ISSN: 1449-1907International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(11):1163-1172. doi:10.7150/ijms.20285 This issue Cite
Research Paper
Neonatal Lipopolysaccharide Exposure Gender-Dependently Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Male Rats
Peng Zhang1, 2, Juanxiu Lv1, Yong Li1, Lubo Zhang1, Daliao Xiao1 ![]()
1. Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, California, USA;
2. The First Affiliated Hospital, Chongqing Medical University, Chongqing, China.
Citation:
Zhang P, Lv J, Li Y, Zhang L, Xiao D. Neonatal Lipopolysaccharide Exposure Gender-Dependently Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Male Rats. Int J Med Sci 2017; 14(11):1163-1172. doi:10.7150/ijms.20285. https://www.medsci.org/v14p1163.htm
Other stylesAbstract
Background: Adverse stress exposure during the early neonatal period has been shown to cause aberrant development, resulting in an increased risk of adult disease. We tested the hypothesis that neonatal exposure to lipopolysaccharide (LPS) does not alter heart function at rest condition but causes heart dysfunction under stress stimulation later in life. Methods: Saline control or LPS were administered to neonatal rats via intraperitoneal injection. Experiments were conducted in 6 week-old male and female rats. Isolated hearts were perfused in a Langendorff preparation. Results: Neonatal LPS exposure exhibited no effects on the body weight of the developing rats, but induced decreases in the left ventricle (LV) to the body weight ratio in male rats. Neonatal LPS exposure showed no effects on the baseline heart function determined by in vivo and ex vivo experiments, but caused decreases in the post-ischemic recovery of the LV function in male but not female rats. Neonatal LPS-mediated LV dysfunction was associated with an increase in myocardial infarct size and the LDH release in the male rats. Conclusion: The present study provides novel evidence that neonatal immune challenges could induce gender-dependent long-term effects on cardiac development and heart function, which reinforces the notion that adverse stress exposure during the early neonatal period can aggravate heart functions and the development of a heart ischemia-sensitive phenotype later in life.
Keywords: lipopolysaccharide, neonatal exposure, ischemia/reperfusion injury.
Introduction
Cardiovascular diseases (CVDs) are the number 1 cause of death globally. About 17.3 million people die annually from CVDs with the number expected to increase to more than 23.6 million by 2030 [1-3]. CVDs are admitted as one of the most costly diseases to the health care system [4]. Therefore, it is very important to understand the underlying molecular mechanisms of CVD for prevention/treatment. It is well-known that the traditional behavioral risk factors such as unhealthy diet, physical inactivity, tobacco use and harmful use of alcohol can lead to CVDs. However, recent studies suggest that some of the risk factors exposed during pregnancy or in early life stage may cause a programming of CVDs later in life [5-7]. Indeed, inflammatory stimulus during early life, such as a bacterial or virus infection, has been shown to increase the incidence of CVD in adulthood [8-10]. Lipopolysaccharide (LPS) from Gram-negative bacteria acting as an endotoxin is a major component of the bacterial outer membrane, and serves a crucial function in the initiation of the pathophysiological cascades [11]. Recent studies in different animal models have demonstrated that maternal exposure to LPS leads to sepsis in rat offspring at an early age, and gradually develops into hypertension and cardiac remodeling later in adulthoods [12, 13]. Perinatal LPS exposure up-regulates the TNFβ-1 and TNFβ-2 protein expression in the offspring and induces myocardial fibrosis later in life [14]. These findings suggest that the maternal inflammatory exposure plays a key role in the fetal programming of cardiovascular disease later in life.
The neonatal period represents a unique developmental stage during which the immune, central nervous system (CNS), and cardiovascular systems are highly plastic. During this vulnerable period of development, any adverse environmental stimuli may significantly affect the maturation of those organ systems. Previous studies in different animal models have shown that neonatal LPS exposure causes long-term alteration in the immune and central nervous activity later in life [15-17]. Recently, we have also demonstrated that neonatal LPS exposure sensitizes the neonatal brain to hypoxic-ischemic injury in rat model [18]. Of particular interest, neonatal LPS treatment in rodents has been reported to produce acute cardiac dysfunction [19]. However, there is less information about the long-term impact of neonatal LPS exposure on cardiac function later in life. Therefore, in present study we examined the cardiac function later in life after the exposure of LPS in early life and tested the novel hypothesis that neonatal exposure to LPS does not alter heart function at rest condition but causes heart dysfunction under stress stimulation later in life. To test this hypothesis, first we examined the effect of neonatal LPS exposure on the baseline heart function of in vivo via echocardiography analysis and ex vivo via Langendorff apparatus preparation in the 6 week-old rats. Then we measured the ex vivo heart function after ischemia/reperfusion (I/R) stimulation between the saline control group and the neonatal LPS-exposed group to see whether neonatal LPS exposure increased heart I/R injury and heart dysfunction in the 6 week-old rats. In addition, to see if there is a gender-different effect, we examined the heart function both in male and female animals.
