Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(11):1143-1153. doi:10.7150/ijms.21353 This issue Cite
Research Paper
MEF2C loss-of-function mutation contributes to congenital heart defects
Xiao-Hui Qiao1*, Fei Wang2*, Xian-Ling Zhang3*, Ri-Tai Huang4, Song Xue4, Juan Wang5, Xing-Biao Qiu6, Xing-Yuan Liu7 ![]() , Yi-Qing Yang6,8,9
, Yi-Qing Yang6,8,9 ![]()
1. Department of Pediatric Internal Medicine, Ningbo Women & Children's Hospital, 339 Liuding Street, Ningbo 315012, China;
2. Department of Neurosurgery, Tongji Hospital, Tongji University School of Medicine, 389 Xincun Road, Shanghai 200065, China;
3. Department of Cardiology, Shanghai Tenth People's Hospital, Tongji University School of Medicine, 301 Middle Yanchang Road, Shanghai 200072, China;
4. Department of Cardiovascular Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 1630 Dongfang Road, Shanghai 200127, China;
5. Department of Cardiovascular Medicine, East Hospital, Tongji University School of Medicine, 150 Jimo Road, Shanghai 200120, China;
6. Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China;
7. Department of Pediatrics, Tongji Hospital, Tongji University School of Medicine, 389 Xincun Road, Shanghai 200065, China;
8. Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China;
9. Department of Central Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China.
* These three authors contributed equally to this work.
Received 2017-6-6; Accepted 2017-8-22; Published 2017-9-8
Abstract
Congenital heart disease (CHD) is the most common type of developmental abnormality in humans, and is a leading cause for substantially increased morbidity and mortality in affected individuals. Increasing studies demonstrates a pivotal role of genetic defects in the pathogenesis of CHD, and presently mutations in more than 60 genes have been associated with CHD. Nevertheless, CHD is of pronounced genetic heterogeneity, and the genetic basis underpinning CHD in a large proportion of patients remains unclear. In the present study, the whole coding exons and splicing donors/acceptors of the MEF2C gene, which codes for a transcription factor essential for normal cardiovascular development, were sequenced in 200 unrelated patients affected with CHD, and a novel heterozygous missense mutation, p.L38P, was identified in an index patient with patent ductus arteriosus (PDA) and ventricular septal defect (VSD). Genetic scan of the mutation carrier's family members available showed that the mutation was present in all affected family members but absent in unaffected family members. Analysis of the proband's pedigree revealed that the mutation co-segregated with PDA, which was transmitted as an autosomal dominant trait with complete penetrance. The mutation changed the amino acid that was completely conserved evolutionarily, and did not exist in 300 unrelated, ethnically-matched healthy individuals used as controls. Functional deciphers by using a dual-luciferase reporter assay system unveiled that the mutant MEF2C protein had a significantly reduced transcriptional activity. Furthermore, the mutation significantly diminished the synergistic activation between MEF2C and GATA4, another cardiac core transcription factor that has been causally linked to CHD. In conclusion, this is the first report on the association of a MEF2C loss-of-function mutation with an increased vulnerability to CHD in humans, which provides novel insight into the molecular mechanisms underlying CHD, implying potential implications for early diagnosis and timely prophylaxis of CHD.
Keywords: Congenital heart disease, Genetics, Transcription factor, MEF2C, Reporter gene assay
Introduction
Congenital heart disease (CHD) is the most common type of developmental defect in humans, occurring in almost 3% of neonates when including bicuspid aortic valve, and accounting for approximately one-third of all major birth defects [1-3]. Each year about 1.35 million newborns are born with CHD worldwide, with an estimated incidence of 1% in live births and as high as 10% in still births [1,2]. As a group of structural abnormalities of the heart, CHD is usually classified into 25 different clinical types, including ventricular septal defect (VSD), atrial septal defect, atrioventricular canal defect, patent ductus arteriosus (PDA), tetralogy of Fallot, double outlet right ventricle, transposition of the great arteries, interrupted aortic arch, aortic stenosis, truncus arteriosus, coarctation of the aorta, tricuspid atresia, pulmonary stenosis (PS), pulmonary atresia, hypoplastic left heart, single ventricle, and abnormal pulmonary venous connection [1]. Although mild cardiovascular anomalies can resolve spontaneously [1], major defects may require timely surgical treatment and otherwise can result in degraded health-related quality of life [4], reduced exercise performance [5], retarded central neural development and brain injury [6-9], cerebral and pulmonary thromboembolism [10,11], infective endocarditis [12-16], pulmonary arterial hypertension [17-22], chronic heart failure [23-26], supraventricular and ventricular arrhythmias [27-31] and sudden cardiac death [32-37]. Therefore, CHD represents the most prevalent cause of infant birth defect-related demises, with roughly 24% of infants who died of a birth defect having a cardiovascular deformity [1]. Due to vast advances in medical care and surgical management of CHD during the last 50 years, today 95 % of CHD patients survive to adulthood, which has resulted in a growing population of adults living with CHD, and now there are more adults living with CHD than children [38-41]. However, the morbidity and mortality rates in adult CHD patients are much higher compared with the general population [38-41]. Despite important clinical significance, the causes responsible for CHD in the majority of patients remain unknown.
Previous studies have demonstrated that the etiology of CHD is complex and is associated with both environmental and genetic causes [2,3,41-44]. The well-established environmental risk factors contributing to CHD encompass maternal exposures to toxic chemicals, drugs or ionizing radiation during the first trimester of pregnancy and maternal conditions such as viral infection, immune disorder and diabetes mellitus [41,42]. However, epidemiologic studies strongly suggest genetic defects as the predominant cause of CHD, which is predominantly transmitted in an autosomal dominant pattern in the family, though familial transmission of CHD is also observed in other inheritance modes, including autosomal recessive and X-linked fashions [41-44]. Genetically, regardless of chromosomal duplications and deletions including trisomy 21, trisomy 18 and 22q11.2 microdeletion [45], more than 60 genes, including those encoding cardiac transcription factors, cardiac structural proteins, signaling transducer molecules and chromatin modifiers, have been causally linked to CHD in humans [41-89]. Of these CHD-associated genes, most code for cardiac transcription factors, including the homeobox-containing protein NKX2-5, zinc-finger proteins GATA4, GATA5 and GATA6, and T-box transcription factors TBX1, TBX5 and TBX20 [89,90]. Theses transcription factors show partially overlapping expression profiles and functional characteristics during embryonic cardiogenesis, suggesting that they constitute a regulatory network crucial for normal cardiovascular development [90]. Nevertheless, CHD is a very heterogeneous disease, and the genetic determinants for CHD in the majority of patients remain unclear.
As a member of the MADS (MCM1, agamous, deficiens, serum response factor)-box containing transcription factor family, the myocyte enhancer factor 2C (MEF2C) is highly expressed in various cells, and plays an important role in transcriptional regulation of all three muscle lineages, including cardiac precursor cells and differentiated cardiomyocytes during embryogenesis [91]. In mice, targeted disruption of the Mef2c gene led to embryonic death due to loss of the right ventricle of the heart, failure of the heart to undergo rightward looping morphogenesis [91]. Additionally, in fetal mice ablation of the Mef2c gene in the anterior second heart field, a late differentiating population of cardiac progenitors, caused a spectrum of outflow tract malformations ranging from overriding aorta to double outlet right ventricle and transposition of the great arteries [92]. These findings make it reasonable to screen MEF2C as a preferred candidate gene for CHD in different cohorts of patients.
