the viral genome [29]. The DENV genome (~11 kb) constitutes a single open reading frame (ORF), encoding three structural proteins (C, prM/M and E proteins) followed by seven non-structural (NS) proteins (NS1, NS2A and 2B, NS3, NS4A and 4B, NS5) (Figure 1) [30]. The translated polyprotein is then cleaved by cellular signal peptidases and virally encoded protease (NS2B and NS3) to generate individual proteins. The structural proteins form the viral particle while the non-structural proteins participate in replication and invasion of the immune system [30]. To design peptides with therapeutic potential against dengue virus, it is necessary to understand the viral replication cycle.
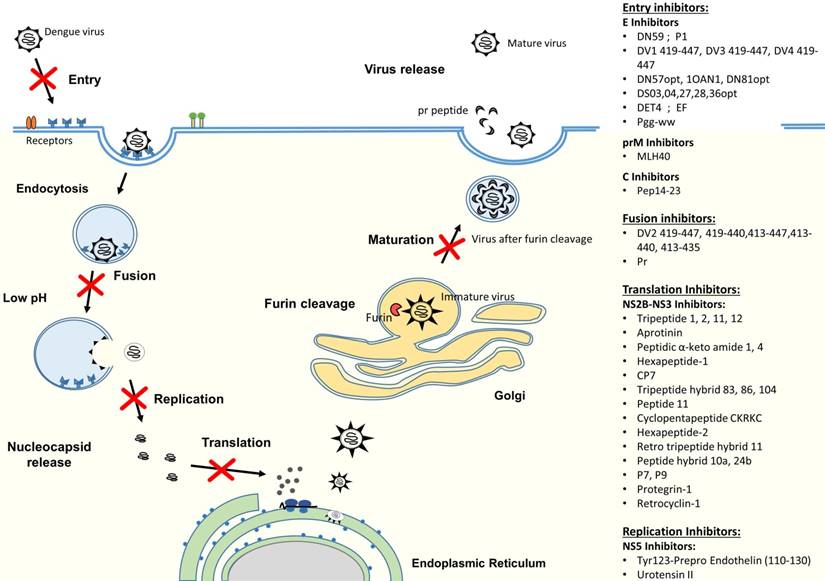
DENV infection in humans starts with a DENV-infected mosquito bite. DENV can replicate in a wide spectrum of cells, including liver, spleen, lymph node, kidney and other organs [31, 32], but monocytes, macrophages and dendritic cells (DC) have been shown to be the major targets for DENV [33, 34]. The life cycle of dengue virus is initiated by the virus attachment through the interaction between viral surface proteins and attachment/receptor molecules on the surface of the target cell (Figure 2). Receptor recognition is believed to be mediated by the domain III of E protein to enable the virus to enter into host cells by receptor-mediated, clathrin-dependent endocytosis (primary method) [35, 36]. However, studies have also shown that viral entry could occur by the direct fusion of the virus and host cells [37-39]. After internalization, dissociation of the E homodimers takes place as a result of the acidic environment in the endosome. Subsequently, domain II of the E protein will project outwardly and the hydrophobic fusion loop in domain II will insert itself into the endosomal membrane [40, 41]. This will then trigger domain III to fold back and force the virus particle and endosomal membrane to move towards each other and fuse together [42, 43]. The fusion of the virus with vesicular membranes would then release the nucleocapsid into cytoplasm, resulting in genome uncoating [44]. Subsequently, the viral RNA genome is released. The viral RNA is translated into a single polyprotein and processed co- and post-translationally by cellular and virus-derived proteases into three structural proteins and seven NS proteins (Figure 1). Upon protein translation, the NS proteins initiate viral genome replication at the intracellular membranes, resulting in the production of more viral RNA strands [45]. Then, the newly synthesized RNA is packed by C proteins to form the nucleocapsid [46]. The prM and E proteins, on the other hand, form heterodimers that oriented into the lumen of ER and are believed to induce a curved surface lattice which guides virion budding [47]. Hence, the virus assembles and buds from the ER before migrating to the trans-Golgi for maturation process. The slightly acidic pH of the trans-Golgi network prompts the dissociation of prM/E heterodimers to form 90 dimers with prM capping the fusion peptide located at the domain II of the E protein [48]. This is followed by the cleavage of the prM at Arg-X-(Lys/Arg)-Arg by cellular endoprotease (furin), (where X is any amino acid) to produce membrane-associated M and “pr” peptide [49, 50]. Both prM and the “pr” will act as chaperones to stabilize the E protein during the secretory pathway by preventing premature membrane fusion. At the end, the “pr” peptide will dissociate upon the release of the progeny by exocytosis [45].
Schematic diagram of the DENV genome showing structural and non-structural polyproteins that are encoded by the DENV genome.
Development of Peptides as Therapeutics
Peptides are biologically active molecules comprising the combination of at least two amino acids through a peptide bond. In contrast to large proteins, they are smaller in size and are considered to contain less than 100 amino acid residues. Peptides are known to have attractive pharmacological profiles due to their highly selective and relatively safe characteristics [27]. They readily exist in the human body and exert diverse biological roles, predominantly as signalling and regulatory molecules in a variety of physiological processes [51]. In the past, peptides were held back in the drug development pipelines due to their instability, whereby they could be easily degraded by at least 569 proteases in the human body [52]. Nevertheless, a number of technological breakthroughs and advancements have reversed the situation. This has resulted in the spark of interest in peptide drug development. Current technologies have allowed the modification of peptides to create artificial variants with improved stability and overcome pharmacodynamic weaknesses. For instance, advances in automated-liquid handling devices, synthetic peptide synthesis, mass spectrometry and in silico drug design have allowed high-throughput drug screening. In addition, advances in peptide manipulations such as synthesis of D-amino acids, cyclic peptides, incorporation of chemicals and nanocarriers have further increased the bioavailability of peptides [53]. Currently, ample examples of efficacious and safe peptide drugs are available in the market [54-57]. Great success has been achieved for the peptide drug FuzeonTM (enfuvirtide), a synthetic peptide that blocks viral fusion by binding to the gp41 (polypeptide chain) of the human immunodeficiency virus (HIV) type-1 envelope protein [55]. It is the only antiviral peptide which has been commercialised. Other antimicrobial peptide candidates, such as MU1140, Arenicin, IMX924, Novexatin and Lytixar, are being evaluated in the preclinical and clinical trials [58, 59]. Myrcludex B, an anti-Hepatitis B and Hepatitis D peptide targeting sodium taurocholate co-transporting polypeptide (NTCP) of liver cells, is also being studied in a phase II clinical trial [60].
At present, the value of global peptide therapeutics market is predicted to increase from US$21.3 billion (2015) to US$46.6 billion in the year 2024 [61]. There are at least 60 therapeutic peptides that have been approved by the US Food and Drug Administration (FDA) and approximately 140 peptide therapeutics are being evaluated in clinical trials [62]. In 2011, 25 of the US-approved peptide drugs accounted for the global sale of over US$14.7 billion, while Victoza®, Zoladex®, Sandostatin®, Lupron® and Copaxone® each had global sales of over US$1,000 million [63]. Some other examples of therapeutic peptides include glucagon-like peptide-1 (GLP-1) and analogues [57], deletion peptides of insulin [56] and a deletion peptide of the heat shock protein 60 [54] that have been used widely in the treatment of diabetes. This has demonstrated the potential and importance of peptides as pharmacological agents. Additionally, as the number of new entities approved by the FDA rapidly decreases over the years [64] and the number of publicities about the side effects of popular small molecules increases (such as the cancer chemotherapeutic or COX-2 inhibitors) [65-67], the pharmaceutical industry is now reviving their interest in peptides as potential drug candidates. With good pharmacology properties and new technologies to mitigate the weakness of peptides, the number of therapeutic peptide candidates will continue to grow.
Mode of Action for Therapeutic Peptides
Antiviral peptides that either interact with the virus particles or target at critical viral replication steps of the life cycle can potentially be used as treatment or prophylaxis for dengue. Several approaches have been explored thus far to inhibit dengue virus infection, including targeting the host cell receptors or attachment factors, viral structural proteins and non-structural (NS) proteins. Drugs that were designed against these three main targets employ different mechanisms of action to stop virus infection. By targeting the host cellular receptors or attachment factors, it will prevent the attachment and binding of viral proteins with the host cell, hence stopping the subsequent entry of DENV. Drug candidates directing at the viral structural proteins [capsid (C), pre-membrane (prM/M) and envelope (E)], on the other hand, might be able to interfere with the binding of viruses to host cells, thereby inhibiting the viral attachment/fusion and viral entry. Lastly, as non-structural proteins are essential components of replication machinery, designing drug candidates against NS proteins will interfere with the viral replication cycle to effectively ameliorate dengue.
Schematic diagram of DENV replication cycle and summary of antiviral peptides. The antiviral peptides are classified according to their mechanism of actions, which include entry inhibitors, fusion inhibitors, translation inhibitors and replication inhibitors.
Entry inhibitors: Targeting host cells
One of the attractive approaches to inhibiting virus infection is by blocking the cellular receptors or attachment factors, or mimicking the cellular receptors, hence preventing the virus from attaching and entering host cells. This will form the first barrier to block viral infection. Studies have shown that this is a feasible approach to halting viral infections [68-70]. Pugach et al. (2008) and Lieberman-Blum et al. (2008) demonstrated that a small molecule, CCR5 inhibitor, Maraviroc, successfully inhibited human immunodeficiency virus type 1 (HIV-1) infection by binding to the CCR5 co-receptor of host cells [68, 69]. On the other hand, Myrcludex B, a lipomyristolated peptide containing 47 homologous amino acid residues of hepatitis B virus pre-S1 protein, was able to bind to the NTCP of host cells and resulted in the restriction of virion uptake in the host cells [70]. The identified DENV receptors or attachment factors on mammalian cells were reviewed by Perera-Lecoin et al. (2014) and Cruz-Oliveira et al. (2015) [71, 72]. Some of the important attachment factors or receptors are the dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN) [34], heparan sulfate [73], mannose receptor [74], HSP90/HSP70 [75], laminin receptor [76], and the TIM and TAM proteins [77]. To date, several small molecules were identified as receptor antagonists or mimics for DENV. For instance, CC-chemokine receptor 5 (CCR5) antagonists, Met-R and UK484900 (a Maraviroc analogue) prevented CCR5 activation and reduced DENV load [78], while heparin sulfate mimetics, such as PI-88 [79], fucoidan [80] and CF-238 [81], were shown to block viral entry. Interestingly, to the best of our knowledge, no peptide inhibitors were found to block DENV infection by binding to cellular attachment factors or receptors. This represents a big research gap that should prompt researchers to investigate.