Materials and Methods
Experimental animals
Time-dated pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Portage, MI). Animals were allowed to give birth and were kept with their pups in a room maintained at 24°C with a 12-h light/dark cycle. They were provided ad libitum access to normal rat chow, filtered treatment and were randomized to receive saline (control group) or 100 µg/kg LPS (Sigma-Aldrich; catalog #L4524; lipopolysaccharides from Escherichia coli 055:B5, purified by ion-exchange manner, respectively, TLR ligand tested). LPS was given via an intraperitoneal (IP) injection on days P3 and P5. (male: controls, n=12; LPS-treated, n=14; female: controls, n=9, LPS treated, n=10). The rationale for the selected dose of LPS was based on previous studies reported that LPS can induce obvious systemic pro-inflammatory effects and functional changes in neonatal rats [18]. The pups in each group were randomly chosen from different litters. The procedures and protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Echocardiography
At 6 weeks of age, rats were then subjected to transthoracic echocardiography using the LOGIQ e Ultrasound (GE Medical System, Jiangsu) as previously described [20]. Briefly, the rat was shaved in the chest area, and a layer of acoustic-coupling gel was applied to the thorax. Then the rat was placed in the left lateral decubitus position. An M-mode recording of the LV was obtained at the level of the mitral valve in the parasternal view using two-dimensional (2D) echocardiographic guidance in both the short and long axis views. Cardiac function and heart dimensions were evaluated by 2D echocardiography on the anesthetized (2% isoflurane) rat. M-mode tracing was used to measure interventricular septal end diastole (IVSd), interventricular septal end systole (IVSs), posterior wall thickness at end diastole (LVPWd), and end systole (LVPWs). LV mass and functional parameters such as LV end-diastolic dimension (LVEDD), LV end-systolic dimension (LVESD), LV end-diastolic volume (LVEDV) and LV end-systolic volume (LVESV) were calculated using the above primary measurements and accompanying software. Left ventricular ejection fraction (EF) was calculated as (LVEDV-LVESV)/LVEDV and the percentage of left ventricular fractional shortening (FS) was calculated as (LVEDD-LVESD)/LVEDD. The echocardiography data was recorded and analyzed blinded to the different treatments.
Measurement of cardiac function and ischemia-reperfusion injury
Rats were anesthetized with isoflurane (5% for induction, 2% for maintenance) in oxygen (2 L/min for induction, 1 L/min for maintenance). The adequacy of anesthesia was determined by the loss of a pedal withdrawal reflexes and any other reactions from the animal in response to pinching the tail or ear. The hearts were removed from the rats and were retrogradely perfused via the aorta in a modified Langendorff apparatus under constant pressure (70 mmHg) with gassed (95% O2, 5% CO2) Krebs-Henseleit buffer at 37°C, as described previously [21]. A pressure transducer was connected to a saline-filled balloon and inserted into the left ventricular (LV). This was used to assess the ventricular function by measuring ventricular pressure (mmHg) and its first derivative (dP/dt). LV end diastolic pressure (LVEDP) was set at approximately 5 mm Hg. After the baseline recording at 60 minutes, hearts were subjected to 30 minutes of global ischemia followed by 30 minutes of reperfusion. The left ventricular developed pressure (LVDP), heart rate (HR), dp/dtmax, dp/dtmin, and LV end-diastolic pressure (LVEDP) were continuously recorded. Myocardial infarct size was measured as described previously [21]. Briefly, at the end of reperfusion, the left ventricles were collected, cut into four slices, incubated with 1% triphenyltetrazolium chloride solution for 15 minutes at 37°C, and immersed in formalin for 30 minutes. Each slice was then photographed separately, the areas of myocardial infarction in each slice were analyzed by computerized planimetry, corrected for the tissue weight, summed for each heart, and expressed as a percentage of the total left ventricle weight. Lactate dehydrogenase (LDH) activity was measured in coronary effluent that was collected at the end of I/R, using a TOX 7 assay kit (Sigma Aldrich) following the manufacturer's instructions.
Statistical analysis
All data are expressed as the mean ± SEM. Experimental number (n) represents pups from multiple dams. The difference between the groups was compared by the Student's t-test or the analysis of variance (ANOVA) using the Graph-Pad Prism software (GraphPad Software Version 4, San Diego, CA, USA). For all comparisons, P-values less than 0.05 indicated statistical significance.
Results
Effect of LPS on body and heart weight
As shown in the Figure 1, the neonatal LPS treatment had no effects on growth body weights in both male and female rats. In addition, the heart weights (Figure 2A) and left ventricular weights (Figure 2B) that were isolated from the 6 week-old rats did not have differences between the LPS-treated and saline control groups both in male and female rats. However, the LPS treatment slightly decreased the whole heart to body weight ratio (Figure 2C), but significantly decreased the LV to body weight ratio (Figure 2D) in male but not female rats.
Figure 1
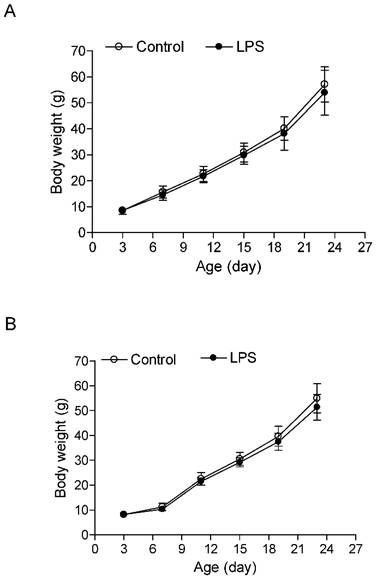
Effect of neonatal LPS exposure on rat body weight. LPS was administered to neonatal rats, as described under Materials and Methods. The control rats received saline. Body weight was measured in both male (A) (n = 11 for control, n = 14 for LPS) and female (B) (n = 11 for control, n = 14 for LPS) rats from 3 to 24 days of age. Data are means ± SEM. Data were analyzed by Student's t-test.
Effect of LPS on baseline heart function
The echocardiographic assessment on in vivo animals indicated that neonatal LPS exposure exhibited no effects on baseline heart function in both male and female rats at the age of 6 week-old (Figure 3 and Table 1). Consistent with the results of the echocardiographic analysis, the ex vivo baseline LV functions before ischemia were also not changed between the LPS-treated and saline control groups in both male and female rats in an isolated heart with Langendorff preparation (Table 2).