Materials and methods
Ethics
This study was conducted in compliance with the ethical principles of the Declaration of Helsinki. The study protocol was approved by the Medical Ethics Committee of Tongji Hospital, Tongji University, Shanghai, China [approval no. LL(H)‑09‑07]. Prior to the study, written informed consent was obtained from the guardians of the CHD patients and control individuals.
Study participants
In this study, a consecutive cohort of 200 unrelated patients suffered from CHD was recruited from the Chinese Han population from January 2014 to December 2016, including 110 males and 90 females with an average age of 4.2 ± 3.5 years, ranging from 0 to 15 years of age. The available family members of the index patient harboring an identified MEF2C mutation were also enrolled. A total of 300 healthy, ethnicity- and geographic-matched individuals were registered as controls, including 162 males and 138 females at a mean age of 4.1 ± 3.3 years, ranging from 1 to 14 years of age. Study participants underwent comprehensive clinical evaluation including individual and familial histories, medical records, electrocardiogram and echocardiography. Magnetic resonance imaging, cardiac angiography or cardiac surgery was performed when indicated. Diagnosis of CHD was made according to the echocardiography, magnetic resonance imaging, angiography or direct view during cardiac surgery. The patients with known chromosomal abnormalities or syndromic CHD, such as DiGeorge syndrome, Turner syndrome, Marfan syndrome, Holt-Oram syndrome and Down syndrome, were excluded from the study.
Genetic screening of the human MEF2C gene
Peripheral venous whole-blood samples were collected from all the study participants and genomic DNA was isolated from blood leukocytes using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer's instructions for DNA isolation. The coding exons and flanking exon-intron boundaries of the MEF2C gene (transcript variant 1) were sequenced in 200 unrelated patients with CHD and in 300 ethnicity- and geographic-matched control individuals. The referential genomic DNA sequence of the human MEF2C gene (accession no. NC_000005.10) was derived from the Nucleotide database at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/nuccore/NC_000005.10?from=88718241&to=88904105&report=genbank&strand=true). The primers utilized to amplify the coding regions and splice junction sites of MEF2C by polymerase chain reaction (PCR) were designed as shown in Table 1, with the online Primer-BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast). PCR was performed in a 25 μL mixture containing 20-40 ng of genomic DNA, 1× Buffer (Qiagen, Hilden, Germany), 0.5 μM of each primer, 0.2 mM dNTPs and 0.02 U/μL of HotStar Taq DNA Polymerase (Qiagen). PCR cycling was carried out on Veriti Thermal Cycler (Applied Biosystems, Waltham, MA, USA) using a pre-denaturation cycle at 95°C for 15 min followed by 35 cycles of denaturation at 95 °C for 30 s, annealing of primers at 62 °C for 30 s and extension at 72 °C for 1 min as well as a final extension step at 72 °C for 6 min. Amplicons were fractionated by electrophoresis on a 1.5% agarose gel and purified with the QIAquick Gel Extraction Kit (Qiagen). Both strands of each PCR product were sequenced with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) under an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems) according to the protocols. The identified MEF2C variant was confirmed by sequencing two independent PCR-generated amplicons from the same patient. The position of an exonic sequence variation was numbered according to the reference sequence of the MEF2C mRNA transcript variant 1 at the Nucleotide database (accession no. NM_002397.4). Additionally, the identified sequence variation was queried in the single nucleotide polymorphism database (http://www.ncbi.nlm.nih.gov/SNP), the human gene mutation database (http://www.hgmd.org), the 1000 Genome Project database (http://www.1000genomes.org), and the Exome Aggregation Consortium (http://exac.broadinstitute.org) to confirm its novelty.
Primers to amplify the coding and adjacent non-coding sequences of the MEF2C gene.
| Coding exon | Forward primer (5´→3´) | Backward primer (5´→3´) | Amplicon (bp) |
|---|---|---|---|
| 1 | ACGAATGCAGGAATTTGGGAACTG | CTTTGTTTGATGATGCGTGCAATGT | 525 |
| 2 | AGGGCTCTGACAACTACACACC | TCCAAACTCCCCTGCTTGCG | 371 |
| 3 | CCCTGAATGTCTCTTACAGCCTT | CTAAATGAGCTGCCCGTGGGA | 542 |
| 4 | CTGAACGTCTTGAGAGCTCTTCTT | GACCTAGCTCGGGGGTGGAT | 483 |
| 5 | TGTGGGCCTTTAGCCTTTCTCG | TTGTGTCCAAGGATGGTATCCAG | 387 |
| 6 | TTGGGTTACTTGTCCCATGGAGG | AAGCAAGGCTCTGTCAATGGC | 472 |
| 7 | GCTACAGCCAAGCAGTGTGG | CATGCCATTTGAGGGAAGCG | 510 |
| 8 | GTTACTATGGCAACAGCTGGTCT | AATGCTGCAGTGCTGTGGACG | 456 |
| 9 | TGCGGACTATGCTACTTCAGAG | ATTATTACAGAGCTCGCTGTCC | 577 |
| 10 | TGCTCTGGTGTCTATGCGAGTC | ACGGCACATATAATGCATATCG | 527 |
Sequence alignment of MEF2C across species
Conservation of the amino acid altered by a missense allelic variant was evaluated by aligning the protein of human MEF2C to those of chimpanzee, monkey, dog, cattle, mouse, rat, fowl, zebrafish, fruit fly, mosquito and frog using the online MUSCLE software (https://www.ncbi.nlm.nih.gov/homologe ne?cmd=Retrieve&dopt=MultipleAlignment&list_uids=31087).
Prediction of the causative potential of a new MEF2C variation in silico
The disease-causing potential of a new MEF2C variation was predicted by MutationTaster (http://www.mutationtaster.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), PROVEAN (http://provean.jcvi.org/index.php) and SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html).
Plasmids and site-targeted mutagenesis
Human heart cDNAs were prepared as described previously [57]. With the human heart cDNAs as templates, the full-length coding sequences for human MEF2C gene (transcript variant 1; accession no. NM_002397.4) were amplified by PCR using the pfuUltra high-fidelity DNA polymerase (Stratagene, Santa Clara, CA, USA) and a pair of primers (forward primer: 5'-TGGGCTAGCAGAGAGAGAAGAAAAACGGG-3'; reverse primer: 5'-CCAGCGGCCGCACTAGTAAGTAATAATCTGA-3'). The amplified products were doubly digested by restriction enzymes NheI and NotI (TaKaRa, Dalian, Liaoning, China). The resultant product with a length of 1493 base pairs was separated by 1.5% agarose gel electrophoresis, extracted with the QIAquick Gel Extraction Kit (Qiagen), and then inserted into the NheI-NotI sites of the pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA) to produce a recombinant expression plasmid MEF2C-pcDNA3.1. The identified mutation was introduced into the wild-type MEF2C-pcDNA3.1 plasmid by site-directed mutagenesis using the QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene) with a complementary pair of primers (Forward: 5′-ATGAGCTGAGCGTGCCGTGTGACTGTGAGAT-3′; Reverse: 5'-ATCTCACAGTCACACGGCACGCTCAGCTCAT-3′) according to the manufacturer's protocols, and was validated by sequencing. The recombinant expression plasmid GATA4-pSSRa and the reporter plasmid ANF-luciferase (ANF-luc), which contains the 2600-bp 5'-untranslated region of the ANF gene and expresses firefly luciferase, were kindly provided by Dr. Ichiro Shiojima from Chiba University School of Medicine, Japan.