The DC-SIGN is a type II transmembrane protein that falls into the category of C-type lectin with an extracellular domain that can bind specifically to carbohydrates [82]. DC-SIGN has been shown to be an essential cellular factor required for the infection of ebola virus [83, 84], HIV-1 [85, 86] and human cytomegalovirus (CMV) [87] into dendritic cells. Studies have also shown that dendritic cells that abundantly express DC-SIGN are highly susceptible to DENV infection [33, 88, 89]. Tassaneetrithep et al. (2003) further validated the importance of DC-SIGN as a DENV receptor [34], whereby the transfection of DC-SIGN into THP-1 cells resulted in DENV infection while dendritic cells blocked with anti-DC-SIGN prevented DENV infection [34]. These results suggest that DC-SIGN is a feasible target for designing therapies that prevent DENV infection. Furthermore, dendritic cells were activated after capturing antigen and resulted in the stimulation of naïve T cells to produce cytokines and chemokines [90]. Blocking the binding of DENV to DC-SIGN can prevent DENV infection, as well as the initiation of immune response which can lead to severe dengue characterized by the cytokine storm. Based on the literature, limited DC-SIGN inhibitors are found to stop DENV infection. In a study, Alen et al. (2011) evaluated the inhibitory properties of various carbohydrate-binding agents (CBAs) which are mannose-specific plant lectins by using the Raji/DC-SIGN+ cell line. Results showed that Hippeastrum hybrid (HHA), Galanthus nivalis (GNA) and Urtica dioica (UDA) were able to bind to the envelope of DENV, hence preventing the subsequent viral attachment [91]. Similarly, pradimicin-s (PRM-S), a small-size non-peptidic CBA, was shown to exert antiviral activity against DENV by binding to the DENV envelope in monocyte-derived dendritic cells [91].
Another important known DENV receptor is the glycosaminoglycans (GAG). Among the GAG family, heparin sulfate (HS) is the most ubiquitous member of the family and is the putative receptor for DENV [92-94]. Studies have shown that HS acted as the first interactive attachment factor to facilitate DENV binding to the second receptor [92, 95]. It was demonstrated that DENV-HS interacted via positively charged E(III) residues, especially Lys291 and Lys 295 binding to the negatively charged HS [73, 96]. Many heparan mimetics were identified to block DENV infection [79, 80, 97]. Lee et al. (2006) showed that a heparin sulfate mimetic, phosphomannopentaose sulfate (PI-88), significantly increased the survival rate of DENV-infected mice [79]. In another study, a sulphated polysaccharide, fucoidan, which was extracted from the marine alga Cladosiphon, was able to inhibit DENV-2 infection by binding to the DENV envelope protein [80]. Interestingly, Talarico et al. (2005) showed that iota-carrageenan and dl-galactan hybrid C2S-3 (sulphated polysaccharides isolated from the red seaweeds Gymnogongrus griffithsiae and Cryptonemia crenulata) inhibited DENV infection in a virus serotype and host cell dependent manner [97]. Many other heparin mimetics, including CF-238 [81], sulphated galactomannan [98], curdlan sulfate [99], sulphated galactan [98], sulphated K5 [100] and chondroitin sulfate [101], were found to inhibit DENV infection but no antiviral peptide was identified to either bind to cellular receptor or act as a receptor mimetic to block DENV entry thus far. Likewise, to the best of our knowledge, no antiviral peptide was found to inhibit DENV infection by targeting other receptors, including mannose receptor [74], HSP 90/70 [75], laminin receptor [76], and the TIM and TAM proteins [77]. Furthermore, inhibitors targeting host cellular receptor(s) are anticipated to be less prone to develop resistance as compared to those targeting viruses. Therefore, this may serve as an interesting research gap to be explored.
Although studies demonstrated that DENV mainly enters host cells via receptor initiated-clathrin mediated endocytosis [102-104], viral entry via clathrin-independent endocytic route has also been observed [104]. In addition, evidence of direct entry via fusion with plasmatic membrane leading to direct penetration into cytoplasm without undergoing endocytosis has also been described [105, 106]. Furthermore, evidence showed that DENV is able to infect a variety of cell types, including those isolated from humans [107, 108], monkeys [92, 93], hamsters [95, 109] and mosquitoes [110, 111] via different receptors. Therefore, the DENV entry pathway is greatly dependent on the cell type and viral strain. Due to the variability in viral entry routes and broad tissue tropism, targeting the viral structural proteins is easier than the vastly different cellular receptors, as DENV possesses the capability to utilize a wide range of cellular receptors and pathways to enter host cells. Viral structural proteins, especially the E protein, is therefore a popular target for antiviral inhibitors to interfere with the virus-host interactions and stop subsequent viral entry.
Entry Inhibitors: Targeting Envelope (E) proteins
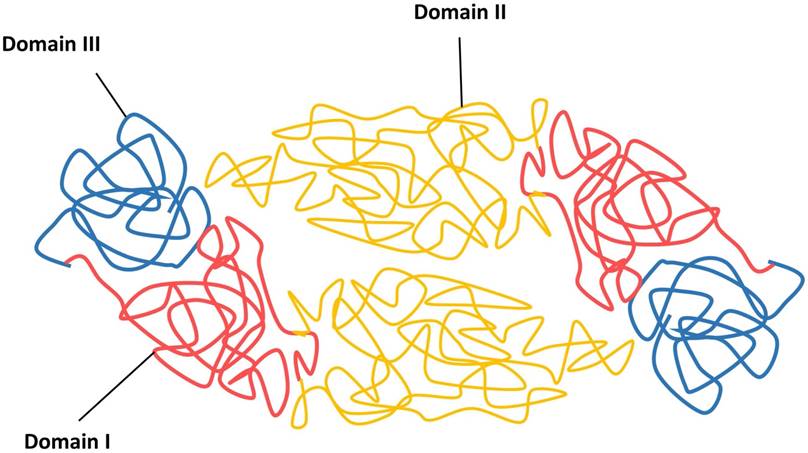
The viral infection cycle starts with the interaction of viral structural proteins, mediated mainly by the E protein with the host cell receptors or attachment factors to facilitate the entry of virus. The DENV E protein is 53 kDa in size and composed of three distinct domains, namely the domain I E(I), flanked by the dimerization domain E(II) containing the fusion peptide and an immunoglobulin-like domain E(III) that protrudes from the virion surface, followed by a membrane proximal stem and a transmembrane anchor (Figure 3) [45, 112].
Schematic diagram of DENV envelope (E) proteins in their dimeric forms.
The function of E(I) has not been fully characterized, although it has been shown to be involved in the structural rearrangement of the E protein during internalization [112]. The E(II) contains a region known as fusion peptide, which is responsible for the viral fusion activity, and the E domain II also contains serotype-conserved epitopes, and contributes to the E protein dimerization [113, 114]. Previous studies have shown the E(III) is responsible for receptor recognition, which is essential for viral attachment to facilitate viral entry into host cells by receptor-mediated, clathrin-dependent endocytosis (primary method) [73, 102, 103]. Additionally, E(III) also harbours the serotype-specific neutralizing epitopes [115, 116]. Because of the involvement of receptor recognition and attachment, as well as its vital role in mediating viral and cellular membrane fusion to release viral genomic RNA for viral replication, the E glycoprotein is the most important protein facilitating viral entry. Hence, this makes the E protein a good antiviral target to stop viral entry.
The DENV E structural proteins have been well determined using nuclear magnetic resonance spectroscopy, X-ray crystallography and cryo-electron microscopy [112, 117, 118]. Recent advancements in the understanding of the high-resolution E structure have allowed researchers to utilize the information in combination with in silico molecular drug designing methods to search for potential antiviral candidates. Several research groups have utilized different strategies including in silico drug design to screen for novel antiviral peptides against the E protein (Table 1). By using Wimley-White interfacial hydrophobicity scale (WWIHS) in combination with known structural data of the E protein, Hrobowski et al. (2005) were the first group to identify a novel peptide DN59, corresponding to the stem region of E, which showed >99% DENV-2 inhibition at <25 µM [119]. The D59 peptide was suggested to function through a sequence of specific mechanisms as a scrambled peptide failed to inhibit DENV infection. It was hypothesized that the DN59 peptide might interfere with the intramolecular interaction, disrupt structural rearrangements of fusion proteins or interact with target cell surface components to exert its inhibitory effects [119]. The mechanism of action for peptide DN59 was further evaluated by a later study (2012) where it was shown that the peptide D59 inhibited DENV infectivity by interacting directly with virus particles, causing the formation of holes at the five-fold vertices in the virus particles [120]. This led to the release of viral genomic RNA and exposure of the viral RNA to exogenous RNase [120].
Likewise, Schmidt et al. (2010) also hypothesized and proved that the stem peptides could inhibit dengue virus infection [121]. In the study, a set of overlapping peptides based on the DV2 stem region (from amino acid residues 396-447) were synthesized and tested for their binding affinities with soluble form of DV2 E (sE, covering only the first 395 residues of E) via fluorescence polarization. Among the set of overlapping peptides, a peptide (DV2419-447) was found to bind selectively to the post-fusion of sE with the concentration of half-maximal change in fluorescence polarization (FP IC50) of 0.125 µM and Kd at approximately 150 nM, while the scrambled peptide DV2419-447 neither bound to pre-fusion nor post-fusion conformers of sE. Schmidt et al. (2010) proposed that the peptide DV2419-447 inhibited DENV infection through a two-step mechanism during late-stage fusion intermediate, whereby the peptide first binds non-specifically to the viral membrane, followed by specific interaction with the E protein when E proteins undergo conformational rearrangement at low pH. Interestingly, they observed that the reduction of the DV2419-447 hydrophobicity (by changing the 441-447 amino acid residues) greatly reduced the inhibitory property of the peptide, but not its binding affinity against dengue virus E proteins. This suggested the importance of peptide hydrophobicity to non-specific host membrane interaction before high binding affinity to the DENV E protein.
The importance of hydrophobicity was further supported by Hrobowski et al. (2005), whereby the novel antiviral peptide was successfully identified using the Wimley-White interfacial hydrophobicity scale (WWIHS) screening method. This screening method calculated a sliding hydrophobicity score to determine the segments of the protein with a propensity to interact with membrane interfaces [119]. By using a similar strategy, another study has identified five hydrophobic regions located on the DENV-2 E protein [122]. Amino acids derived from these regions were screened via WWIHS and further optimized using RAPDF biased Monte Carlo method. This resulted in the identification of several novel peptides, namely DS03/DS04, DS27/DS28 and DS36, which could potentially interrupt protein-protein interactions during DENV fusion. Likewise, many antiviral peptides were identified against other viruses such as type-1 herpes simplex virus (HSV-1) [122], severe acute respiratory syndrome coronavirus (SARS) [123], human cytomegalovirus (HCMV) [124] and rift valley fever virus (RVFV) [125] via the same approach. This again has validated the importance of hydrophobicity property for antiviral peptides.
In addition, it was observed that many of the antiviral peptides known to inhibit entry of enveloped viruses to cells have hydrophobic and/or amphipathic properties to facilitate the interaction with cellular lipid membrane interfaces [126, 127]. Besides using WWIHS, the interfacial helical hydrophobic moment (iHHM) is another physio-chemical determining strategy which can be used to augment the peptide-membrane interfaces. This approach quantified the segregation of hydrophobic and hydrophilic faces of a peptide folded into an α-helix structure [128]. Higher iHHM value indicates stronger membrane interaction with peptides [129, 130]. Additionally, Badani et al. (2014) also suggested hydrophobicity and amphipathicity to be critical properties for peptides in their interaction with cellular membrane and thereby inhibiting viral entry [126]. Therefore, for future drug design and development, researchers could consider incorporating the hydrophobic and amphipathic properties into antiviral peptides to further enhance antiviral efficacies.