Effect of LPS on post-ischemia recovery of LV function
As shown in Figure 4 and 5, global ischemia for 30 minutes caused a damage of LV function in both male and female rats. In male rats, neonatal LPS exposure caused an increase in LVEDP after 30 minutes of ischemia and 30 minutes of reperfusion (Fig 4A). However, neonatal LPS exposure caused decreases in post-ischemic recovery of dP/dtmax and dP/dtmin in the hearts as compared with the saline control groups (Fig 4C-D). As shown in Figure 4, the values of LVDP (Figure 4B), HR (Figure 4E), and CF (Figure 4F) after 30 minutes of ischemia and 30 minutes of reperfusion did not have a difference between the saline control and LPS treated groups. In addition, in female rats the neonatal LPS exposure showed no effects on the post-ischemia recovery of LV function (Figure 5).
Figure 2
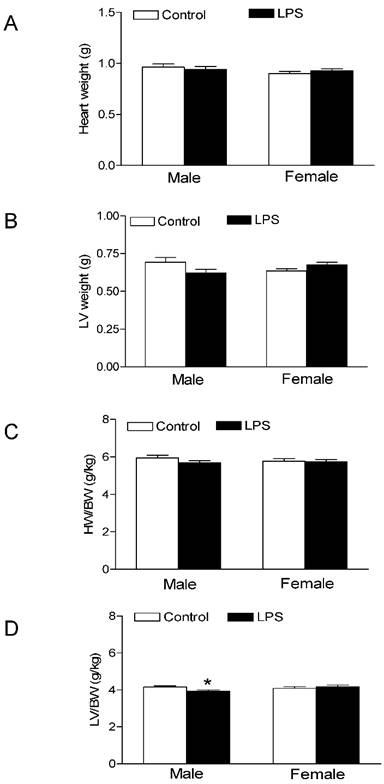
Effect of neonatal LPS exposure on heart weight and heart to body weight ratio. LPS was administered to neonatal rats, as described under Materials and Methods. The control rats received saline. The whole hearts and left ventricle (LV) tissues were isolated from the rats at the age of 6 weeks. The heart weight (A), LV weight (B), heart to body weight ratio (C), and LV to body weight ratio (D) were measured in both male (n = 7 for control, n = 11 for LPS) and female (n = 8 for control, n = 10 for LPS) rats. Data are means ± SEM. * P < 0.05 versus saline control. Data were analyzed by Student's t-test.
Figure 3
Echocardiographic evaluation of cardiac function. LPS was administered to neonatal rats, as described under Materials and Methods. The control rats received saline. At 6 weeks of age, transthoracic echocardiography was performed on the rats after they were anaesthetized with inhaled isoflurane, as described under Materials and Methods. A representative echocardiography shows the measurement of LVSd, LVEDd, LVPWd, LVSs, LVEDs, and LVPWs. A summary of the most relevant cardiac measurements was shown in Table 1. Data were analyzed by Student's t-test.
Table 1
Cardiac function measured by echocardiography.
| Animal groups | C-M | LPS-M | C-F | LPS-F |
|---|---|---|---|---|
| IVSd (cm) | 0.156±0.016 | 0.150±0.022 | 0.147±0.019 | 0.149±0.017 |
| IVSs (cm) | 0.255±0.027 | 0.244±0.035 | 0.235±0.038 | 0.246±0.036 |
| LVEDD (cm) | 0.647±0.037 | 0.639±0.045 | 0.582±0.042 | 0.567±0.048 |
| LVESD (cm) | 0.375±0.039 | 0.385±0.045 | 0.338±0.039 | 0.333±0.040 |
| LVPWd (cm) | 0.139±0.013 | 0.134±0.011 | 0.129±0.008 | 0.143±0.018 |
| LVPWs (cm) | 0.215±0.030 | 0.202±0.035 | 0.213±0.028 | 0.227±0.055 |
| EF (%) | 78.03±5.57 | 75.16±7.47 | 78.15±6.20 | 77.91±4.41 |
| FS (%) | 42.08±5.28 | 39.80±6.50 | 42.01±5.32 | 41.54±4.15 |
| SV (ml) | 2.81±0.38 | 2.78±0.68 | 2.50±0.38 | 2.42±0.40 |
| LV EDV (ul) | 627.9±100.3 | 607.9±111.8 | 467.8±94.9 | 437.6±97.2 |
| LV ESV (ul) | 136.5±41.6 | 147.8±46.9 | 102.1±39.8 | 98.0±33.2 |
| LV mass (mg/g) | 2.75±0.34 | 2.74±0.70 | 2.53±0.23 | 2.71±0.28 |
Note: A summary of the most relevant cardiac measurement that were obtained at 6 weeks of age using echocardiography. LV, left ventricle; IVSd and IVSs, Interventricular septal end diastole and end systole; LVEDD, LV end-diastolic dimension; LVESD, LV end-systolic dimension; LVPWd and LVPWs, left ventricular posterior wall thickness at end diastole and systole; EF, LV ejection fraction; FS, LV fractional shortening; SV, stroke volume; LVEDV, LV end-diastolic volume; LVESV, LV end-systolic volume. Data are means ± SEM. Data were analyzed by Student's t-test. (male: controls, n=12; LPS-treated, n=14; female: controls, n=9, LPS treated, n=10).