Cell transfection and luciferase assays
HeLa cells were cultured in Dulbecco's modified Eagle's media containing 10% fetal bovine serum, 100 μg/ml streptomycin and 100 U/ml penicillin in an incubator with 5% CO2 at 37°C. Cells were seeded in 24-well plates 24 h before transfection with various plasmids using the Lipofectamine 3000® reagent (Invitrogen) according to the manufacturer's instructions. The pGL4.75 (Promega) vector expressing a renilla luciferase was co-transfected into the cells as an internal control to normalize transfection efficiency. For transient transfection experiments, HeLa cells were transfected with 1.0 μg of wild-type MEF2C-pcDNA3.1, 1.0 μg of L38P-mutant MEF2C-pcDNA3.1, 0.5 μg of wild-type MEF2C-pcDNA3.1, or 0.5 μg of wild-type MEF2C-pcDNA3.1 together with 0.5 μg of L38P-mutant MEF2C-pcDNA3.1, in combination with 1.0 μg of ANF-luc and 0.04 μg of pGL4.75 (Promega). To evaluate the synergistic transcriptional activation, the same amount (0.4 μg) of expression plasmid DNA (empty pcDNA3.1, wild-type MEF2C-pcDNA3.1, L38P-mutant MEF2C-pcDNA3.1 or GATA4-pSSRa) was used alone or together, in the presence of 1.0 μg of ANF-luc and 0.04 μg of pGL4.75. The empty plasmid pcDNA3.1 was used as a negative control. Transfections were conducted in three independent experiments in triplicate. Cells were harvested and lysed 36 h after transfection. Luciferase activity of the lysates was measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's manual. Firefly luciferase data was normalized to the renilla luciferase readings, and the results were expressed as mean ± standard deviations (SD).
Statistical analysis
The data was statistically analyzed using the SPSS software package for Windows, version 17.0 (SPSS Inc., Chicago, IL, USA). Continuous variables were expressed as mean ± SD. Categorical variables were expressed as a number and percentage. Comparison of continuous variables between two groups was performed using Student's unpaired t-test; whereas categorical variables were compared with Pearson's χ2 test or Fisher's exact test, when appropriate. A 2-tailed probability value of P < 0.05 was considered to be significantly different.
Results
Clinical features of the study participants
In the present study, 200 unrelated CHD patients was clinically investigated in contrast to 300 unrelated healthy control individuals. The patients and controls were matched in ethnicity, gender, age, and geography. All the patients had echocardiogram- documented CHD, of whom approximately 19% had a positive family history of CHD. The control subjects were healthy with a negative family history of CHD, and their echocardiograms were normal with no evidence of cardiovascular structural deformities. The demographic and clinical characteristics of the cohort of CHD patients that participated in the study are summarized in Table 2.
Demographic and clinical features of the patients with congenital heart disease (n = 200).
| Parameter | n or mean | % or range |
|---|---|---|
| Gender | ||
| Male | 110 | 55 |
| Female | 90 | 45 |
| Age (years) | 4 | 0-15 |
| Positive family history of CHD | 38 | 19 |
| Distribution of different forms of CHD | ||
| Isolated CHD | 108 | 54 |
| VSD | 32 | 16 |
| ASD | 25 | 12.5 |
| PDA | 16 | 8 |
| AS | 8 | 4 |
| PS | 6 | 3 |
| DORV | 5 | 2.5 |
| TGA | 5 | 2.5 |
| TA | 4 | 2 |
| CoA | 4 | 2 |
| PA | 2 | 1 |
| SV | 1 | 0.5 |
| Complex CHD | 92 | 46 |
| TOF | 27 | 13.5 |
| VSD + ASD | 22 | 11 |
| VSD + PDA | 21 | 10.5 |
| DORV + VSD | 7 | 3.5 |
| ASD + PDA | 5 | 2.5 |
| TGA + VSD | 5 | 2.5 |
| TA + VSD | 3 | 1.5 |
| VSD + ASD + PDA | 2 | 1 |
| Incidence of arrhythmias | ||
| Atrioventricular block | 7 | 3.5 |
| Atrial fibrillation | 4 | 2 |
| Treatment | ||
| Surgical treatment | 106 | 53 |
| Catheter-based repair | 69 | 34.5 |
| Follow-up | 25 | 12.5 |
CHD, congenital heart defect; VSD, ventricular septal defect; ASD, atrial septal defect; PDA, patent ductus arteriosus; AS, aortic stenosis; PS, pulmonary stenosis; DORV, double outlet right ventricle; TGA, transposition of the great arteries; TA, truncus arteriosus; CoA, coarctation of the aorta; PA, pulmonary atresia; SV, single ventricle; TOF, tetralogy of Fallot.
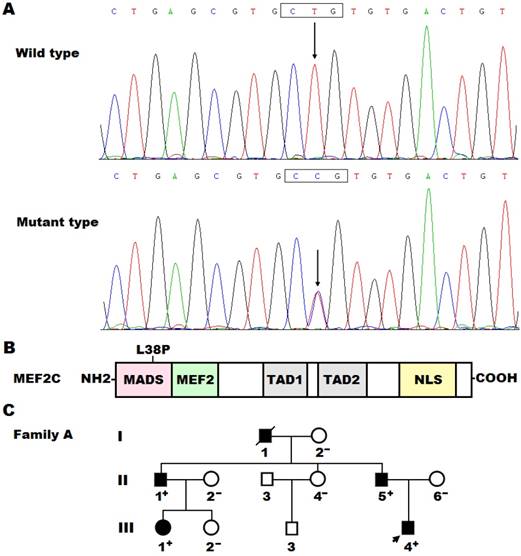
Identification of a novel MEF2C mutation
Sequence analysis of the MEF2C gene in our patient's cohort led to identification of a non-synonymous heterozygous variant in a male patient who was one year old, with a mutational prevalence of 0.5%. Specifically, a substitution of cytosine for thymine at the second nucleotide of codon 38 (c.113T>C), which was predicted to result in the change of leucine at amino acid position 38 into proline (p.L38P), was detected in a boy with PDA and VSD, who had positive family history of CHD. The DNA sequence chromatograms that exhibited the heterozygous allelic mutation of c.113T>C and its homozygous wild-type control base are shown in Figure 1A. A schematic drawing of the MEF2C protein displaying the functionally important structural domains with approximated localization of the mutation found in the current study is illustrated in Figure 1B. The missense mutation was neither discovered in the 300 control subjects nor reported in the single nucleotide polymorphism, human gene mutation, 1000 Genomes Project and Exome Aggregation Consortium database (queried again in June 6, 2017). Genetic scan of the mutation carrier's family members available revealed that the mutation was present in all the affected family members, but absent in unaffected family members. Analysis of the proband's pedigree unveiled that the mutation co-segregated with PDA, which was transmitted as an autosomal dominant trait in the family with complete penetrance. Additionally, the proband's father (II-5) and uncle (II-1) had also VSD and PS, respectively, while his grandfather (I-1) had also VSD and PS and died of heart failure at the age of 58 years. Besides, the affected adult family members had also stereotypic movements, mental retardation, and paroxysmal epilepsy. The pedigree structure of the proband's family is represented in Figure 1C. The clinical characteristics and molecular findings of the proband's affected family members are listed in Table 3.