Previous studies have shown that the lateral loop located on E(III) played an important role in virus-host receptor(s) interaction [73, 131], hence making it an interesting target. A short sequence (380-IGVEPGQLKL-389) in the lateral loop on the DENV-2 E(III) was used as a target to screen for potential antiviral peptides using the BioMoDroid algorithm [132]. Four different peptides were designed and DET4 (one of the peptides) was found to inhibit 85% DENV-2 at 500 µM. TEM images indicated that DET4 inhibited DENV-2 entry by causing structural abnormalities and alteration of the conformational changes of E proteins. On the contrary, Panya et al. (2014) targeted on the n-octyl-β-D-glycoside (βOG) hydrophobic pocket located in the domain I domain II interface of DV E protein [133]. A previous study has shown that the shift of two β-hairpins located at the hydrophobic pocket was essential to cause correct conformational changes during virus fusion step [113]. The importance of the βOG hydrophobic pocket was further validated as several compounds targeting this hydrophobic pocket were able to stop DENV infection [134, 135]. By using AUTODOCK v4.2 and CDOCKER DISCOVERY STUDIO v2.1 software, Panya et al. (2014) found a di-peptide, EF, to be the most effective antiviral peptide as it inhibited DENV-2 infection with the IC50 value of 96 µM [133].
Studies have shown that the E stem region was well conserved among Flaviviruses [including WNV, tick-borne encephalitis virus (TBE), YFV and JEV] [121, 136]. Therefore, an antiviral peptide targeting against one DENV serotype might possess the possibility to inhibit other DENV serotypes and closely related Flaviviruses. To further examine this hypothesis, Schmidt et al. (2010) investigated the antiviral potential of stem peptides derived from DENV-2 and WNV against DENV 1, 2, 3 and 4 [136]. The amino acid residues from 419-447 of the genome of each of the four DENV serotypes were synthesized along with a solubility tag (RGKGR). Results showed that DV2419-447 remained the strongest inhibitor against all four dengue serotypes, followed by DV1419-447, DV3419-447 and DV4419-447. Nonetheless, when stem peptides (residues 419-447) from related flaviviruses were tested against DENV infection, none of these peptides inhibited any of the DENV serotypes. This might be due to the variation in the seven residues located at the C-terminal which could have affected the non-specific interaction with the viral membrane rather than poor affinity against E protein, as WNV had nearly identical binding affinities for trimeric DV2 sE [136]. To further validate the observation, mutagenesis was performed, confirming that residues 442-444 were important in conferring the antiviral activity of the stem peptide whereby increased hydrophobicity would increase inhibitory strength [136]. On the other hand, a similar situation was observed by Hrobowski et al. (2005), whereby the DN 59 peptide (peptide sequence corresponding to the pre-anchor stem of the E protein and highly conserved among flaviviruses) which was shown to inhibit DENV had also demonstrated cross-inhibitory activity against WNV (>99% inhibition with concentration of <25µM) [119].
Bai et al. (2007) found a peptide, P1, which was isolated from the murine brain cDNA phage display library by biopanning against the recombinant WNV E protein [137]. When P1 was tested against DENV-2, it inhibited ~99% DENV-2 at a concentration of 200 µM. Surface plasmon resonance (SPR) showed that P1 bound to the WNV E protein with a Kd of 6 µM. However, the specific binding site on E protein and the mode of action are unknown. Other peptides that blocked DENV infection by binding to the E proteins were DN57 opt and 1OAN1 [138]. In the study, a set of peptides were computationally designed based on the pre-entry dimeric E structure. Peptides DN57 and 1OAN1 specifically designed from the hinge of domain II and the first domain I/domain II connection, was shown to display IC50 of 8 and 7 µM, respectively [138]. Both peptides were shown to bind specifically (affinities ~1 µM) to the purified DENV-2 E protein. Images from cryoEM suggested that these peptides might have caused structural deformations to the DENV-2 surface, hence interfering the virus-host cell binding. Further study of peptide inhibitors 1OAN1 and DN59 has also revealed that both peptide inhibitors were able to inhibit the antibody dependent enhancement (ADE) effect in vitro with an IC50 of 3 µM and 6 µM, respectively [139]. In a recent study, Chew et al. (2015) identified a novel peptide, peptide gg-ww, by biopanning a randomised phage display peptide library against the purified DENV-2 viral particles [140]. Approximately 96% inhibition was achieved at the concentration of 250 µM and the data indicated that peptide gg-ww inhibited DENV-2 entry. On the other hand, screening of commercial cyclic peptides through molecular docking resulted in the identification of a peptide, the brain natriuretic peptide fragment 7-32 (BNP7-32), which could bind to the E protein with a pKi value of 32.7 and ΔG of -44.9 kcalmol-1 [141]. Due to the fact that data were obtained via in silico design, further experiments were required to explore the inhibitory potential of the peptide against DENV.
Entry Inhibitors: Targeting prM/M and C proteins
Many studies have focused on the DENV E protein due to the nature of the virus structure, whereby the E proteins cover most of the surface area of the viral particle [112], and the vast and expanded knowledge on the E protein. Nonetheless, prM and C proteins are feasible targets to look into in the screening for antiviral peptides.
The prM protein (about 21 kDa) is the precursor of the M protein (approximately 8 kDa). The cleavage of the prM protein by the cellular protease (furin) would separate the prM protein into the “pr” peptide (1-91 residues), the ectodomain (92-130 residues) and the M protein (131-166 residues) [45, 142, 143]. The hydrophilic N-terminal region of the protein is responsible for coding the glycosylated “pr” segment of the prM protein. The prM is believed to protect the E protein from conformational changes during the maturation pathway in the acidic environment of the trans-golgi network. A previous study has shown that prM-containing-virus is more resistant to the low pH environment [144]. Upon release of the matured virions, “pr” will be separated, leaving the E and M proteins on the surface of the mature DENV. In a recent study, a peptide inhibitor (MLH40) mimicking the conserved ectodomain of the M protein was designed and it was shown to inhibit all four DENV serotypes with an IC50 of 24-31 µM [145]. Docking results indicated that MLH40 bound to the interior site of E homodimer, which is the same interacting site for the native M protein against the E protein. Additionally, the expression of the pr protein also successfully inhibited virus fusion at low pH and stopped viral infection [146]. Approximately 81-85% inhibition was achieved at 30 µM. Data indicated that the pr peptide interacted with a highly conserved histidine (H244) because the substitution of H244 to alanine had led to the disruption of the pr-E interaction [146]. This disruption could have resulted in a distorted E conformation, therefore interfering the pH-triggered fusion reaction in the endosome. Further optimization could be carried out using truncated pr peptides to identify the amino acid residues that are essential to block viral infection.
On the other hand, the DENV C protein (approximately 11 kDa) is composed of four α-helical regions arranged in antiparallel homodimers [147]. The structure of the C protein contains high net charge with an asymmetric distribution of basic residues which lie along the surface of the C protein to orchestrate RNA binding. In contrast, the opposite surface forms a hydrophobic region which may enable interactions with lipids [147, 148]. This configuration makes the C protein essential in virus assembly as it enables encapsulation of the ssRNA genome to form the nucleocapsid [45]. Despite a general understanding of the viral RNA assembly role, it is believed that the hydrophobic region of the C-terminal capsid protein contains a signal sequence for anchoring the protein into the endoplasmic reticulum (ER) membrane and partitioning the prM protein to the membrane [143, 149]. The C-terminal is then cleaved off by the viral NS2B-NS3 protease to form a mature protein during virus assembly [148]. Faustino et al. (2015) have designed a peptide inhibitor, pep14-23, based on the conserved region of DENV C protein. The pep14-23 was able to interfere with the interaction of the DENV C protein with the host intracellular lipid droplet, which was shown to be essential for viral particle formation [46, 150]. It was found that the binding forces between the C protein and lipid droplets were reduced from 33 pN to 19 pN with the addition of 100 µM pep14-23. Interestingly, despite the importance of the C protein for viral survival, limited antiviral peptide was designed against the C protein. This is a target worth looking into.
Replication and Translation Inhibitors: Targeting NS proteins
Viral proteases have been shown to serve as good inhibitory targets. For instance, protease inhibition was shown to be a successful strategy in treating HIV infection [151]. The HIV-1 protease cleaves the translated polypeptide chain into smaller functional proteins, thereby allowing the virus particle to mature [152]. By inhibiting the protease, the immature virus particles would not be able to transform into the mature virion, hence obstructing the viral replication. Several HIV-1 protease inhibitors were discovered and used clinically, such as saquinavir, ritonavir, indinavir, nelfinavir, amprenavir (and its prodrug, fosamprenavir), lopinavir, atazanavir, and darunavir [153]. Similarly, the NS5, NS3 and NS2B (co-factor) proteins were known to play major roles in enzymatic activities for DENV infection, thus making them ideal antiviral targets [154, 155]. After DENV infection, translation of the viral genome will give rise to a polyprotein containing three structural and seven non-structural proteins. The polyprotein will be cleaved into individual proteins during virus maturation by the host proteases (signalase and furin) on the luminal side of the endoplasmic reticulum, as well as by the viral serine protease (NS2B-NS3 protease) on the cytoplasmic side to ensure the success of viral replication and maturation [49, 154, 156]. DENV NS3 contains a trypsin-like protease and it requires the NS2B cofactor to be active to cleave the DENV polyprotein at the Ser-His-Asp catalytic triad [157-160].
In a previous study, Schuller et al. (2011) synthesized a series of tripeptide aldehyde inhibitors whereby four of them had the IC50 values in the range between ~6.7 and 12.2 µM against the DENV-2 NS2B-NS3 protease [161]. Among the four tripeptide aldehyde inhibitors, tripeptide 1 (benzoyl-n-Lys-Arg-Arg-H) and tripeptide 2 (phenylacetyl-Lys-Arg-Arg-H) were reported to have the most potential as anti-DENV candidates with IC50 of 9.5 µM and 6.7 µM, respectively [162]. Further investigation of the tripeptide1 revealed that it bound covalently to the DENV-3 NS2B-NS3 protease and resulted in the formation of a closed conformation of the NS2B-NS3 protease in which the hydrophilic β-hairpin region of NS2B would wrap around the NS3 core [163]. Structural analysis of this protease-peptide complex further revealed a pocket located on the NS2B-NS3 protease which could act as a new antiviral target for drug development [163]. Another protease inhibitor, aprotinin, a large polypeptide [also known as bovine pancreatic trypsin inhibitor (BPTI)] was hypothesized to form multiple interactions with the NS2B-NS3 protease and gained its inhibitory activity from the steric hindrance of the active site [163]. In contrast to tripeptide 1 which required the interactions with NS2B, no direct binding was observed between aprotinin and NS2B [163].
In a recent study, several cyclic peptides were designed based on the binding loop of aprotinin which is highly similar to the sequence of the native NS2B-NS3 cleavage site and extends from the P3 to P4' position at the active site of the NS2B-NS3 protease [164]. Results indicated that a peptide, CP7, was able to show good inhibitory property (Ki value of 2.9 µM) against the DENV-3 protease. Similar with the binding of aprotinin to the protease, strong hydrogen bonds contributed by the P1 and P2' positions were observed but the inhibitory constant value was not as strong as aprotinin (Ki of 0.026 µM). This might be due to the flexibility of the cyclic peptide which resulted in the decreased affinity against the protease. Nevertheless, this study proved the feasibility of designing inhibitors against both prime and non-prime regions of the protease, and CP7 could act as an alternative candidate for developing a therapeutic against the NS2B-NS3 protease.