Table 2
Pre-ischemic left ventricular functional parameters.
| Animal groups | HR (beat/min) | LVDP (mmHg) | dP/dtmax (mmHg/s) | dP/dtmin (mmHg/s) | CF (ml/min/g) |
|---|---|---|---|---|---|
| C-M | 323.0±14.5 | 80.4±5.2 | 2779.0±96.7 | 1330.6±59.7 | 6.8±0.4 |
| LPS-M | 321.8±16.7 | 81.5±3.6 | 2860.0±143.3 | 1444.9±93.2 | 7.2±0.6 |
| C-F | 307.3±16.0 | 97.5±5.0 | 3314.0±120.2 | 1658.6±79.0 | 8.3±0.8 |
| LPS-F | 309.9±7.6 | 91.1±3.7 | 3092.0±116.0 | 1523.0±89.0 | 6.6±0.5 |
Note: HR, heart rate; LVDP, left ventricular developed pressure; LVEDP, left ventricular end diastolic; dP/dtmax, maximal rate of contraction; dP/dtmin, maximal rate of relaxation; CF, coronary flow; C, control; LPS, lipopolysaccharide; M, male; F, female. Data are means ± SEM. Data were analyzed by Student's t-test. (male: controls, n=12; LPS-treated, n=14; female: controls, n=9, LPS treated, n=10).
Effect of LPS on the heart ischemic/reperfusion injury
As shown in figure 6, global ischemia/reperfusion (I/R) caused LV myocardial damage and increased the LDH (a myocardial injury biomarker in the perfused hearts) release. In addition, the neonatal LPS exposure caused an increase in myocardial infarct size and LDH release of hearts after 30 minutes of I/R in the male but not female rats as compared with the saline control animals group.
Figure 4
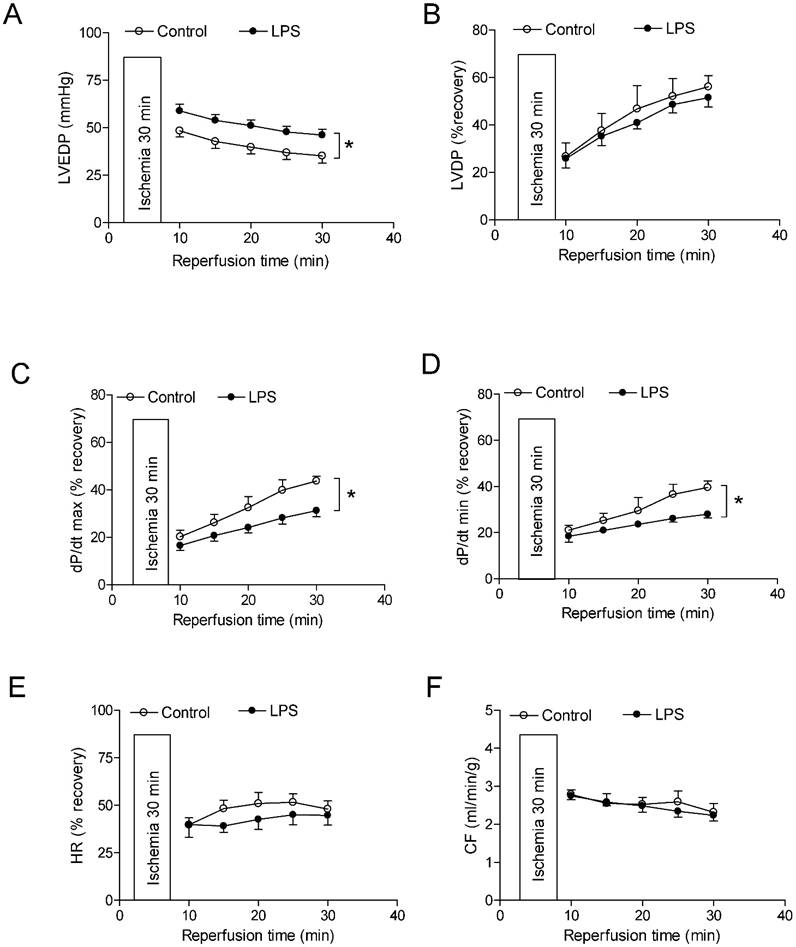
Effects of neonatal LPS exposure on the post-ischemic recovery of LV function in male rats. Hearts were isolated from the 6 week-old male rats that were given the neonatal treatment with saline control or LPS. The hearts were subjected to 30 min of ischemia and 30 min of reperfusion in a langendorff preparation. (A) Post-ischemic recovery of the left ventricular end-diastolic pressure (LVEDP) was determined during the course of reperfusion. (B) Post-ischemic recoveries of the left ventricular developed pressure (LVDP). (C) dP/dpmax. (D) dP/dpmin. (E) Heart rate. (F) Pulmonary artery effluent was collected as an index of coronary flow (milliliters per minute per gram of heart wet weigh). Data are means ± SEM of animals from each group (n = 5-7 for control, n = 8-11 for LPS). Data were analyzed by two way repeated measures ANOVA. *P < 0.05 vs. control.