A novel MEF2C mutation associated with congenital heart disease. (A) DNA sequence chromatograms displaying the heterozygous MEF2C mutation and its corresponding wild-type control. The arrow points to the heterozygous nucleotides of T/C in the proband (mutant type) or the homozygous nucleotides of T/T in a control individual (wild type). The rectangle marks the nucleotides comprising codon 38 of MEF2C. (B) Schematic diagram delineating the structural domains of the MEF2C protein and the location of the mutation involved in congenital heart disease. The mutation identified in patients with congenital heart disease is shown above the structural domains. NH2, amino terminus; MADS, MCM1, agamous, deficiens, serum response factor; MEF2, myocyte enhancer factor 2; TAD1, transcriptional activation domain 1; TAD2, transcriptional activation domain 2; NLS, nuclear localization signal; COOH, carboxyl terminus. (C) Pedigree structure of the family with congenital heart disease. The family was designated as family A. Family members are recognized by generations and numbers. Square indicates male family member; circle, female member; closed symbol, affected member; open symbol, unaffected member; arrow, proband; “+”, carrier of the heterozygous missense mutation; “-”, non-carrier.
Phenotypic characteristics and genotypes for MEF2C of the affected family members.
| Individual | Gender | Age (years) | Cardiac phenotype | MEF2C mutation |
|---|---|---|---|---|
| Family A | L38P | |||
| I-1 | M | 58* | PDA, VSD, PS | NA |
| II-1 | M | 32 | PDA, PS | +/- |
| II-5 | M | 26 | PDA, VSD | +/- |
| III-1 | F | 5 | PDA | +/- |
| III-4 | M | 1 | PDA, VSD | +/- |
M, male; F, female; PDA, patent ductus arteriosus; VSD, ventricular septal defect; PS, pulmonary stenosis; NA, not available; +/-, heterozygote.
*Age at death.
Evolutionary conservation of the altered amino acid
As shown in Figure 2, multiple alignments of the MEF2C protein sequences across species exhibited that the altered leucine at amino acid 38 was completely conserved evolutionarily.
Alignment of multiple MEF2C proteins across species. Multiple alignments of MEF2C proteins among various species exhibited that the altered leucine at amino acid 38 was completely conserved evolutionarily.

A novel MEF2C variation predicted to be pathogenic
The identified MEF2C variation c.113T>C was predicted to be disease-causing with a p value of 1.000 by MutationTaster, probably damaging with a score of 0.999 (sensitivity: 0.09; specificity: 0.99) by PolyPhen-2, deleterious with a PROVEAN score of -6.438 by PROVEAN, and damaging with a SIFT score of 0 and a median information content of 3.41 by SIFT.
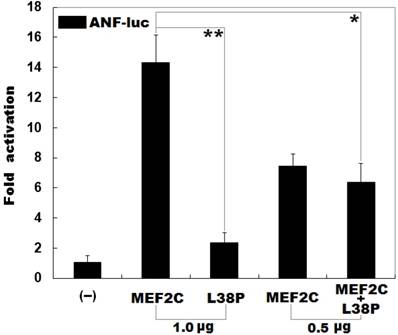
Reduced transcriptional activity of MEF2C caused by the mutation
As shown in Figure 3, the same amount (1.0 μg) of wild-type and L38P-mutant MEF2C-pcDNA3.1 plasmids transcriptionally activated the ANF promoter by ∼14 folds and ∼2 folds, respectively. When 0.5 μg of wild-type MEF2C-pcDNA3.1 was used alone or together with 0.5 μg of L38P-mutant MEF2C-pcDNA3.1, the induced transcriptional activation of the ANF promoter was ∼7-fold or ∼6-fold.
Reduced transcriptional activity of MEF2C caused by the mutation. Activation of the ANF promoter driven luciferase in HeLa cells by wild-type MEF2C or L38P-mutant MEF2C (L38P), alone or together, showed significantly reduced transcriptional activity by the mutant protein. Three independent transfection experiments were conducted in triplicate for each expression plasmid, and mean and standard deviations are shown. ** indicates t = 10.7379, p = 0.00043; * indicates t = 6.29383, p = 0.00326, when compared with wild-type MEF2C (1.0 μg).
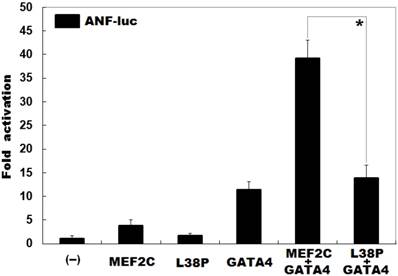
Diminished synergistic activation between MEF2C and GATA4 resulted from the mutation
As shown in Figure 4, in the presence of 0.4 μg of wild-type GATA4, the same amount (0.4 μg) of wild-type and L38P-mutant MEF2C activated the ANF promoter by ∼39 folds and ∼14 folds, respectively; while in the absence of wild-type GATA4, the same amount (0.4 μg) of wild-type and L38P-mutant MEF2C activated the ANF promoter by ∼4 folds and ∼2 folds, respectively.
Diminished synergistic transcriptional activation between MEF2C and GATA4 resulted from the mutation. In the presence of GATA4, activation of the ANF promoter driven luciferase reporter in HeLa cells by wild-type MEF2C or L38P-mutant MEF2C L38P) showed significantly diminished synergistic transcriptional activation between L38P-mutant MEF2C and GATA4. Three independent transfection experiments were done in triplicate for each expression plasmid, and mean and standard deviations are given. * indicates t = 9.37232, p = 0.00072, when compared with the wild-type counterpart.
Discussion
In the present study, a novel heterozygous mutation (c.113T>C, equivalent to p.L38P) in the MEF2C gene was discovered in a family with PDA as well as VSD and PS. The missense mutation, which was absent in the 600 control chromosomes, co-segregated with CHD in the family with complete penetrance. Functional analyses demonstrated that the L38P-mutant MEF2C protein had a significantly diminished transcriptional activity. Furthermore, the mutation significantly decreased the synergistic transcriptional activation between MEF2C and GATA4, another cardiac core transcription factor that has been associated with CHD. Hence, it is very likely that genetically compromised MEF2C contributes to CHD in this family.