On the other hand, the N-benzoyl capped tetrapeptide sequence (Nle-Lys-Arg-Arg) was previously shown to be the favoured amino acid residues for the S1-S4 subsites of the NS2B-NS3 protease binding cavity [165, 166]. Yusof et al. (2000) showed that the Arg-Arg residues in the P1 and P2 positions located next to the cleavage site were responsible for the high binding affinity against the protease, while Li et al. (2005) found that Lys and Nle (norleucine) in the P3 and P4 positions were essential for high binding affinity [165, 166]. Interestingly, Nitsche et al. (2012) showed that the removal of an arginine resulted in better inhibitory activity [167].
Nitsche et al. (2012) showed that a retro-peptide based on the sequence R-Arg-Lys-Nle-NH2 with an arylcyano-acrylamide group as N-terminal cap possessed the best inhibition activity at Ki value of 4.9 µM [167]. It is hypothesized that the arylcyanoacrylamide moiety mimic the first Arg in the P1 position while the Arg-Lys-Nle tripeptide bound to other protein pockets of NS2B-NS3 protease. Unfortunately, even though the drug candidate possessed good binding ability, it did not have significant antiviral activity in the cell culture against DENV. Therefore, Nitsche et al. (2013) further optimized the lead candidate via structure activity relationship assays in a subsequent study and successfully developed a thiazolidinedione-based peptide hybrid (hybrid 24b) containing a hydrophobic group with a better Ki value of 1.5 µM [168]. Nonetheless, drugs designed via structure-based activity faced the limitation due to the differences in protease structures derived from crystallization versus the in vivo protease structures, thereby the antiviral candidate which has high binding affinity against the crystallized protease structure might not have the same binding affinity against the protease in vivo. To overcome this challenge, Nitsche et al. (2013) further modified the N-terminal cap moieties and incorporated membrane-permeable peptide to increase the potential antiviral activities [168]. The peptide hybrid which possessed the best antiviral activity in the cell culture was found to be the rhodanine-based peptide hybrid 10a with an EC50 value of 16.7 µM and Ki value of 9.3 µM. From the study, the Arg-Lys residues were found to be sufficient to create high target affinity with Ki values below 2 µM.
On the other hand, Behnam et al. (2015) utilized the Bz-Arg-Lys-Nle as the core sequence to further optimize the chemical structure to improve its antiviral potential against the NS2B-NS3 protease [169]. Tripeptide hybrid 83 was created by the combination of 4-CF3-benzyl ether and the thiazole cap, while tripeptide hybrid 86 was created by the combination of 4-CF3-benzyl ether and the thien-2-yl cap. Both of the peptide hybrids successfully showed improved Ki values of 12 nM and 19 nM, respectively. Thus far, these are the two peptide hybrids which possess the highest binding affinity against the NS2B-NS3 protease. Nonetheless, the derivative 83 which possessed the highest binding affinity had a lower EC50 value of 20 µM in reducing the virus titer as compared to the derivative 86 (EC50 value of 7 µM) while the derivative 104 and derivative 90 were found to have the most potent EC50 value of 3.42 µM and 4.06 µM, respectively. Theoretically, the inhibitor which has a higher binding affinity against a target would result in a higher antiviral property. But in contrast to the direct in vitro protein-peptide interaction assay, cellular antiviral assay in the cell culture is complicated by factors such as membrane permeability and metabolic stability. Data suggested that the lipophilicity of the tripeptide hybrids correlated well with the observed cellular antiviral activities which might be influenced by the fact that higher polarity (lower lipophilicity) could lead to weaker membrane permeability [169]. In this case, the tripeptide hybrid 104 was shown to be more lipophilic than the tripeptide hybrid 83. Moreover, data also indicated that the tripeptide hybrid 104 possessed a much higher half-life of 175 minutes compared to the tripeptide hybrid 83 (half-life of 45 minutes) in metabolic clearance via rat liver microsomes. These observations might offer the possible explanation of the higher antiviral activity of the tripeptide hybrid 104 in cellular assays as compared to the analogue of tripeptide hybrid 83 which possessed higher binding ability against DENV-2 protease in vitro.
In another study, 15 peptide inhibitors against the NS2B-NS3 protease were modified via molecular modelling method [170]. Results from the ADME/Tox calculation indicated that the peptide inhibitor 11 (Bz-Nle-Lys-Arg-Bip) possessed the best pharmacokinetic properties among all inhibitors due to its ability to be better absorbed into the intestine and to remain inactive in the central nervous system [170]. This results supported the use of Bz-Nle-Lys-Arg as the lead candidate for drug development against DENV by targeting the NS2B-NS3 protease. On the other hand, several hexapeptides derived from the NS2B-NS3 protease cleavage sites were synthesized and they were shown to inhibit DENV in a low micromolar range [171]. Among all candidates, the best hexapeptide inhibitor was derived from the NS2A/NS2B region with a Ki value of 12 µM. Likewise, several peptidic α-keto amide inhibitors mimicking the NS3/NS4 region were designed and two of the peptidic inhibitors were found to inhibit the NS3 protease with Ki values of 16 and 47 µM, respectively [172].
Tambunan and Alamudi (2010) designed several cyclopentapeptides based on the substrate specificity for the NS2B-NS3 protease and a cyclopentapeptide, CKRKC, was found to be the best candidate with the estimated free binding energy of -8.39 kcal/mol and Ki of 0.707 μM [173]. Nonetheless, the cyclopentapeptide was designed in silico, further in vitro and cell-based experiments were required to further exploit the inhibitory potential of the peptide against DENV. Similarly, Velmurugan et al. (2014) found a hexapeptide with the highest binding energy of -80.4 kcal/mol against the NS2B-NS3 protease, but further investigation is required to verify its antiviral potential via in vitro studies [174]. In another study, a natural peptide library composed of conotoxins (a mixture of peptide neurotoxins produced by cone snails) was used to screen against the DENV NS2B-NS3 protease [175]. A 13-mer cyclic peptide inhibitor, MrIA (derived from Conus marmoreus), was shown to possess a Ki value of 9 µM. Mutagenesis study further revealed that the inhibitory activity was mainly mediated by a disulphide bond loop with a Lys residue at the active site. As the NS2B-NS3 protease was previously shown to have a preference for Ser at the P' position, further optimization was carried out. By changing the Leu to Ser, the resultant peptide 7 (P7) and peptide 9 (P9) were shown to have stronger inhibitory activities with improved Ki values of 1.4 µM and 2.2 µM, respectively. Nonetheless, stability assays showed that about half of the inhibitory activity of P7 was lost after 2.5h, while only ~25% inhibitory activity of P9 was lost after incubating with protease for 2.5h. Although P9 had a higher Ki value of 2.2 µM as compared to P7 (1.4 µM), the stability of P9 to withstand protease degradation and its efficient cell permeability in BHK-21 and Vero cells had shown that P9 is a better drug candidate. Rothan et al. (2012a and 2012b) identified two antiviral peptides, namely protegrin-1 and retrocyclin-1 against DENV NS2B-NS3 protease [176, 177]. It was previously shown that protegrin-1, a cationic cyclic peptide, possessed broad antimicrobial properties against different microorganisms [178]. In addition to having good binding affinity (Ki value of 5.85 μM) against the DENV NS2B-NS3 protease, protegrin-1 was able to inhibit DENV replication in Rhesus monkey kidney (MK2) cells with an IC50 of 11.7 μM [176]. On the contrary, retrocyclin-1, a circular cationic peptide, was shown to inhibit the NS2B-NS3 protease at an IC50 of 21.4 μM [177]. Results indicated that retrocyclin-1 worked best when incubated with DENV-2 during simultaneous treatment, giving 85% reduction in viral replication after 75 hours. These findings suggest that both protegrin-1 and retrocyclin-1 are feasible candidates to be potential therapeutic drugs for dengue treatment.
NS5 is a large multifunctional protein which plays an important role in viral replication and modulation of the host immune response. It contains RNA-dependent RNA polymerase domain at the C-terminal end which is essential for viral replication and methyltransferase at the N-terminal region for RNA capping [155, 179]. These functions are essential for viral replication, thereby making NS5 a promising target for antiviral drug development. Inhibiting the polymerase or methyltransferase function can actively suppress virus growth and propagation in host cells [180]. Both NITD-008 and Balapiravir are examples of nucleoside inhibitors, whereby NITD-008 corresponds to an adenosine analogue while balapiravir is another nucleoside analogue originally developed for the treatment of hepatitis C virus. However, NITD-008 was terminated due to severe side effects while balapiravir failed due to its low efficiency in clinical trials [21, 22]. To search for novel antiviral peptide against the NS5 methyltranserase, Tambunan and colleagues (2014) screened over 300 commercial cyclic peptides and molecular docking results revealed two potential ligands, namely, tyr123-Prepro Endothelin (110-130) and urotensin II [181]. Both peptides were shown to bind to the NS5 methyltranserase and formed stable complexes. Docking results suggested that tyr123-Prepro Endothelin (110-130) was found to bind to the S-adenosyl-L-methionine (SAM) site of NS5 with a ΔG of -24.73 kkal/mol while urotensin II bound to the RNA-cap site of NS5 with a ΔG of -19.04 kkal/mol. Nevertheless, further in vitro verification is required to elucidate the antiviral potential of these inhibitors. Despite the importance of the NS5 protein, there are limited antiviral peptide screening studies targeting the NS5 protein. Perhaps this is due to the lack of a crystal structure that contains the full length of the NS5 protein. With the recent publication of the crystal structure of the NS5 protein [182], development of antiviral peptide targeting the NS5 protein can be expected.
Limitations of Antiviral Peptides
There are a number of limitations that hinder the use of peptides as therapeutic drugs. The main challenges are due to the poor stability and bioavailability of peptides. Unmodified peptides were shown to commonly degrade quickly in human serum, resulting in low in vivo activities [183, 184]. Nonetheless, various chemical modifications can be applied to manipulate the physicochemical properties of peptides, thereby increasing the stability and bioavailability of the peptides. These chemical modification approaches have been reviewed by Gentilucci et al. (2010) [53]. For instance, conjugation to polymers such as polyethylene glycol (PEG) increased the molecular weight of peptide, hence enhancing its peripheral stability to undergo hydrolysis in the brain [185]. A previous study also showed that the half-lives of unmodified peptides of less than 0.5 hour could be increased to approximately 1.5 hour after C-amidation and N-acetylation, while cyclization successfully increased the half-lives of the peptides to 6.5 hours [186]. On the other hand, the replacement of arginine residues with an amino acid derivate of arginine, α-amino-3-guanidino-propionic acid (Agp), could dramatically reduce the cleavage of peptide by trypsin [187]. The substitution of L-amino acids to D-amino acids of pandinin-2 increased its resistance to the degradation by bovine pancreatic trypsin and human elastase. The stability of the D-amino acid substituted pandinin-2 was increased to 4 hours as compared to L-pandinin-2 which was rapidly cleaved by these two enzymes [188].