Figure 5
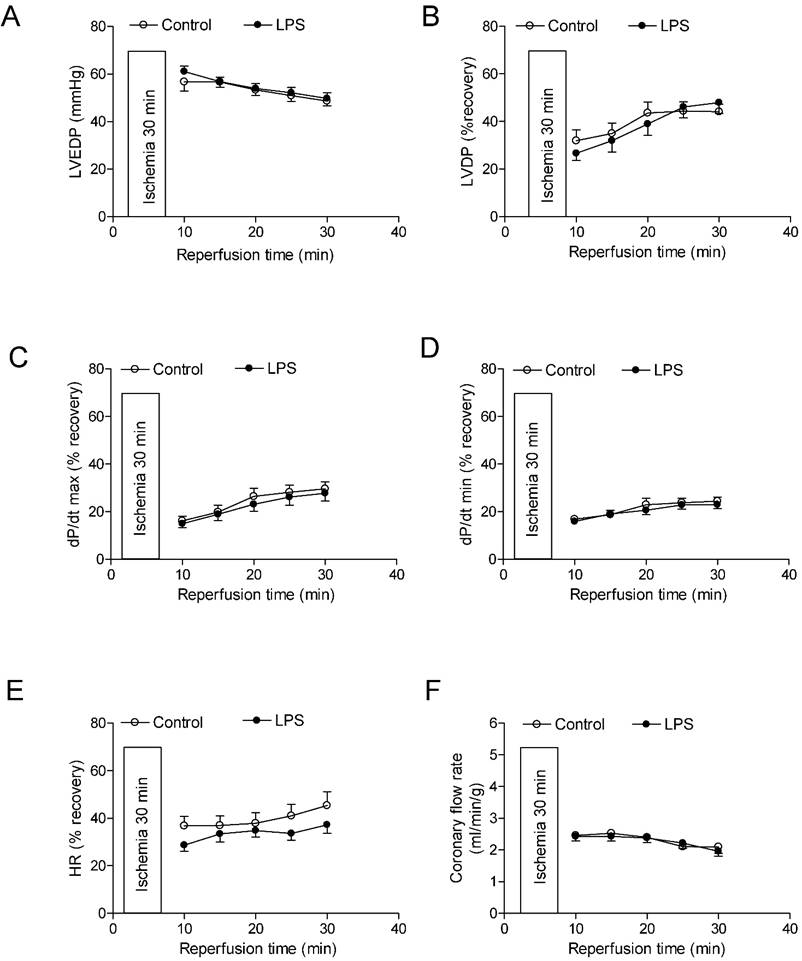
Effects of neonatal LPS exposure on the post-ischemic recovery of LV function in female rats. Hearts were isolated from the 6 week-old female rats that were given the neonatal treatment with saline control or LPS. The hearts were subjected to 30 min of ischemia and 30 min of reperfusion in a langendorff preparation. (A) Post-ischemic recovery of the left ventricular end-diastolic pressure (LVEDP) was determined during the course of reperfusion. (B) Post-ischemic recoveries of the left ventricular developed pressure (LVDP). (C) dP/dpmax. (D) dP/dpmin. (E) Heart rate. (F) Pulmonary artery effluent was collected as an index of coronary flow (milliliters per minute per gram of heart wet weigh). Data are means ± SEM of animals from each group (n = 7-8 for control, n = 10 for LPS). Data were analyzed by two way repeated measures ANOVA.
Figure 6
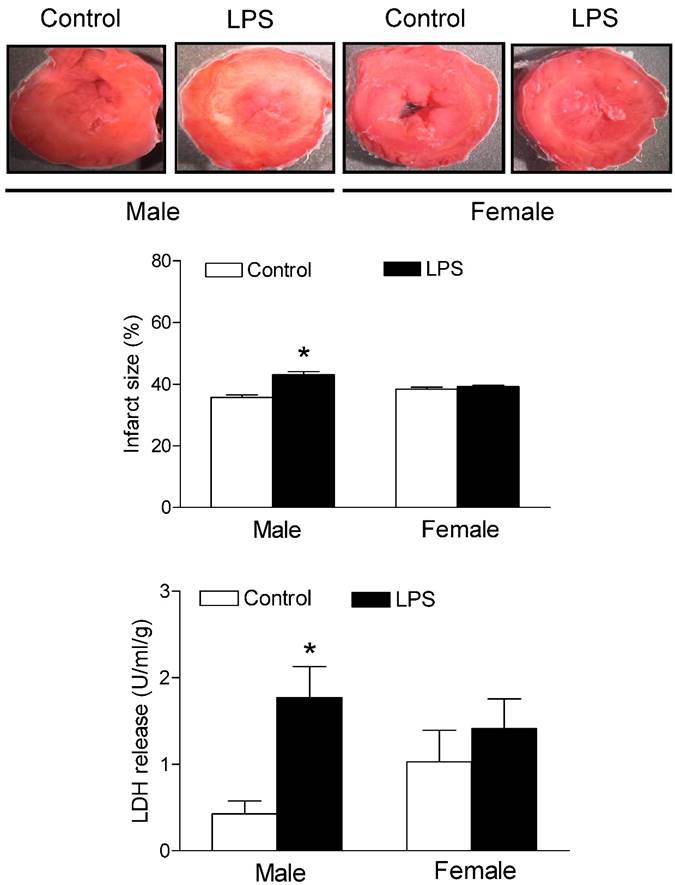
Effects of neonatal LPS exposure on the I/R-induced myocardial injury. Hearts were isolated from the 6 week-old female rats that were given the neonatal treatment with saline control or LPS. The hearts were subjected to 30 min of ischemia and 30 min of reperfusion in a langendorff preparation. The left ventricular tissues were collected at the end of reperfusion, and the myocardial infarct size was determined with 1% triphenyltrazolium chloride (TTC) staining and expressed as a percentage of the total ventricular weight. The lactate dehydrogenase (LDH) activity was measured in coronary effluent that was collected at end of I/R. Data are means ± SEM of animals from each group (male n = 7 for control, n = 11 for LPS; female n = 8 for control, n = 10 for LPS). Data were analyzed by Student's t-test. *P<0.05 vs. control.
Discussion
The present study shows that neonatal LPS exposure induces a gender-dependent development of the ischemic sensitive phenotype of the heart in male rats. The major findings in the present study are that: 1) neonatal LPS exposure exhibited no effects on the body weight of the developing rats, but decreased LV to body weight ratio in male rats; 2) neonatal LPS exposure showed no effects on baseline heart function determined by in vivo and ex vivo experiments; 3) neonatal LPS exposure caused decreases in post-ischemic recovery of LV function in male but not in female rats; 4) the neonatal LPS-mediated LV dysfunction was associated with an increase in myocardial infarct size and LDH release in the male rats.