To date, there are four members of the MEF2 family found in vertebrates, encompassing MEF2A, MEF2B, MEF2C and MEF2D, of which MEF2B and MEF2C are firstly activated in the heart mesoderm at approximately embryonic day 7.5; while MEF2A and MEF2D are expressed after birth for 24 hours [93]. MEF2C is widely expressed in many types of cells in vivo to regulate tissue-specific gene expression during embryonic period of eukaryote organisms, including cardiac muscle, skeletal muscle, neural, chondroid, immune and endothelial cells [93]. In humans, MEF2C maps on chromosome 5q14.3, which codes for several isoforms of proteins, including isoform 1 with 473 amino acids. The human MEF2C protein contains five key structural domains, including MADS, MEF2, transcriptional activation domain 1 (TAD1), transcriptional activation domain 2 (TAD2), and nuclear localization signal (NLS) [93]. The highly conserved MADS domain at the amino-terminus of MEF2C consists of 56 amino acids, and its main role is to mediate DNA binding, dimerization, and interactions with co-factors. The MEF2 domain is adjoining the MADS domain and comprises 30 highly conserved amino acids, starting from amino acid 57 to amino acid 86. In combination with the MADS domain, the MEF2 domain plays an important role in mediating dimerization and DNA binding. The TAD1 and TAD2 domains function as the transcriptional activators; while the NLS domain is located at the Carboxyl-terminus of MEF2C, which is responsible for nuclear translocation of the protein [93]. In the current study, the mutation detected in CHD patients was located at the MADS domain of MEF2C, and thus was anticipated to impair the transcriptional activity of MEF2C mainly by interfering with its binding to the promoters of target genes, including the ANF gene highly expressed in the heart during embryogenesis [94]. Functional deciphers demonstrated that the L38P-mutant MEF2C protein had significantly reduceded transcriptional activation of the ANF promoter alone or in synergy with GATA4. These results suggest that haploinsufficiency for MEF2C is potentially an alternative pathological mechanism underlying CHD.
Association of MEF2C loss-of-function mutation with enhanced susceptibility to CHD may be partially explained by abnormal cardiovascular development. In fruit fly, there is a single Mef2 gene that encodes a protein with extensive homology to the MADS domains of the mammalian MEF2 proteins, and during embryonic development, its expression is initiated at gastrulation within mesodermal precursor cells in the ventral furrow and then becomes restricted to the somatic, cardiac, and visceral muscle lineages [95]. In the embryos of fruit fly, loss-of-function mutation of Mef2 gives rise to a block in the development of all three types of muscle cells, including cardiac, somatic and visceral muscle cells [95]. In zebrafish, there are two Mef2c genes, namely Mef2ca and Mef2cb, which are expressed similarly in the bilateral heart fields at the primitive heart tube stage, and knockdown of a single Mef2ca leads to delayed heart development, but mutants with loss of a single Mef2cb have a functional heart [96]. In frog, knocking down either Mef2c or Mef2d by corresponding antisense morpholino oligonucleotides results in congenital heart defects including morphological anomalies, pericardial edema, and brachycardia [97]. In mice, MEF2C is the first member of the MEF2 family expressed during embryogenesis in cells of the cardiac mesoderm that form the primitive heart tube [98], and targeted deletion of the Mef2c gene causes severe cardiac structural abnormalities and embryonic lethality in homozygous mutants due to the failure of heart tube to undergo looping morphogenesis and the absence of the right ventricular region of the heart [91]. Furthermore, in mice ablation of Mef2c in the anterior second heart field causes outflow tract defects, such as overriding aorta, double outlet right ventricle and transposition of the great arteries [92]. In humans, all four MEF2 transcript variants (MEF2A, MEF2B, MEF2C and MEF2D) are expressed in all developmental stages of the heart [99]. More importantly, Kodo and colleagues made a sequence analysis of the MEF2C gene in 256 non-syndromic, non-familial patients with cardiac outflow tract defects, and identified a novel sequence variant (p.A103V) in a female patient with pulmonary atresia and VSD [100]. Functional assays using a luciferase reporter showed that the A103V-mutant MEF2C protein had a significantly increased transcriptional activity, and furthermore, overexpression of the A103V-mutant MEF2C in a fish model system disturbed early cardiac development [100]. Taken collectively, these findings together with the results of the current study indicate that genetically compromised MEF2C predisposes to CHD in humans.
Interestingly, in the present study all the affected adult family members harboring the identified MEF2C mutation had also mental retardation, stereotypic movements, and paroxysmal epilepsy. Factually, the MEF2C gene plays a crucial role not only in myogenesis but also in neurogenesis, and MEF2C haploinsufficiency has been involved in the pathogenesis of central nervous system disease [101]. Clinically, the MEF2C haploinsufficiency syndrome has been well recognized as a neurodevelopmental disorder, which is characterized mainly by intellectual disability with inability to speak, limited walking ability, hypotonia, stereotypic movement, epilepsy, and minor brain malformation [101]. Therefore, this study expands the phenotypic spectrum linked to MEF2C mutation.
In conclusion, this is the first report on the association of MEF2C loss-of-function mutation with increased vulnerability to CHD in humans, which provides novel insight into the molecular mechanism of CHD, suggesting potential implications for an improved strategy in early diagnosis and individualized treatment of CHD.
Acknowledgements
The authors are grateful to the study participants for their participation in the study. This work was financially supported by grants from the National Natural Science Foundation of China (grant nos. 81641014 and 81470372), the Natural Science Foundation of Shanghai, China (grant no. 16ZR1432500), and the key program for Basic Research of Shanghai, China (grant no. 14JC1405500).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation. 2017;135:e146-603