Additionally, multiple strategies could also be incorporated into peptide drug development to enhance the antiviral properties of peptides. These include the mutagenesis assays to identify the vital amino acids which are responsible for the inhibitory effects and the addition of cell penetrating peptides to increase the cell permeability of peptides. For example, the IC50 of two antiviral peptides (DN57 opt and DN81 opt) were successfully reduced to 8 μM and 40 μM, respectively, after amino acid optimization was performed via residue-specific all-atom probability discriminatory function approach [119, 138]. Similarly, Schmidt and co-workers (2010a, 2010b) successfully enhanced the antiviral properties of the identified peptides after optimizations via mutagenesis assays and the addition of a solubility tag [121, 136]. On the other hand, due to the hydrophilicity and conformation properties of peptides, cellular uptake of peptides is constrained [189, 190]. Oral bioavailability of peptides is therefore limited by the membrane barrier. To overcome this challenge, cell-penetrating peptides (CPP) can be incorporated into the peptide sequence to act as a cargo delivery in carrying antiviral peptide across the permeability barrier and entering cells to exert its inhibitory activity [191]. For instance, by conjugating the tat peptide (derived from the HIV-1 transcriptional activator protein) to galactosidase protein, it enabled the delivery of the fusion protein to all the tissues in mice while those without tat peptide-conjugated were restrained [192]. With the advancement of technology, the drawbacks of peptides serving as antiviral agents can therefore be overcome by various strategies. This, in turn, will aid the development and use of peptides as therapeutics.
Conclusions
The development of peptides as therapeutic drugs against viruses is a promising field for drug discovery. Peptides are known to be highly specific and selective against targets, have lower toxicity and low accumulation in tissues, all of which make them good candidates for drug development [193]. This phenomenon is in contrast to small molecule drugs, whereby off-target and toxicity are some of the biggest drawbacks for small molecule drugs [194, 195]. Although peptides possess several limitations, these could be overcome via chemical modifications.
Many peptides are shown to be effective as they demonstrated good in silico and in vitro binding affinity against viral targets (Table 1 and 2). However, many of these antiviral peptides against DENV have not been tested in cell-based assays and none of them has been evaluated in any in vivo study. Therefore, further studies should focus on elucidating the antiviral potential of these peptides in animal models, such as the AG129 mice which are highly susceptible to DENV infection as they lack both type I and type II IFN receptors [196-198]. Furthermore, an ideal DENV inhibitor will need to be effective against all four DENV serotypes. The inhibitory potential against all DENV serotypes have not been explored by many of the studies described in this review. With just a few exceptions, the mechanisms of inhibition of most of the peptides are not well defined. These are the areas where further studies should be performed to exploit the potential of these antiviral peptide candidates. Moreover, the commercially available vaccine (CYD-TDV) is unable to achieve the maximal protection against all four DENV serotypes, while the vector control strategy has failed to reduce the spread of DENV. Peptide inhibitors targeting host cells, viral structural proteins or non-structural proteins may help to overcome dengue infection. With effective antivirals, vaccination and vector controls such as fogging, dengue in endemic countries can be better controlled and the state of public health can be improved.
List of antiviral peptides against DENV structural proteins
| Peptides | DENV serotypes | Sequences | Inhibitory activities | Target | References |
|---|---|---|---|---|---|
| DN59 | All serotypes | MAILGDTAWDFGSLGGVFTSIGKALHQVFGAIY | IC50 less than 10µM for all serotypes | Envelope | [119,120] |
| P1 | DENV-2 | DTRACDVIALLCHLNT | 200 µM (~99.3% inhibition) | Envelope | [137] |
| DV2419-447 | All serotypes | AWDFGSLGGVFTSIGKALHQVFGAIYGAA (solubility tag-RGKGR) | FP IC50 of 0.125µM (DENV-2) | Envelope | [121, 136] |
| DV2419-440 | All serotypes | AWDFGSLGGVFTSIGKALHQVF | FP IC50 of 0.25µM (DENV-2) | Envelope | [121] |
| DV2413-447 | All serotypes | AILGDTAWDFGSLGGVFTSIGKALHQVFGAIYGAA | FP IC50 of 0.275µM (DENV-2) | Envelope | [121] |
| DV2413-440 | All serotypes | AILGDTAWDFGSLGGVFTSIGKALHQVF | FP IC50 of 0.25µM (DENV-2) | Envelope | [121] |
| DV2413-435 | All serotypes | AILGDTAWDFGSLGGVFTSIGKA | FP IC50 of 2 µM (DENV-2) | Envelope | [121] |
| DV1419-447 | All serotypes | AWDFGSIGGVFTSVGKLVHQVFGTAYGVL (solubility tag-RGKGR) | IC 90: DV1- 1.5 µM; DV2- 2 µM DV3- >6 µM; DV4- >6 µM | Envelope | [136] |
| DV3419-447 | All serotypes | AWDFGSVGGVLNSLGKMVHQIFGSAYTAL (solubility tag-RGKGR) | IC 90: DV1- 0.1 µM; DV2- 2 µM DV3- 4 µM; DV4- 1.5 µM | Envelope | [136] |
| DV4419-447 | All serotypes | AWDFGSVGGLFTSLGKAVHQVFGSVYTTM (solubility tag-RGKGR) | IC 90: DV1- 5 µM; DV2- 6 µM DV3- >6 µM; DV4- 6 µM | Envelope | [136] |
| DN57opt | DENV-2 | RWMVWRHWFHRLRLPYNPGKNKQNQQWP | 8 µM | Envelope | [138] |
| 1OAN1 | DENV-2 | FWFTLIKTQAKQPARYRRFC | 7 µM | Envelope | [138] |
| DN81 opt | DENV-2 | RQMRAWGQDYQHGGMGYSC | 36 µM | Envelope | [138] |
| DS03opt | DENV-2 | FPFDFHHDRYYHFHWKRYQH | na | Envelope | [122] |
| DS04opt | DENV-2 | IWWRPRDWPTFIFYFREWRW | na | Envelope | [122] |
| DS27opt | DENV-2 | KEYFRRFFHCHNHQREWHWH | na | Envelope | [122] |
| DS28opt | DENV-2 | KEKRREWEWRFRWEFRLYFE | na | Envelope | [122] |
| DS36opt | DENV-2 | RHWEQFYFRRRERKFWLFFW | na | Envelope | [122] |
| DET4 | DENV-2 | AGVKDGKLDF | 35 µM | Envelope | [132] |
| EF | DENV-2 | EF | 96 µM | Envelope | [133] |
| Pgg-ww | DVEN-2 | GGARDAGKAEWW | IC50 of ~77 -91 µM | Envelope | [140] |
| MLH40 | DENV | SVALVPHVGMGLETRTETWMSSEGAWKHVQRIETWILRHPG | IC50 of 24-31µM | Pre-Membrane | [145] |
| pr | DENV-1 and DENV-2 | pr protein | 30 µM (81-85% inhibitions) | Pre-Membrane | [146] |
| Pep14-23 | DENV | NMLKRARNRV | Binding forces were reduced to 19pN from 33pN with the addition of 100 µM pep14-23 | Capsid | [150] |
DENV: Dengue virus; na: not available
List of antiviral peptides against DENV non-structural proteins
| Peptides | DENV Serotypes | Sequences | Inhibitory activities | Target | References |
|---|---|---|---|---|---|
| Tripeptide 2 | DENV-2 | Phenylacetyl-K-R-R-H | IC50 of 6.7 µM | NS2B-NS3 | [161] |
| Tripeptide 1 | DENV-2 | Benzoyl-n-K-R-R-H (n=norleucine) | IC50 of 9.5 µM | NS2B-NS3 | [161-163] |
| Tripeptide 12 | DENV-2 | 4-Aminophenylacetyl-K-R-R-H | IC50 of 11.2 µM | NS2B-NS3 | [161] |
| Tripeptide 11 | DVEN-2 | 4-Phenylphenylacetyl-K-K-R-H | IC50 of 12.2 µM | NS2B-NS3 | [161] |
| Aprotinin | DENV-3 | RPDFC LEPPY TGPCK ARIIR YFYNA KAGLC QTFVY GGCRA KRNNF KSAED CMRTC GGA | Ki of 0.026 µM | NS2B-NS3 | [163] |
| Peptidic α-keto amide 4 | DENV-2 | Ac-FAAGRR-CHO | Ki of 16 µM | NS2B-NS3 | [172] |
| Peptidic α-keto amide 1 | DENV-2 | Ac-FAAGRR-αketo-SL-CONH2 | Ki of 47 µM | NS2B-NS3 | [172] |
| Hexapeptide-1 | DENV | Ac-RTSKKR-CONH2 | Ki of 12 µM | NS2B-NS3 | [171] |
| CP7 | DENV-3 | PCRARIYGGCA | Ki of 2.9 µM | NS2B-NS3 | [164] |
| Tripeptide hybrid 83 | DENV-2 | Bz-Arg-Lys-L-Phg-NH2 by the combination of 4-CF3-benzyl ether and thiazole cap | Ki value of 12 nM; EC50 value of 20 µM | NS2B-NS3 | [169] |
| Tripeptide hybrid 86 | DENV-2 | Bz-Arg-Lys-L-Phg-NH2 by the combination of 4-CF3-benzyl ether and thien-2-yl cap | Ki value of 19 nM; EC50 value of 7 µM | NS2B-NS3 | [169] |
| Tripeptide hybrid 104 | DENV-2 | Bz-Arg-Lys-L-Phg-NH2 by the combination of 3-OCH3-benzyl ether and bithiophene cap | EC50 value of 3.42 µM | NS2B-NS3 | [169] |
| Peptide inhibitor 11 (BDBM50175978) | DENV-2 | Bz-Nle-Lys-Arg-Bip | Ki value of 1.16E+4 nM | NS2B-NS3 | [162, 170] |
| Cyclopentapeptide CKRKC | DENV-2 | CKRKC | Ki of 0.707 μM | NS2B-NS3 | [173] |
| Hexapeptide-2 | DENV-2 | AIKKFS | Glide energy -80.4kcal/mol | NS2B-NS3 | [174] |
| Retro tripeptide hybrid 11 | DENV-2 | R-Arg-Lys-Nle-NH2 with an arylcyano-acrylamide group as N-terminal cap | Ki value of 4.9 µM | NS2B-NS3 | [167] |
| peptide hybrid 10a | DENV-2 | Rhodanine-based peptide hybrid bearing a cyclohexyl moiety at the heterocycle | EC50 value of 16.7 µM | NS2B-NS3 | [168] |
| peptide hybrid 24b | DENV-2 | thiazolidinedione-based peptide hybrid | Ki value of 1.5 µM; IC50 value of 2.9 µM | NS2B-NS3 | [168] |
| P7 | DENV-2 | CGKRKSC | Ki value of 1.4 µM | NS2B-NS3 | [175] |
| P9 | DENV-2 | CAGKRKSG | Ki value of 2.2 µM | NS2B-NS3 | [175] |
| Protegrin-1 | DENV-2 | RGGRLCYCRRRFCVCVGR | IC50 of 11.7 μM | NS2B-NS3 | [176] |
| Retrocyclin-1 | DENV-2 | GICRCICGRGICRCICGRIGGRVPGVGVPGVGHHHHHH | IC50 of 21.4 μM | NS2B-NS3 | [177] |
| tyr123-Prepro Endothelin (110-130) | DENV-2 | CQCASQKDKKWSYCQAGKEI | ΔG of -24.73 kkal/mol | NS5 | [181] |
| Urotensin II | DENV-2 | ETPDCFWKYCV | ΔG of -19.04 kkal/mol | NS5 | [181] |
DENV: Dengue virus; NS: non-structure
Acknowledgements
This work was supported by Sunway University Internal Grant 2017 - INTM-2017-SST-RCBS-01.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL. et al. The global distribution and burden of dengue. Nature. 2013;496:504-7
2. WHO. Dengue control. 2017. http://www.who.int/denguecontrol/epidemiology/en/
3. Sayce AC, Miller JL, Zitzmann N. Targeting a host process as an antiviral approach against dengue virus. Trends Microbiol. 2010;18:323-30
4. Halstead SB. Dengue. Lancet. 2007;370:1644-52
5. Midgley CM, Bajwa-Joseph M, Vasanawathana S, Limpitikul W, Wills B, Flanagan A. et al. An in-depth analysis of original antigenic sin in dengue virus infection. J Virol. 2011;85:410-21
6. Zompi S, Harris E. Original antigenic sin in dengue revisited. Proc Natl Acad Sci U S A. 2013;110:8761-2
7. Flipse J, Diosa-Toro MA, Hoornweg TE, van de Pol DP, Urcuqui-Inchima S, Smit JM. Antibody-dependent enhancement of dengue virus infection in primary human macrophages; balancing higher fusion against antiviral responses. Sci Rep. 2016;6:29201
8. Murray NE, Quam MB, Wilder-Smith A. Epidemiology of dengue: past, present and future prospects. Clin Epidemiol. 2013;5:299-309