Growing evidence has shown that adverse perinatal environmental stimuli can alter fetal and neonatal organogenesis and increase the risk of cardiovascular disease later in life [22]. Specifically, fetal and neonatal inflammation is one of the most common risk factors in the developmental programming of cardiovascular disease later in life [11-13, 23, 24]. LPS, a specific inflammatory stimulator, is widely used in different animal models to investigate the effect of perinatal inflammation in fetal and neonatal programming of cardiovascular disease later in life [12-14, 25]. Previous studies have shown that perinatal LPS exposure produces a differential effect on postnatal growth [13, 26]. For example, Wei et al. [13] reported an increase in body weight of the 24 week-old rat offspring prenatally exposed to LPS (0.79 mg/kg, i.p.). In contrast to the increased body weight, a decrease in body weight has been reported in the 3 week-old rats that were exposed to LPS (1 mg/kg, i.c.) at the age of 5 days-old [26]. In the present study, we found that treatment with a low dose of LPS (0.1 mg/kg, i.p.) during the postnatal period (day 3 and 5) did not affect the body weights of the rats at the age of 6 weeks-old. These observations suggest that the effect of neonatal LPS exposure on the animal growth may be dependent on the doses and routes of administration, or the time period of treatment. In our current animal model, our results also indicate that neonatal LPS treatment did not affect the whole heart weight but decreased the left ventricle to body weight ratio in the rat. This suggests that neonatal LPS may cause an asymmetric inhibition of the left ventricle heart development. However, in contrast to our current results, previous studies have demonstrated that prenatal LPS exposure results in myocardial fibrosis and induces myocardial remodeling and cardiac hypertrophy in the adult offspring [12, 14]. More interesting, Wei et al. [12] reported that neonatal LPS exposed hearts showed a normal mass index at the age of 4 months old, but an increased mass index at the age of 8 months old. In present study, the neonatal LPS exposed hearts show a smaller LV size at the age of 1 month old. These findings suggest that the effect of neonatal LPS exposure on cardiac size may be age-dependent.
The present study showed that neonatal LPS exposure had no effect on pre-ischemic baseline values of heart function but significantly increased the LV myocardial infarct size and decreased the post-ischemic recovery of LV function after 30 minutes of I/R in 6 week-old male rats. In addition, our results also indicate that neonatal LPS treatment had no effect on basal cardiac function and heart dimensions evaluated by 2D echocardiography. These data suggest that neonatal LPS treatment with a lower dose (0.1 mg/kg, i.p.) does not impair heart function at a resting condition but alters the heart function when it encounters an ischemic stress challenge later in life. Similar findings have been reported in different animal models where perinatal exposure to adverse stimuli have had no effect on the cardiac function at resting condition but enhance the heart ischemic injury and dysfunction after the ischemia stimulation in adult offspring [27-29]. Our current findings that neonatal LPS treatment caused an increased heart ischemia/reperfusion injury and dysfunction are consistent with previous studies showing a direct link between infection and an epigenetically increased risk of cardiovascular disease later in life [12, 13]. In addition, previous studies have shown that immune stimulation in early life has potentially long-term effects on the neurobehavioral development and can also affect the susceptibility to disease later in life [30, 31]. The molecular mechanisms underlying the neonatal LPS-induced increase in the susceptibility to disease later in life is largely unknown. LPS activates the immune system to release proinflammatory cytokines such as interleukin-1β (IL-1β) and IL-6. These cytokines are considered to be mainly responsible for neuro- and cardiovascular-developmental alterations and the response to disease later in life [12, 26, 32]. Additionally, neonatal LPS exposure is associated with an elevation of IL-1β protein expression in the brain following emotional stress in adulthood [17]. Furthermore, Wei et al. [12] has demonstrated that prenatal exposure to LPS causes a left ventricle hypertrophy and LV diastolic dysfunction associated with an over-expression of the NF-kB protein in the myocardium of LPS-treated adult rat offspring. This study further demonstrated that the LPS-induced cardiac hypertrophy and dysfunction can be rescued by the prenatal treatment with the NF-kB inhibitor [12]. These findings suggest that long-term alteration of the inflammatory cytokine protein expression may be one of the vital molecular mechanisms underlying the fetal and neonatal programming of adult disease. In our future studies, we will need to investigate the effect of neonatal LPS exposure on the specific cytokine cascade and its role in developmental programming of heart ischemia-sensitive phenotype later in life.