2. Postma AV, Bezzina CR, Christoffels VM. Genetics of congenital heart disease: the contribution of the noncoding regulatory genome. J Hum Genet. 2016;61:13-9
3. Gelb BD, Chung WK. Complex genetics and the etiology of human congenital heart disease. Cold Spring Harb Perspect Med. 2014;4:a013953
4. Kahr PC, Radke RM, Orwat S, Baumgartner H, Diller GP. Analysis of associations between congenital heart defect complexity and health-related quality of life using a meta-analytic strategy. Int J Cardiol. 2015;199:197-203
5. Rosenblum O, Katz U, Reuveny R, Williams CA, Dubnov-Raz G. Exercise performance in children and young adults after complete and incomplete repair of congenital heart disease. Pediatr Cardiol. 2015;36:1573-81
6. Peyvandi S, De Santiago V, Chakkarapani E, Chau V, Campbell A, Poskitt KJ, Xu D, Barkovich AJ, Miller S, McQuillen P. Association of prenatal diagnosis of critical congenital heart disease with postnatal brain development and the risk of brain injury. JAMA Pediatr. 2016;170:e154450
7. Marelli A, Miller SP, Marino BS, Jefferson AL, Newburger JW. Brain in congenital heart disease across the lifespan: the cumulative burden of injury. Circulation. 2016;133:1951-62
8. Morton PD, Ishibashi N, Jonas RA. Neurodevelopmental abnormalities and congenital heart disease: insights into altered brain maturation. Circ Res. 2017;120:960-77
9. Pironkova RP, Giamelli J, Seiden H, Parnell VA, Gruber D, Sison CP, Kowal C, Ojamaa K. Brain injury with systemic inflammation in newborns with congenital heart disease undergoing heart surgery. Exp Ther Med. 2017;14:228-38
10. Jensen AS, Idorn L, Thomsen C, von der Recke P, Mortensen J, Sørensen KE, Thilén U, Nagy E, Kofoed KF, Ostrowski SR, Søndergaard L. Prevalence of cerebral and pulmonary thrombosis in patients with cyanotic congenital heart disease. Heart. 2015;101:1540-6
11. Masuda K, Ishizu T, Niwa K, Takechi F, Tateno S, Horigome H, Aonuma K. Increased risk of thromboembolic events in adult congenital heart disease patients with atrial tachyarrhythmias. Int J Cardiol. 2017;234:69-75
12. Rushani D, Kaufman JS, Ionescu-Ittu R, Mackie AS, Pilote L, Therrien J, Marelli AJ. Infective endocarditis in children with congenital heart disease: cumulative incidence and predictors. Circulation. 2013;128:1412-9
13. Kuijpers JM, Koolbergen DR, Groenink M, Peels KCH, Reichert CLA, Post MC, Bosker HA, Wajon EMCJ, Zwinderman AH, Mulder BJM, Bouma BJ. Incidence, risk factors, and predictors of infective endocarditis in adult congenital heart disease: focus on the use of prosthetic material. Eur Heart J. 2017;38:2048-56
14. Diller GP, Baumgartner H. Endocarditis in adults with congenital heart disease: new answers-new questions. Eur Heart J. 2017;38:2057-9
15. Sun LC, Lai CC, Wang CY, Wang YH, Wang JY, Hsu YL, Hu YL, Wu ET, Lin MT, Sy LB, Chen L. Risk factors for infective endocarditis in children with congenital heart diseases - A nationwide population-based case control study. Int J Cardiol. 2017 doi: 10.1016/j.ijcard.2017.08.009
16. Tutarel O, Alonso-Gonzalez R, Montanaro C, Schiff R, Uribarri A, Kempny A, Grübler MR, Uebing A, Swan L, Diller GP, Dimopoulos K, Gatzoulis MA. Infective endocarditis in adults with congenital heart disease remains a lethal disease. Heart. 2017 doi: 10.1136/heartjnl-2017-311650
17. Alonso-Gonzalez R, Lopez-Guarch CJ, Subirana-Domenech MT, Ruíz JM, González IO, Cubero JS, del Cerro MJ, Salvador ML, Dos Subira L, Gallego P, Escribano-Subias P; REHAP investigators. Pulmonary hypertension and congenital heart disease: An insight from the REHAP National Registry. Int J Cardiol. 2015;184:717-23
18. Opotowsky AR. Clinical evaluation and management of pulmonary hypertension in the adult with congenital heart disease. Circulation. 2015;131:200-10
19. Lüscher TF. Frontiers in congenital heart disease: pulmonary hypertension, heart failure, and arrhythmias. Eur Heart J. 2016;37:1407-9
20. Zorzanelli L, Maeda N, Clavé M, Thomaz A, Galas F, Rabinovitch M, Lopes A. Relation of cytokine profile to clinical and hemodynamic features in young patients with congenital heart disease and pulmonary hypertension. Am J Cardiol. 2017;119:119-25
21. Müller J, Heck PB, Ewert P, Hager A. Noninvasive screening for pulmonary hypertension by exercise testing in congenital heart disease. Ann Thorac Surg. 2017;103:1544-9
22. van der Feen DE, Bartelds B, de Boer RA, Berger RM. Pulmonary arterial hypertension in congenital heart disease: translational opportunities to study the reversibility of pulmonary vascular disease. Eur Heart J. 2017;38:2034-2041
23. Stout KK, Broberg CS, Book WM, Cecchin F, Chen JM, Dimopoulos K, Everitt MD, Gatzoulis M, Harris L, Hsu DT, Kuvin JT, Law Y, Martin CM, Murphy AM, Ross HJ, Singh G, Spray TL; American Heart Association Council on Clinical Cardiology, Council on Functional Genomics and Translational Biology, and Council on Cardiovascular Radiology and Imaging. Chronic heart failure in congenital heart disease: a scientific statement from the American Heart Association. Circulation. 2016;133:770-801
24. Ohuchi H, Hayama Y, Negishi J, Noritake K, Iwasa T, Miyazaki A, Yamada O, Shiraishi I. Heart failure with preserved right ventricular ejection fraction in postoperative adults with congenital heart disease: A subtype of severe right ventricular pathophysiology. Int J Cardiol. 2016;212:223-31
25. Budts W, Roos-Hesselink J, Rädle-Hurst T, Eicken A, McDonagh TA, Lambrinou E, Crespo-Leiro MG, Walker F, Frogoudaki AA. Treatment of heart failure in adult congenital heart disease: a position paper of the Working Group of Grown-Up Congenital Heart Disease and the Heart Failure Association of the European Society of Cardiology. Eur Heart J. 2016;37:1419-27
26. Hinton RB, Ware SM. Heart failure in pediatric patients with congenital heart disease. Circ Res. 2017;120:978-94
27. McLeod CJ, Warnes C. Recognition and management of arrhythmias in adult congenital heart disease. Curr Opin Cardiol. 2016;31:117-23
28. El-Assaad I, Al-Kindi SG, Abraham J, Sanatani S, Bradley DJ, Halsey C, Law IH, Balaji S, Shetty I, Aziz PF. Use of dofetilide in adult patients with atrial arrhythmias and congenital heart disease: A PACES collaborative study. Heart Rhythm. 2016;13:2034-9