9. Phuc HK, Andreasen MH, Burton RS, Vass C, Epton MJ, Pape G. et al. Late-acting dominant lethal genetic systems and mosquito control. BMC Biol. 2007;5:11
10. Gubler DJ. The changing epidemiology of yellow fever and dengue, 1900 to 2003: full circle?. Comp Immunol Microbiol Infect Dis. 2004;27:319-30
11. Bouri N, Sell TK, Franco C, Adalja AA, Henderson DA, Hynes NA. Return of epidemic dengue in the United States: implications for the public health practitioner. Public Health Rep. 2012;127:259-66
12. Ebi KL, Nealon J. Dengue in a changing climate. Environ Res. 2016;151:115-23
13. Rothman AL, Ennis FA. Dengue vaccine: the need, the challenges, and progress. J Infect Dis. 2016;214:825-7
14. Guirakhoo F, Weltzin R, Chambers TJ, Zhang ZX, Soike K, Ratterree M. et al. Recombinant chimeric yellow fever-dengue type 2 virus is immunogenic and protective in nonhuman primates. J Virol. 2000;74:5477-85
15. Guirakhoo F, Pugachev K, Zhang Z, Myers G, Levenbook I, Draper K. et al. Safety and efficacy of chimeric yellow fever-dengue virus tetravalent vaccine formulations in nonhuman primates. J Virol. 2004;78:4761-75
16. Capeding MR, Tran NH, Hadinegoro SR, Ismail HI, Chotpitayasunondh T, Chua MN. et al. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet. 2014;384:1358-65
17. Villar L, Dayan GH, Arredondo-Garcia JL, Rivera DM, Cunha R, Deseda C. et al. Efficacy of a tetravalent dengue vaccine in children in Latin America. N Engl J Med. 2015;372:113-23
18. Hadinegoro SR, Arredondo-Garcia JL, Capeding MR, Deseda C, Chotpitayasunondh T, Dietze R. et al. Efficacy and long-term safety of a dengue vaccine in regions of endemic disease. N Engl J Med. 2015;373:1195-206
19. WHO. Dengue vaccine: WHO position paper. July 2016. http://www.who.int/immunization/newsroom/press/dengue_first_position_paper/en/
20. Lim SP, Wang QY, Noble CG, Chen YL, Dong H, Zou B. et al. Ten years of dengue drug discovery: progress and prospects. Antiviral Res. 2013;100:500-19
21. Nguyen NM, Tran CN, Phung LK, Duong KT, Huynh Hle A, Farrar J. et al. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis. 2013;207:1442-50
22. Yin Z, Chen YL, Schul W, Wang QY, Gu F, Duraiswamy J. et al. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A. 2009;106:20435-9
23. Tricou V, Minh NN, Van TP, Lee SJ, Farrar J, Wills B. et al. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl Trop Dis. 2010;4:e785
24. Tam DT, Ngoc TV, Tien NT, Kieu NT, Thuy TT, Thanh LT. et al. Effects of short-course oral corticosteroid therapy in early dengue infection in Vietnamese patients: a randomized, placebo-controlled trial. Clin Infect Dis. 2012;55:1216-24
25. Low JG, Sung C, Wijaya L, Wei Y, Rathore AP, Watanabe S. et al. Efficacy and safety of celgosivir in patients with dengue fever (CELADEN): a phase 1b, randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet Infect Dis. 2014;14:706-15
26. Whitehorn J, Nguyen CVV, Khanh LP, Kien DTH, Quyen NTH, Tran NTT. et al. Lovastatin for the treatment of adult patients with dengue: a randomized, double-blind, placebo-controlled trial. Clin Infect Dis. 2016;62:468-76
27. Uhlig T, Kyprianou T, Martinelli FG, Oppici CA, Heiligers D, Hills D. et al. The emergence of peptides in the pharmaceutical business: From exploration to exploitation. EuPA Open Proteomics. 2014;4:58-69
28. Kuno G, Chang GJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus flavivirus. J Virol. 1998;72:73-83
29. Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E. et al. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108:717-25
30. Lindenbach BD, Rice CM. Molecular biology of flaviviruses. Adv Virus Res. 2003;59:23-61
31. Wahid SF, Sanusi S, Zawawi MM, Ali RA. A comparison of the pattern of liver involvement in dengue hemorrhagic fever with classic dengue fever. Southeast Asian J Trop Med Public Health. 2000;31:259-63
32. Upanan S, Kuadkitkan A, Smith DR. Identification of dengue virus binding proteins using affinity chromatography. J Virol Methods. 2008;151:325-8
33. Marovich M, Grouard-Vogel G, Louder M, Eller M, Sun W, Wu SJ. et al. Human dendritic cells as targets of dengue virus infection. J Investig Dermatol Symp Proc. 2001;6:219-24
34. Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W. et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. 2003;197:823-9
35. Swaminathan S, Khanna N. Dengue: recent advances in biology and current status of translational research. Curr Mol Med. 2009;9:152-73
36. Huerta V, Chinea G, Fleitas N, Sarria M, Sanchez J, Toledo P. et al. Characterization of the interaction of domain III of the envelope protein of dengue virus with putative receptors from CHO cells. Virus Res. 2008;137:225-34
37. Chu JJ, Ng ML. Infectious entry of west nile virus occurs through a clathrin-mediated endocytic pathway. J Virol. 2004;78:10543-55
38. Chu JJ, Leong PW, Ng ML. Analysis of the endocytic pathway mediating the infectious entry of mosquito-borne flavivirus west nile into Aedes albopictus mosquito (C6/36) cells. Virology. 2006;349:463-75
39. Krishnan MN, Sukumaran B, Pal U, Agaisse H, Murray JL, Hodge TW. et al. Rab 5 is required for the cellular entry of dengue and West Nile viruses. J Virol. 2007;81:4881-5
40. Stiasny K, Allison SL, Schalich J, Heinz FX. Membrane interactions of the tick-borne encephalitis virus fusion protein E at low pH. J Virol. 2002;76:3784-90
41. Heinz FX, Stiasny K, Allison SL. The entry machinery of flaviviruses. Arch Virol Suppl. 2004:133-7
42. Kielian M, Rey FA. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol. 2006;4:67-76
43. Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690-8
44. Clyde K, Kyle JL, Harris E. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol. 2006;80:11418-31
45. Rodenhuis-Zybert IA, Wilschut J, Smit JM. Dengue virus life cycle: viral and host factors modulating infectivity. Cell Mol Life Sci. 2010;67:2773-86
46. Samsa MM, Mondotte JA, Iglesias NG, Assuncao-Miranda I, Barbosa-Lima G, Da Poian AT. et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009;5:e1000632
47. Zhang Y, Zhang W, Ogata S, Clements D, Strauss JH, Baker TS. et al. Conformational changes of the flavivirus E glycoprotein. Structure. 2004;12:1607-18
48. Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J. et al. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004;23:728-38
49. Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997;71:8475-81
50. Zybert IA, van der Ende-Metselaar H, Wilschut J, Smit JM. Functional importance of dengue virus maturation: infectious properties of immature virions. J Gen Virol. 2008;89:3047-51
51. Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551-7
52. Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800-8
53. Gentilucci L, De Marco R, Cerisoli L. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr Pharm Des. 2010;16:3185-203
54. Cohen IR. Peptide therapy for Type I diabetes: the immunological homunculus and the rationale for vaccination. Diabetologia. 2002;45:1468-74
55. Dando TM, Perry CM. Enfuvirtide. Drugs. 2003;63:2755-66 discussion 67-8
56. Martinez NR, Augstein P, Moustakas AK, Papadopoulos GK, Gregori S, Adorini L. et al. Disabling an integral CTL epitope allows suppression of autoimmune diabetes by intranasal proinsulin peptide. J Clin Invest. 2003;111:1365-71
57. Gallwitz B. New therapeutic strategies for the treatment of type 2 diabetes mellitus based on incretins. Rev Diabet Stud. 2005;2:61-9
58. Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. 2011;11:37-51
59. Fox JL. Antimicrobial peptides stage a comeback. Nat Biotechnol. 2013;31:379-82
60. Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M. et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol. 2016;65:490-8
61. Research TM. Peptide therapeutics market-Global industry analysis, size, share, growth, trends and forecast 2016-2024. 26 April. 2017:1-236
62. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20:122-8
63. Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18:807-17
64. Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM. et al. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:155-64
65. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A. et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337-45
66. Widakowich C, de Castro G Jr, de Azambuja E, Dinh P, Awada A. Review: side effects of approved molecular targeted therapies in solid cancers. Oncologist. 2007;12:1443-55
67. Greenstein AJ, Wahed AS, Adeniji A, Courcoulas AP, Dakin G, Flum DR. et al. Prevalence of adverse intraoperative events during obesity surgery and their sequelae. J Am Coll Surg. 2012;215:271-7 e3
68. Pugach P, Ketas TJ, Michael E, Moore JP. Neutralizing antibody and anti-retroviral drug sensitivities of HIV-1 isolates resistant to small molecule CCR5 inhibitors. Virology. 2008;377:401-7
69. Lieberman-Blum SS, Fung HB, Bandres JC. Maraviroc: a CCR5-receptor antagonist for the treatment of HIV-1 infection. Clin Ther. 2008;30:1228-50
70. Urban S, Bartenschlager R, Kubitz R, Zoulim F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology. 2014;147:48-64
71. Perera-Lecoin M, Meertens L, Carnec X, Amara A. Flavivirus entry receptors: an update. Viruses. 2014;6:69
72. Cruz-Oliveira C, Freire JM, Conceição TM, Higa LM, Castanho MARB, Da Poian AT. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiology Reviews. 2015;39:155-70
73. Hung JJ, Hsieh MT, Young MJ, Kao CL, King CC, Chang W. An external loop region of domain III of dengue virus type 2 envelope protein is involved in serotype-specific binding to mosquito but not mammalian cells. J Virol. 2004;78:378-88
74. Miller JL, de Wet BJ, Martinez-Pomares L, Radcliffe CM, Dwek RA, Rudd PM. et al. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008;4:e17
75. Reyes-Del Valle J, Chavez-Salinas S, Medina F, Del Angel RM. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J Virol. 2005;79:4557-67
76. Thepparit C, Smith DR. Serotype-specific entry of dengue virus into liver cells: identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J Virol. 2004;78:12647-56
77. Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E. et al. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe. 2012;12:544-57
78. Marques RE, Guabiraba R, Del Sarto JL, Rocha RF, Queiroz AL, Cisalpino D. et al. Dengue virus requires the CC-chemokine receptor CCR5 for replication and infection development. Immunology. 2015;145:583-96