In the present study, we found that neonatal LPS exposure significantly increased the I/R-induced heart injury and LV dysfunction in the male but not female rats at the age of 6 week-old. It suggests a sexually dimorphic effect of inflammatory stimuli during the neonatal period on cardiac development and the susceptibility to heart ischemic injury later in life. Consistent with the present study, the gender different response to neonatal LPS treatment has also been reported in different animal models [30, 33]. Tenk et al. [33] examined the effect of the neonatal LPS treatment on exploratory behavior in male and female rats after a challenge with LPS in adulthood and found that adult male but not female rats exhibited less activity in response to the LPS challenge compared with the saline control groups. Furthermore, a recent study has demonstrated that neonatal LPS exposure induces gender-dependent behavioral, neuroendocrine, and immune effects after a LPS challenge in adulthood [30]. Although the precise mechanisms underlying the neonatal LPS exposure-induced sexually dimorphic effects later in life are not completely understood, the thrifty phenotype hypothesis may at least partly explain the gender difference. The thrifty phenotype hypothesis proposes that early life stresses can induce specific adaptation responses to the environmental stimuli [34, 35]. Male and female have different reproductive systems which could produce different responses to environmental cues. For example, a previous study has shown that, in response to neonatal LPS exposure, males exhibit an increase in cell proliferation, and females exhibit a decrease in corticosterone levels [30]. This leads to differential changes in the susceptibility to disease later in life in a gender-dependent manner. Growing evidence shows that steroid hormones such as estrogen may contribute to sex differences in fetal and neonatal programming of cardiovascular disease later in life [36, 37]. Estrogen can serve as an anti-oxidant and an NOS stimulator to protect against increased cardiovascular dysfunction in females that were prenatally exposed to adverse stresses. Furthermore, previous reports have identified estrogen to be an important immune modulator. Yet, estrogen can have either immune-stimulant or immuno-suppressive effects [38, 39]. Our present data supports the postulated protective effects of estrogen in the females exposed to LPS. However, whether and how estrogen protects against the neonatal LPS exposure-mediated I/R heart injury in females later in life warrant future studies.
In summary, the present study provides novel evidence that the neonatal immune challenge induces the long-term detrimental effects of cardiac development and heart function later in life. Our data suggest that adverse stress exposure during the early neonatal period can aggravate heart function and the development of a heart ischemia-sensitive phenotype later in life. Our results also suggest that, at the lower dose of neonatal LPS exposure, the exposed rats may not display apparent heart developmental defect at the basal condition but exhibit heart dysfunction after an ischemia challenge later in life. As most previous studies on perinatal infection models are limited to male animals, our current study included female animals and extended our knowledge to understand the gender differences in neonatal LPS exposure-induced heart ischemia-sensitive phenotypes. However, the epigenetic molecular mechanisms underlying the neonatal LPS exposure-induced gender-related increase in heart susceptibility to I/R injury later in life remain to be determined.
Acknowledgements
This work was supported by National Institutes of Health Grants R01HL135623 (D.X.), R01HD088039 (D.X.), R03DA041492 (D.X.), R01HL118861 (L.Z.), and by the Regents of the University of Califorinia Tobacco Related Disease Research Program (TRDRP) grant 22XT-0022 (D.X.). The author (Peng Zhang) was supported by China scholarship council (CSC, 201608500088). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Townsend N, Wilson L, Bhatnagar P. et al. Cardiovascular disease in Europe: epidemiological update 2016. European heart journal. 2016;37:3232-45
2. Mozaffarian D, Benjamin EJ, Go AS. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38-360
3. GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117-71
4. Laslett LJ, Alagona P Jr, Clark BA. et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. Journal of the American College of Cardiology. 2012;60:S1-49
5. Ramirez-Velez R. In utero fetal programming and its impact on health in adulthood. Endocrinol Nutr. 2012;59:383-93
6. Elmes MJ, Gardner DS, Langley-Evans SC. Fetal exposure to a maternal low-protein diet is associated with altered left ventricular pressure response to ischaemia-reperfusion injury. The British journal of nutrition. 2007;98:93-100
7. Turdi S, Ge W, Hu N. et al. Interaction between maternal and postnatal high fat diet leads to a greater risk of myocardial dysfunction in offspring via enhanced lipotoxicity, IRS-1 serine phosphorylation and mitochondrial defects. Journal of molecular and cellular cardiology. 2013;55:117-29
8. Hay PE, Lamont RF, Taylor-Robinson D. et al. Abnormal bacterial colonisation of the genital tract and subsequent preterm delivery and late miscarriage. Bmj. 1994;308:295-8
9. DeBoer MD, Lima AA, Oria RB. et al. Early childhood growth failure and the developmental origins of adult disease: do enteric infections and malnutrition increase risk for the metabolic syndrome?. Nutrition reviews. 2012;70:642-53
10. Martin A, Emery S. Metabolic disorders and cardiovascular consequences of HIV infection and antiretroviral therapy. Expert review of clinical pharmacology. 2009;2:381-90
11. Liao W, Wei Y, Yu C. et al. Prenatal exposure to zymosan results in hypertension in adult offspring rats. Clinical and experimental pharmacology & physiology. 2008;35:1413-8
12. Wei Y, Du W, Xiong X. et al. Prenatal exposure to lipopolysaccharide results in myocardial remodelling in adult murine offspring. Journal of inflammation. 2013;10:35
13. Wei YL, Li XH, Zhou JZ. Prenatal exposure to lipopolysaccharide results in increases in blood pressure and body weight in rats. Acta pharmacologica Sinica. 2007;28:651-6