29. Holst KA, Said SM, Nelson TJ, Cannon BC, Dearani JA. Current interventional and surgical management of congenital heart disease: specific focus on valvular disease and cardiac arrhythmias. Circ Res. 2017;120:1027-44
30. Labombarda F, Hamilton R, Shohoudi A. et al. Increasing prevalence of atrial fibrillation and permanent atrial arrhythmias in congenital heart disease. J Am Coll Cardiol. 2017;70:857-65
31. McLeod CJ. Acute arrhythmias in adults with congenital heart disease. Heart. 2017;103:1380-8
32. Diller GP, Kempny A, Alonso-Gonzalez R, Swan L, Uebing A, Li W, Babu-Narayan S, Wort SJ, Dimopoulos K, Gatzoulis MA. Survival prospects and circumstances of death in contemporary adult congenital heart disease patients under follow-up at a large tertiary center. Circulation. 2015;13:2118-25
33. Khairy P. Ventricular arrhythmias and sudden cardiac death in adults with congenital heart disease. Heart. 2016;102:1703-9
34. Diller GP, Baumgartner H. Sudden cardiac death during exercise in patients with congenital heart disease: the exercise paradox and the challenge of appropriate counselling. Eur Heart J. 2016;37:627-9
35. Engelings CC, Helm PC, Abdul-Khaliq H, Asfour B, Bauer UM, Baumgartner H, Kececioglu D, Körten MA, Diller GP, Tutarel O. Cause of death in adults with congenital heart disease—an analysis of the German National Register for Congenital Heart Defects. Int J Cardiol. 2016;211:31-6
36. Jortveit J, Eskedal L, Hirth A, Fomina T, Døhlen G, Hagemo P, Tell GS, Birkeland S, Øyen N, Holmstrøm H. Sudden unexpected death in children with congenital heart defects. Eur Heart J. 2016;37:621-6
37. Koyak Z, de Groot JR, Bouma BJ, Zwinderman AH, Silversides CK, Oechslin EN, Budts W, Van Gelder IC, Mulder BJ, Harris L. Sudden cardiac death in adult congenital heart disease: can the unpredictable be foreseen? Europace. 2017;19:401-6
38. Bouma BJ, Mulder BJ. Changing landscape of congenital heart disease. Circ Res. 2017;120:908-22
39. Mandalenakis Z, Rosengren A, Skoglund K, Lappas G, Eriksson P, Dellborg M. Survivorship in children and young adults with congenital heart disease in Sweden. JAMA Intern. 2017;177:224-30
40. Williams RG. Late causes of death after congenital heart defects: a population-based study from Finland. J Am Coll Cardiol. 2016;68:499-501
41. Andersen TA, Troelsen Kde L, Larsen LA. Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci. 2014;71:1327-52
42. Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112:707-20
43. Edwards JJ, Gelb BD. Genetics of congenital heart disease. Curr Opin Cardiol. 2016;31:235-41
44. Blue GM, Kirk EP, Giannoulatou E, Sholler GF, Dunwoodie SL, Harvey RP, Winlaw DS. Advances in the genetics of congenital heart disease: a clinician's guide. J Am Coll Cardiol. 2017;69:859-70
45. Calcagni G, Unolt M, Digilio MC, Baban A, Versacci P, Tartaglia M, Baldini A, Marino B. Congenital heart disease and genetic syndromes: new insights into molecular mechanisms. Expert Rev Mol Diagn. 2017;17:861-70
46. Abou Hassan OK, Fahed AC, Batrawi M, Arabi M, Refaat MM, DePalma SR, Seidman JG, Seidman CE, Bitar FF, Nemer GM. NKX2-5 mutations in an inbred consanguineous population: genetic and phenotypic diversity. Sci Rep. 2015;5:8848
47. Zheng J, Li F, Liu J, Xu Z, Zhang H, Fu Q, Wang J, Sun K. Investigation of somatic NKX2-5 mutations in Chinese children with congenital heart disease. Int J Med Sci. 2015;12:538-43
48. Cao Y, Wang J, Wei C, Hou Z, Li Y, Zou H, Meng M, Wang W, Jiang L. Genetic variations of NKX2-5 in sporadic atrial septal defect and ventricular septal defect in Chinese Yunnan population. Gene. 2016;575:29-33
49. Ellesøe SG, Johansen MM, Bjerre JV, Hjortdal VE, Brunak S, Larsen LA. Familial atrial septal defect and sudden cardiac death: identification of a novel NKX2-5 mutation and a review of the literature. Congenit Heart Dis. 2016;11:283-90
50. Xu YJ, Qiu XB, Yuan F, Shi HY, Xu L, Hou XM, Qu XK, Liu X, Huang RT, Xue S, Yang YQ, Li RG. Prevalence and spectrum of NKX2.5 mutations in patients with congenital atrial septal defect and atrioventricular block. Mol Med Rep. 2017;15:2247-54
51. Tong YF. Mutations of NKX2.5 and GATA4 genes in the development of congenital heart disease. Gene. 2016;588:86-94
52. Chen J, Qi B, Zhao J, Liu W, Duan R, Zhang M. A novel mutation of GATA4 (K300T) associated with familial atrial septal defect. Gene. 2016;575:473-7
53. Kassab K, Hariri H, Gharibeh L, Fahed AC, Zein M, El-Rassy I, Nemer M, El-Rassi I, Bitar F, Nemer G. GATA5 mutation homozygosity linked to a double outlet right ventricle phenotype in a Lebanese patient. Mol Genet Genomic Med. 2015;4:160-71
54. Wang J, Mao JH, Ding KK, Xu WJ, Liu XY, Qiu XB, Li RG, Qu XK, Xu YJ, Huang RT, Xue S, Yang YQ. A novel NKX2.6 mutation associated with congenital ventricular septal defect. Pediatr Cardiol. 2015;36:646-56
55. Zhao CM, Sun B, Song HM, Wang J, Xu WJ, Jiang JF, Qiu XB, Yuan F, Xu JH, Yang YQ. TBX20 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med. 2016;54:325-32
56. Zhou YM, Dai XY, Huang RT, Xue S, Xu YJ, Qiu XB, Yang YQ. A novel TBX20 loss-of-function mutation contributes to adult-onset dilated cardiomyopathy or congenital atrial septal defect. Mol Med Rep. 2016;14:3307-14
57. Huang RT, Wang J, Xue S, Qiu XB, Shi HY, Li RG, Qu XK, Yang XX, Liu H, Li N, Li YJ, Xu YJ, Yang YQ. TBX20 loss-of-function mutation responsible for familial tetralogy of Fallot or sporadic persistent truncus arteriosus. Int J Med Sci. 2017;14:323-32
58. Pan Y, Wang ZG, Liu XY, Zhao H, Zhou N, Zheng GF, Qiu XB, Li RG, Yuan F, Shi HY, Hou XM, Yang YQ. A novel TBX1 loss-of-function mutation associated with congenital heart disease. Pediatr Cardiol. 2015;36:1400-10
59. Zhou YM, Dai XY, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Huang RT, Xue S, Yang YQ. HAND1 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med. 2016;54:1161-7
60. Li L, Wang J, Liu XY, Liu H, Shi HY, Yang XX, Li N, Li YJ, Huang RT, Xue S, Qiu XB, Yang YQ. HAND1 loss-of-function mutation contributes to congenital double outlet right ventricle. Int J Mol Med. 2017;39:711-8
61. Sun YM, Wang J, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Shi HY, Hou XM, Huang RT, Xue S, Yang YQ. A HAND2 loss-of-function mutation causes familial ventricular septal defect and pulmonary stenosis. G3 (Bethesda). 2016;6:987-92