79. Lee E, Pavy M, Young N, Freeman C, Lobigs M. Antiviral effect of the heparan sulfate mimetic, PI-88, against dengue and encephalitic flaviviruses. Antiviral Res. 2006;69:31-8
80. Hidari KI, Takahashi N, Arihara M, Nagaoka M, Morita K, Suzuki T. Structure and anti-dengue virus activity of sulfated polysaccharide from a marine alga. Biochem Biophys Res Commun. 2008;376:91-5
81. Rees CR, Costin JM, Fink RC, McMichael M, Fontaine KA, Isern S. et al. In vitro inhibition of dengue virus entry by p-sulfoxy-cinnamic acid and structurally related combinatorial chemistries. Antiviral Res. 2008;80:135-42
82. Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol. 2002;2:77-84
83. Alvarez CP, Lasala F, Carrillo J, Muniz O, Corbi AL, Delgado R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by ebola virus in cis and in trans. J Virol. 2002;76:6841-4
84. Baribaud F, Pohlmann S, Leslie G, Mortari F, Doms RW. Quantitative expression and virus transmission analysis of DC-SIGN on monocyte-derived dendritic cells. J Virol. 2002;76:9135-42
85. Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J. et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587-97
86. Trumpfheller C, Park CG, Finke J, Steinman RM, Granelli-Piperno A. Cell type-dependent retention and transmission of HIV-1 by DC-SIGN. Int Immunol. 2003;15:289-98
87. Halary F, Amara A, Lortat-Jacob H, Messerle M, Delaunay T, Houles C. et al. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity. 2002;17:653-64
88. Libraty DH, Pichyangkul S, Ajariyakhajorn C, Endy TP, Ennis FA. Human dendritic cells are activated by dengue virus infection: enhancement by gamma interferon and implications for disease pathogenesis. J Virol. 2001;75:3501-8
89. Ho LJ, Wang JJ, Shaio MF, Kao CL, Chang DM, Han SW. et al. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J Immunol. 2001;166:1499-506
90. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245-52
91. Alen MM, De Burghgraeve T, Kaptein SJ, Balzarini J, Neyts J, Schols D. Broad antiviral activity of carbohydrate-binding agents against the four serotypes of dengue virus in monocyte-derived dendritic cells. PLoS One. 2011;6:e21658
92. Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ. et al. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat Med. 1997;3:866-71
93. Germi R, Crance JM, Garin D, Guimet J, Lortat-Jacob H, Ruigrok RW. et al. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology. 2002;292:162-8
94. Hilgard P, Stockert R. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology. 2000;32:1069-77
95. Hung SL, Lee PL, Chen HW, Chen LK, Kao CL, King CC. Analysis of the steps involved in dengue virus entry into host cells. Virology. 1999;257:156-67
96. Watterson D, Kobe B, Young PR. Residues in domain III of the dengue virus envelope glycoprotein involved in cell-surface glycosaminoglycan binding. J Gen Virol. 2012;93:72-82
97. Talarico LB, Pujol CA, Zibetti RG, Faria PC, Noseda MD, Duarte ME. et al. The antiviral activity of sulfated polysaccharides against dengue virus is dependent on virus serotype and host cell. Antiviral Res. 2005;66:103-10
98. Pujol CA, Ray S, Ray B, Damonte EB. Antiviral activity against dengue virus of diverse classes of algal sulfated polysaccharides. Int J Biol Macromol. 2012;51:412-6
99. Ichiyama K, Gopala Reddy SB, Zhang LF, Chin WX, Muschin T, Heinig L. et al. Sulfated polysaccharide, curdlan sulfate, efficiently prevents entry/fusion and restricts antibody-dependent enhancement of dengue virus infection in vitro: a possible candidate for clinical application. PLoS Negl Trop Dis. 2013;7:e2188
100. Vervaeke P, Alen M, Noppen S, Schols D, Oreste P, Liekens S. Sulfated Escherichia coli K5 polysaccharide derivatives inhibit dengue virus infection of human microvascular endothelial cells by interacting with the viral envelope protein E domain III. PLoS One. 2013;8:e74035
101. Kato D, Era S, Watanabe I, Arihara M, Sugiura N, Kimata K. et al. Antiviral activity of chondroitin sulphate E targeting dengue virus envelope protein. Antiviral Res. 2010;88:236-43
102. Acosta EG, Castilla V, Damonte EB. Functional entry of dengue virus into Aedes albopictus mosquito cells is dependent on clathrin-mediated endocytosis. J Gen Virol. 2008;89:474-84
103. van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X. et al. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008;4:e1000244
104. Acosta EG, Castilla V, Damonte EB. Alternative infectious entry pathways for dengue virus serotypes into mammalian cells. Cell Microbiol. 2009;11:1533-49
105. Hase T, Summers PL, Eckels KH. Flavivirus entry into cultured mosquito cells and human peripheral blood monocytes. Arch Virol. 1989;104:129-43
106. Lim HY, Ng ML. A different mode of entry by dengue-2 neutralisation escape mutant virus. Arch Virol. 1999;144:989-95
107. Navarro-Sanchez E, Altmeyer R, Amara A, Schwartz O, Fieschi F, Virelizier JL. et al. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003;4:723-8
108. Takada A, Kawaoka Y. Antibody-dependent enhancement of viral infection: molecular mechanisms and in vivo implications. Rev Med Virol. 2003;13:387-98
109. Aoki C, Hidari KI, Itonori S, Yamada A, Takahashi N, Kasama T. et al. Identification and characterization of carbohydrate molecules in mammalian cells recognized by dengue virus type 2. J Biochem. 2006;139:607-14
110. Sakoonwatanyoo P, Boonsanay V, Smith DR. Growth and production of the dengue virus in C6/36 cells and identification of a laminin-binding protein as a candidate serotype 3 and 4 receptor protein. Intervirology. 2006;49:161-72
111. Wichit S, Jittmittraphap A, Hidari KI, Thaisomboonsuk B, Petmitr S, Ubol S. et al. Dengue virus type 2 recognizes the carbohydrate moiety of neutral glycosphingolipids in mammalian and mosquito cells. Microbiol Immunol. 2011;55:135-40
112. Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313-9
113. Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A. 2003;100:6986-91
114. Modis Y, Ogata S, Clements D, Harrison SC. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol. 2005;79:1223-31
115. Sukupolvi-Petty S, Austin SK, Purtha WE, Oliphant T, Nybakken GE, Schlesinger JJ. et al. Type- and subcomplex-specific neutralizing antibodies against domain III of dengue virus type 2 envelope protein recognize adjacent epitopes. Journal of Virology. 2007;81:12816-26
116. Ji GH, Deng YQ, Yu XJ, Jiang T, Wang HJ, Shi X. et al. Characterization of a novel dengue serotype 4 virus-specific neutralizing epitope on the envelope protein domain III. PLoS One. 2015;10:e0139741
117. Volk DE, Lee YC, Li X, Thiviyanathan V, Gromowski GD, Li L. et al. Solution structure of the envelope protein domain III of dengue-4 virus. Virology. 2007;364:147-54
118. Fibriansah G, Ibarra KD, Ng TS, Smith SA, Tan JL, Lim XN. et al. Cryo-EM structure of an antibody that neutralizes dengue virus type 2 by locking E protein dimers. Science. 2015;349:88-91
119. Hrobowski YM, Garry RF, Michael SF. Peptide inhibitors of dengue virus and west nile virus infectivity. Virol J. 2005;2:49
120. Lok SM, Costin JM, Hrobowski YM, Hoffmann AR, Rowe DK, Kukkaro P. et al. Release of dengue virus genome induced by a peptide inhibitor. PLoS One. 2012;7:e50995
121. Schmidt AG, Yang PL, Harrison SC. Peptide inhibitors of dengue-virus entry target a late-stage fusion intermediate. PLoS Pathog. 2010;6:e1000851
122. Xu Y, Rahman NA, Othman R, Hu P, Huang M. Computational identification of self-inhibitory peptides from envelope proteins. Proteins. 2012;80:2154-68
123. Sainz B Jr, Mossel EC, Gallaher WR, Wimley WC, Peters CJ, Wilson RB. et al. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) infectivity by peptides analogous to the viral spike protein. Virus Res. 2006;120:146-55
124. Melnik LI, Garry RF, Morris CA. Peptide inhibition of human cytomegalovirus infection. Virol J. 2011;8:76
125. Koehler JW, Smith JM, Ripoll DR, Spik KW, Taylor SL, Badger CV. et al. A fusion-inhibiting peptide against rift valley fever virus inhibits multiple, diverse viruses. PLoS Negl Trop Dis. 2013;7:e2430
126. Badani H, Garry RF, Wimley WC. Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim Biophys Acta. 2014;1838:2180-97
127. Galdiero S, Falanga A, Morelli G, Galdiero M. gH625: a milestone in understanding the many roles of membranotropic peptides. Biochim Biophys Acta. 2015;1848:16-25
128. Segrest JP, De Loof H, Dohlman JG, Brouillette CG, Anantharamaiah GM. Amphipathic helix motif: classes and properties. Proteins. 1990;8:103-17
129. Wimley WC, Hristova K, Ladokhin AS, Silvestro L, Axelsen PH, White SH. Folding of beta-sheet membrane proteins: a hydrophobic hexapeptide model. J Mol Biol. 1998;277:1091-110
130. Ladokhin AS, White SH. Folding of amphipathic alpha-helices on membranes: energetics of helix formation by melittin. J Mol Biol. 1999;285:1363-9
131. Mazumder R, Hu ZZ, Vinayaka CR, Sagripanti JL, Frost SD, Kosakovsky Pond SL. et al. Computational analysis and identification of amino acid sites in dengue E proteins relevant to development of diagnostics and vaccines. Virus Genes. 2007;35:175-86
132. Alhoot MA, Rathinam AK, Wang SM, Manikam R, Sekaran SD. Inhibition of dengue virus entry into target cells using synthetic antiviral peptides. Int J Med Sci. 2013;10:719-29
133. Panya A, Bangphoomi K, Choowongkomon K, Yenchitsomanus PT. Peptide inhibitors against dengue virus infection. Chem Biol Drug Des. 2014;84:148-57
134. Kampmann T, Yennamalli R, Campbell P, Stoermer MJ, Fairlie DP, Kobe B. et al. In silico screening of small molecule libraries using the dengue virus envelope E protein has identified compounds with antiviral activity against multiple flaviviruses. Antiviral Res. 2009;84:234-41
135. Poh MK, Yip A, Zhang S, Priestle JP, Ma NL, Smit JM. et al. A small molecule fusion inhibitor of dengue virus. Antiviral Res. 2009;84:260-6
136. Schmidt AG, Yang PL, Harrison SC. Peptide inhibitors of flavivirus entry derived from the E protein stem. J Virol. 2010;84:12549-54
137. Bai F, Town T, Pradhan D, Cox J, Ashish, Ledizet M. et al. Antiviral peptides targeting the West Nile virus envelope protein. J Virol. 2007;81:2047-55
138. Costin JM, Jenwitheesuk E, Lok SM, Hunsperger E, Conrads KA, Fontaine KA. et al. Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl Trop Dis. 2010;4:e721
139. Nicholson CO, Costin JM, Rowe DK, Lin L, Jenwitheesuk E, Samudrala R. et al. Viral entry inhibitors block dengue antibody-dependent enhancement in vitro. Antiviral Res. 2011;89:71-4