14. Chen X, Tang Y, Gao M. et al. Prenatal exposure to lipopolysaccharide results in myocardial fibrosis in rat offspring. International journal of molecular sciences. 2015;16:10986-96
15. Ellis S, Mouihate A, Pittman QJ. Early life immune challenge alters innate immune responses to lipopolysaccharide: implications for host defense as adults. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1519-21
16. Hodgson DM, Knott B, Walker FR. Neonatal endotoxin exposure influences HPA responsivity and impairs tumor immunity in Fischer 344 rats in adulthood. Pediatric research. 2001;50:750-5
17. Walker AK, Nakamura T, Hodgson DM. Neonatal lipopolysaccharide exposure alters central cytokine responses to stress in adulthood in Wistar rats. Stress. 2010;13:506-15
18. Harding B, Conception K, Li Y. et al. Glucocorticoids Protect Neonatal Rat Brain in Model of Hypoxic-Ischemic Encephalopathy (HIE). International journal of molecular sciences. 2016:18
19. Mukherjee R, McQuinn TC, Dugan MA. et al. Cardiac function and circulating cytokines after endotoxin exposure in neonatal mice. Pediatric research. 2010;68:381-6
20. Wang H, Bei Y, Shen S. et al. miR-21-3p controls sepsis-associated cardiac dysfunction via regulating SORBS2. Journal of molecular and cellular cardiology. 2016;94:43-53
21. Xiao D, Dasgupta C, Chen M. et al. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovascular research. 2014;101:373-82
22. Fernandez-Twinn DS, Ozanne SE. Early life nutrition and metabolic programming. Annals of the New York Academy of Sciences. 2010;1212:78-96
23. Stoll LL, Denning GM, Weintraub NL. Endotoxin, TLR4 signaling and vascular inflammation: potential therapeutic targets in cardiovascular disease. Current pharmaceutical design. 2006;12:4229-45
24. Velten M, Gorr MW, Youtz DJ. et al. Adverse perinatal environment contributes to altered cardiac development and function. American journal of physiology Heart and circulatory physiology. 2014;306:H1334-40
25. Nilsson C, Larsson BM, Jennische E. et al. Maternal endotoxemia results in obesity and insulin resistance in adult male offspring. Endocrinology. 2001;142:2622-30
26. Wang KC, Fan LW, Kaizaki A. et al. Neonatal lipopolysaccharide exposure induces long-lasting learning impairment, less anxiety-like response and hippocampal injury in adult rats. Neuroscience. 2013;234:146-57
27. Bae S, Gilbert RD, Ducsay CA. et al. Prenatal cocaine exposure increases heart susceptibility to ischaemia-reperfusion injury in adult male but not female rats. The Journal of physiology. 2005;565:149-58
28. Lawrence J, Xiao D, Xue Q. et al. Prenatal nicotine exposure increases heart susceptibility to ischemia/reperfusion injury in adult offspring. The Journal of pharmacology and experimental therapeutics. 2008;324:331-41
29. Xiong F, Lin T, Song M. et al. Antenatal hypoxia induces epigenetic repression of glucocorticoid receptor and promotes ischemic-sensitive phenotype in the developing heart. Journal of molecular and cellular cardiology. 2016;91:160-71
30. Bernardi MM, Teixeira LP, Ligeiro-de-Oliveira AP. et al. Neonatal lipopolysaccharide exposure induces sexually dimorphic sickness behavior in adult rats. Psychology & Neuroscience. 2014;7:113-23
31. Stoll BJ, Hansen NI, Adams-Chapman I. et al. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. Jama. 2004;292:2357-65
32. Boisse L, Mouihate A, Ellis S. et al. Long-term alterations in neuroimmune responses after neonatal exposure to lipopolysaccharide. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:4928-34
33. Tenk CM, Kavaliers M, Ossenkopp KP. Sexually dimorphic effects of neonatal immune system activation with lipopolysaccharide on the behavioural response to a homotypic adult immune challenge. International journal of developmental neuroscience: the official journal of the International Society for Developmental Neuroscience. 2008;26:331-8
34. Wells JC. The thrifty phenotype as an adaptive maternal effect. Biological reviews of the Cambridge Philosophical Society. 2007;82:143-72
35. Wells JC. The thrifty phenotype: An adaptation in growth or metabolism?. American journal of human biology: the official journal of the Human Biology Council. 2011;23:65-75
36. Ojeda NB, Grigore D, Robertson EB. et al. Estrogen protects against increased blood pressure in postpubertal female growth restricted offspring. Hypertension. 2007;50:679-85
37. Xiao D, Huang X, Yang S. et al. Estrogen normalizes perinatal nicotine-induced hypertensive responses in adult female rat offspring. Hypertension. 2013;61:1246-54
38. Calippe B, Douin-Echinard V, Delpy L. et al. 17Beta-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor alpha signaling in macrophages in vivo. Journal of immunology. 2010;185:1169-76
39. Gourdy P, Calippe B, Laurell H. et al. Role of inflammatory cytokines in the effect of estradiol on atheroma. Clinical and experimental pharmacology & physiology. 2008;35:396-401
Author contact
![]() Corresponding author: DaLiao Xiao, PhD, Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University, School of Medicine, Loma Linda, CA 92350 Tel: 909-558-4325 Fax: 909-558-4029 E-mail: Dxiaoedu
Corresponding author: DaLiao Xiao, PhD, Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University, School of Medicine, Loma Linda, CA 92350 Tel: 909-558-4325 Fax: 909-558-4029 E-mail: Dxiaoedu
Received 2017-3-27
Accepted 2017-7-24
Published 2017-9-19
Citation styles
APA
Zhang, P., Lv, J., Li, Y., Zhang, L., Xiao, D. (2017). Neonatal Lipopolysaccharide Exposure Gender-Dependently Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Male Rats. International Journal of Medical Sciences, 14(11), 1163-1172. https://doi.org/10.7150/ijms.20285.
ACS
Zhang, P.; Lv, J.; Li, Y.; Zhang, L.; Xiao, D. Neonatal Lipopolysaccharide Exposure Gender-Dependently Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Male Rats. Int. J. Med. Sci. 2017, 14 (11), 1163-1172. DOI: 10.7150/ijms.20285.
NLM
Zhang P, Lv J, Li Y, Zhang L, Xiao D. Neonatal Lipopolysaccharide Exposure Gender-Dependently Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Male Rats. Int J Med Sci 2017; 14(11):1163-1172. doi:10.7150/ijms.20285. https://www.medsci.org/v14p1163.htm
CSE
Zhang P, Lv J, Li Y, Zhang L, Xiao D. 2017. Neonatal Lipopolysaccharide Exposure Gender-Dependently Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Male Rats. Int J Med Sci. 14(11):1163-1172.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.