140. Chew MF, Tham HW, Rajik M, Sharifah SH. Anti-dengue virus serotype 2 activity and mode of action of a novel peptide. J Appl Microbiol. 2015;119:1170-80
141. Parikesit AA, Kinanty, Tambunan US. Screening of commercial cyclic peptides as inhibitor envelope protein dengue virus (DENV) through molecular docking and molecular dynamics. Pak J Biol Sci. 2013;16:1836-48
142. Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J. et al. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science. 2008;319:1830-4
143. Tomlinson SM, Malmstrom RD, Watowich SJ. New approaches to structure-based discovery of dengue protease inhibitors. Infect Disord Drug Targets. 2009;9:327-43
144. Guirakhoo F, Bolin RA, Roehrig JT. The Murray Valley Encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology. 1992;191:921-31
145. Panya A, Sawasdee N, Junking M, Srisawat C, Choowongkomon K, Yenchitsomanus PT. A peptide inhibitor derived from the conserved ectodomain region of DENV membrane (M) protein with activity against dengue virus infection. Chem Biol Drug Des. 2015;86:1093-104
146. Zheng A, Umashankar M, Kielian M. In vitro and in vivo studies identify important features of dengue virus pr-E protein interactions. PLoS Pathog. 2010;6:e1001157
147. Ma L, Jones CT, Groesch TD, Kuhn RJ, Post CB. Solution structure of dengue virus capsid protein reveals another fold. Proc Natl Acad Sci U S A. 2004;101:3414-9
148. Balinsky CA, Schmeisser H, Ganesan S, Singh K, Pierson TC, Zoon KC. Nucleolin interacts with the dengue virus capsid protein and plays a role in formation of infectious virus particles. J Virol. 2013;87:13094-106
149. Nowak T, Farber PM, Wengler G, Wengler G. Analyses of the terminal sequences of west nile virus structural proteins and of the in vitro translation of these proteins allow the proposal of a complete scheme of the proteolytic cleavages involved in their synthesis. Virology. 1989;169:365-76
150. Faustino AF, Guerra GM, Huber RG, Hollmann A, Domingues MM, Barbosa GM. et al. Understanding dengue virus capsid protein disordered N-Terminus and pep14-23-based inhibition. ACS Chem Biol. 2015;10:517-26
151. De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents. 2009;33:307-20
152. Sundquist WI, Krausslich HG. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med. 2012;2:a006924
153. Menendez-Arias L. Molecular basis of human immunodeficiency virus drug resistance: an update. Antiviral Res. 2010;85:210-31
154. Stocks CE, Lobigs M. Signal peptidase cleavage at the flavivirus C-prM junction: dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J Virol. 1998;72:2141-9
155. Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757-68
156. Keelapang P, Sriburi R, Supasa S, Panyadee N, Songjaeng A, Jairungsri A. et al. Alterations of pr-M cleavage and virus export in pr-M junction chimeric dengue viruses. J Virol. 2004;78:2367-81
157. Falgout B, Pethel M, Zhang YM, Lai CJ. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. Journal of Virology. 1991;65:2467-75
158. Lin C, Amberg SM, Chambers TJ, Rice CM. Cleavage at a novel site in the NS4A region by the yellow fever virus NS2B-3 proteinase is a prerequisite for processing at the downstream 4A/4B signalase site. Journal of Virology. 1993;67:2327-35
159. Lobigs M. Flavivirus premembrane protein cleavage and spike heterodimer secretion require the function of the viral proteinase NS3. Proc Natl Acad Sci U S A. 1993;90:6218-22
160. Teo KF, Wright PJ. Internal proteolysis of the NS3 protein specified by dengue virus 2. J Gen Virol. 1997;78( Pt 2):337-41
161. Schuller A, Yin Z, Brian Chia CS, Doan DN, Kim HK, Shang L. et al. Tripeptide inhibitors of dengue and west nile virus NS2B-NS3 protease. Antiviral Res. 2011;92:96-101
162. Yin Z, Patel SJ, Wang WL, Chan WL, Ranga Rao KR, Wang G. et al. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg Med Chem Lett. 2006;16:40-3
163. Noble CG, Seh CC, Chao AT, Shi PY. Ligand-bound structures of the dengue virus protease reveal the active conformation. J Virol. 2012;86:438-46
164. Lin KH, Ali A, Rusere L, Soumana DI, Kurt Yilmaz N, Schiffer CA. Dengue virus NS2B/NS3 protease inhibitors exploiting the prime side. J Virol. 2017:91
165. Yusof R, Clum S, Wetzel M, Murthy HM, Padmanabhan R. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J Biol Chem. 2000;275:9963-9
166. Li J, Lim SP, Beer D, Patel V, Wen D, Tumanut C. et al. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J Biol Chem. 2005;280:28766-74
167. Nitsche C, Behnam MA, Steuer C, Klein CD. Retro peptide-hybrids as selective inhibitors of the dengue virus NS2B-NS3 protease. Antiviral Res. 2012;94:72-9
168. Nitsche C, Schreier VN, Behnam MA, Kumar A, Bartenschlager R, Klein CD. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J Med Chem. 2013;56:8389-403
169. Behnam MA, Graf D, Bartenschlager R, Zlotos DP, Klein CD. Discovery of nanomolar dengue and west nile virus protease inhibitors containing a 4-benzyloxyphenylglycine residue. J Med Chem. 2015;58:9354-70
170. César FdS, Anna EdSeS, Aline MLM, Carlton AT, Carlos HTdPdS, Cleydson BRdS. et al. Molecular modeling of peptide derivatives NS3 protease inhibitors of the type 2 dengue virus. Current Physical Chemistry. 2016;6:28-39
171. Katzenmeier G. Inhibition of the NS2B-NS3 protease- towards a causative therapy for dengue virus diseases. Dengue Bulletin. 2004;28:58-67
172. Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G. et al. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J Biol Chem. 2001;276:45762-71
173. Tambunan US, Alamudi S. Designing cyclic peptide inhibitor of dengue virus NS3-NS2B protease by using molecular docking approach. Bioinformation. 2010;5:250-4
174. Velmurugan D, Mythily U, Rao K. Design and docking studies of peptide inhibitors as potential antiviral drugs for dengue virus ns2b/ns3 protease. Protein Pept Lett. 2014;21:815-27
175. Xu S, Li H, Shao X, Fan C, Ericksen B, Liu J. et al. Critical effect of peptide cyclization on the potency of peptide inhibitors against dengue virus NS2B-NS3 protease. J Med Chem. 2012;55:6881-7
176. Rothan HA, Abdulrahman AY, Sasikumer PG, Othman S, Rahman NA, Yusof R. Protegrin-1 inhibits dengue NS2B-NS3 serine protease and viral replication in MK2 cells. J Biomed Biotechnol. 2012;2012:251482
177. Rothan HA, Han HC, Ramasamy TS, Othman S, Rahman NA, Yusof R. Inhibition of dengue NS2B-NS3 protease and viral replication in Vero cells by recombinant retrocyclin-1. BMC Infect Dis. 2012;12:314
178. Miyasaki KT, Lehrer RI. b-sheet antibiotic peptides as potential dental therapeutics. International Journal of Antimicrobial Agents. 1998;9:269-80
179. Ackermann M, Padmanabhan R. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J Biol Chem. 2001;276:39926-37
180. Pryor MJ, Rawlinson SM, Butcher RE, Barton CL, Waterhouse TA, Vasudevan SG. et al. Nuclear localization of dengue virus nonstructural protein 5 through its importin alpha/beta-recognized nuclear localization sequences is integral to viral infection. Traffic. 2007;8:795-807
181. Tambunan US, Zahroh H, Utomo BB, Parikesit AA. Screening of commercial cyclic peptide as inhibitor NS5 methyltransferase of dengue virus through molecular docking and molecular dynamics simulation. Bioinformation. 2014;10:23-7
182. Zhao Y, Soh TS, Zheng J, Chan KW, Phoo WW, Lee CC. et al. A crystal structure of the dengue virus NS5 protein reveals a novel inter-domain interface essential for protein flexibility and virus replication. PLoS Pathog. 2015;11:e1004682
183. Bruno BJ, Miller GD, Lim CS. Basics and recent advances in peptide and protein drug delivery. Ther Deliv. 2013;4:1443-67
184. Diao L, Meibohm B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet. 2013;52:855-68
185. Witt KA, Huber JD, Egleton RD, Roberts MJ, Bentley MD, Guo L. et al. Pharmacodynamic and pharmacokinetic characterization of poly(ethylene glycol) conjugation to met-enkephalin analog [D-Pen2, D-Pen5]-enkephalin (DPDPE). J Pharmacol Exp Ther. 2001;298:848-56
186. Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SA, Vogel HJ. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One. 2010:5
187. Knappe D, Henklein P, Hoffmann R, Hilpert K. Easy strategy to protect antimicrobial peptides from fast degradation in serum. Antimicrob Agents Chemother. 2010;54:4003-5
188. Carmona G, Rodriguez A, Juarez D, Corzo G, Villegas E. Improved protease stability of the antimicrobial peptide Pin2 substituted with D-amino acids. Protein J. 2013;32:456-66
189. Delcroix M, Riley LW. Cell-penetrating peptides for antiviral drug development. Pharmaceuticals (Basel). 2010;3:448-70
190. Bock JE, Gavenonis J, Kritzer JA. Getting in shape: controlling peptide bioactivity and bioavailability using conformational constraints. ACS Chem Biol. 2013;8:488-99
191. Kauffman WB, Fuselier T, He J, Wimley WC. Mechanism matters: a taxonomy of cell penetrating peptides. Trends Biochem Sci. 2015;40:749-64
192. Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569-72
193. Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136-47
194. Patterson AD, Idle JR. A metabolomic perspective of small molecule toxicity. General, Applied and Systems Toxicology: John Wiley & Sons, Ltd. 2009
195. Mócsai A, Kovács L, Gergely P. What is the future of targeted therapy in rheumatology: biologics or small molecules?. BMC Medicine. 2014;12:43
196. van den Broek MF, Muller U, Huang S, Zinkernagel RM, Aguet M. Immune defence in mice lacking type I and/or type II interferon receptors. Immunol Rev. 1995;148:5-18
197. Watanabe S, Vasudevan SG. Evaluation of dengue antiviral candidates in vivo in mouse model. Methods Mol Biol. 2014;1138:391-400
198. Chan KW, Watanabe S, Kavishna R, Alonso S, Vasudevan SG. Animal models for studying dengue pathogenesis and therapy. Antiviral Res. 2015;123:5-14
Author contact
![]() Corresponding author: Chit-Laa Poh, Address: Research Centre for Biomedical Sciences, Sunway University, 5, Jalan Universiti, Malaysia. Phone No.: +6 (03) 7491 8622 (ext. 7338) Fax No: +6 (03) 5635 8630 Email address: pohcledu.my
Corresponding author: Chit-Laa Poh, Address: Research Centre for Biomedical Sciences, Sunway University, 5, Jalan Universiti, Malaysia. Phone No.: +6 (03) 7491 8622 (ext. 7338) Fax No: +6 (03) 5635 8630 Email address: pohcledu.my
Received 2017-7-12
Accepted 2017-9-1
Published 2017-10-15