62. Lu CX, Gong HR, Liu XY, Wang J, Zhao CM, Huang RT, Xue S, Yang YQ. A novel HAND2 loss-of-function mutation responsible for tetralogy of Fallot. Int J Mol Med. 2016;37:445-51
63. Sun YM, Wang J, Qiu XB, Yuan F, Xu YJ, Li RG, Qu XK, Huang RT, Xue S, Yang YQ. PITX2 loss-of-function mutation contributes to tetralogy of Fallot. Gene. 2016;577:258-64
64. Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, Jin SC, Deanfield J, Giardini A, Porter GA Jr, Kim R, Bilguvar K, López-Giráldez F, Tikhonova I, Mane S, Romano-Adesman A, Qi H, Vardarajan B, Ma L, Daly M, Roberts AE, Russell MW, Mital S, Newburger JW, Gaynor JW, Breitbart RE, Iossifov I, Ronemus M, Sanders SJ, Kaltman JR, Seidman JG, Brueckner M, Gelb BD, Goldmuntz E, Lifton RP, Seidman CE, Chung WK. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262-6
65. Guimier A, Gabriel GC, Bajolle F, Tsang M, Liu H, Noll A, Schwartz M, El Malti R, Smith LD, Klena NT, Jimenez G, Miller NA, Oufadem M, Moreau de Bellaing A, Yagi H, Saunders CJ, Baker CN, Di Filippo S, Peterson KA, Thiffault I, Bole-Feysot C, Cooley LD, Farrow EG, Masson C, Schoen P, Deleuze JF, Nitschké P, Lyonnet S, de Pontual L, Murray SA, Bonnet D, Kingsmore SF, Amiel J, Bouvagnet P, Lo CW, Gordon CT. MMP21 is mutated in human heterotaxy and is required for normal left-right asymmetry in vertebrates. Nat Genet. 2015;47:1260-3
66. Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, Chang C, Yagi H, San Agustin JT, Thahir M, Anderton S, Lawhead C, Vescovi A, Pratt H, Morgan J, Haynes L, Smith CL, Eppig JT, Reinholdt L, Francis R, Leatherbury L, Ganapathiraju MK, Tobita K, Pazour GJ, Lo CW. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521:520-4
67. Racedo SE, McDonald-McGinn DM, Chung JH, Goldmuntz E, Zackai E, Emanuel BS, Zhou B, Funke B, Morrow BE. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am J Hum Genet. 2015;96:235-44
68. Sanchez-Castro M, Pichon O, Briand A, Poulain D, Gournay V, David A, Le Caignec C. Disruption of the SEMA3D gene in a patient with congenital heart defects. Hum Mutat. 2015;36:30-3
69. Mlynarski EE, Sheridan MB, Xie M, Guo T, Racedo SE, McDonald-McGinn DM, Gai X, Chow EW, Vorstman J, Swillen A, Devriendt K, Breckpot J, Digilio MC, Marino B, Dallapiccola B, Philip N, Simon TJ, Roberts AE, Piotrowicz M, Bearden CE, Eliez S, Gothelf D, Coleman K, Kates WR, Devoto M, Zackai E, Heine-Suñer D, Shaikh TH, Bassett AS, Goldmuntz E, Morrow BE, Emanuel BS; International Chromosome 22q11.2 Consortium. Copy-number variation of the glucose transporter gene SLC2A3 and congenital heart defects in the 22q11.2 deletion syndrome. Am J Hum Genet. 2015;96:753-64
70. Quintero-Rivera F, Xi QJ, Keppler-Noreuil KM, Lee JH, Higgins AW, Anchan RM, Roberts AE, Seong IS, Fan X, Lage K, Lu LY, Tao J, Hu X, Berezney R, Gelb BD, Kamp A, Moskowitz IP, Lacro RV, Lu W, Morton CC, Gusella JF, Maas RL. MATR3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum Mol Genet. 2015;24:2375-89
71. Theis JL, Hrstka SC, Evans JM, O'Byrne MM, de Andrade M, O'Leary PW, Nelson TJ, Olson TM. Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum Genet. 2015;134:1003-11
72. Theis JL, Zimmermann MT, Evans JM, Eckloff BW, Wieben ED, Qureshi MY, O'Leary PW, Olson T.M. Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circ Cardiovasc Genet. 2015;8:564-71
73. Boyle L, Wamelink MM, Salomons GS, Roos B, Pop A, Dauber A, Hwa V, Andrew M, Douglas J, Feingold M, Kramer N, Saitta S, Retterer K, Cho MT, Begtrup A, Monaghan KG, Wynn J, Chung WK. Mutations in TKT are the cause of a syndrome including short stature, developmental delay, and congenital heart defects. Am J Hum Genet. 2016;98:1235-42
74. Vetrini F, D'Alessandro LC, Akdemir ZC, Braxton A, Azamian MS, Eldomery MK, Miller K, Kois C, Sack V, Shur N, Rijhsinghani A, Chandarana J, Ding Y, Holtzman J, Jhangiani SN, Muzny DM, Gibbs RA, Eng CM, Hanchard NA, Harel T, Rosenfeld JA, Belmont JW, Lupski JR, Yang Y. Bi-allelic mutations in PKD1L1 are associated with laterality defects in Humans. Am J Hum Genet. 2016;99:886-93
75. Fregeau B, Kim BJ, Hernández-García A, Jordan VK, Cho MT, Schnur RE, Monaghan KG, Juusola J, Rosenfeld JA, Bhoj E, Zackai EH, Sacharow S, Barañano K, Bosch DG, de Vries BB, Lindstrom K, Schroeder A, James P, Kulch P, Lalani SR, van Haelst MM, van Gassen KL, van Binsbergen E, Barkovich AJ, Scott DA, Sherr EH. De novo mutations of RERE cause a genetic syndrome with features that overlap those associated with proximal 1p36 deletions. Am J Hum Genet. 2016;98:963-70
76. Huang RT, Xue S, Wang J, Gu JY, Xu JH, Li YJ, Li N, Yang XX, Liu H, Zhang XD, Qu XK, Xu YJ, Qiu XB, Li RG, Yang YQ. CASZ1 loss-of-function mutation associated with congenital heart disease. Gene. 2016;595:62-8
77. Li N, Subrahmanyan L, Smith E, Yu X, Zaidi S, Choi M, Mane S, Nelson-Williams C, Bahjati M, Kazemi M, Hashemi M, Fathzadeh M, Narayanan A, Tian L, Montazeri F, Mani M, Begleiter ML, Coon BG, Lynch HT, Olson EN, Zhao H, Ruland J, Lifton RP, Mani A. Mutations in the histone modifier PRDM6 are associated with isolated nonsyndromic patent ductus arteriosus. Am J Hum Genet. 2016;98:1082-91
78. Reijnders MR, Zachariadis V, Latour B, Jolly L, Mancini GM, Pfundt R, Wu KM, van Ravenswaaij-Arts CM, Veenstra-Knol HE, Anderlid BM, Wood SA, Cheung SW, Barnicoat A, Probst F, Magoulas P, Brooks AS, Malmgren H, Harila-Saari A, Marcelis CM, Vreeburg M, Hobson E, Sutton VR, Stark Z, Vogt J, Cooper N, Lim JY, Price S, Lai AH, Domingo D, Reversade B; DDD Study, Gecz J, Gilissen C, Brunner HG, Kini U, Roepman R, Nordgren A, Kleefstra T. De novo loss-of-function mutations in USP9X cause a female-specific recognizable syndrome with developmental delay and congenital malformations. Am J Hum Genet. 2016;98:373-81
79. Shaheen R, Anazi S, Ben-Omran T, Seidahmed MZ, Caddle LB, Palmer K, Ali R, Alshidi T, Hagos S, Goodwin L, Hashem M, Wakil SM, Abouelhoda M, Colak D, Murray SA, Alkuraya FS. Mutations in SMG9, encoding an essential component of nonsense-mediated decay machinery, cause a multiple congenital anomaly syndrome in humans and mice. Am J Hum Genet. 2016;98:643-52
80. Liu D, Liu QQ, Guan LH, Jiang X, Zhou DX, Beghetti M, Qu JM, Jing ZC. BMPR2 mutation is a potential predisposing genetic risk factor for congenital heart disease associated pulmonary vascular disease. Int J Cardiol. 2016;211:132-6
81. Werner P, Latney B, Deardorff MA, Goldmuntz E. MESP1 mutations in patients with congenital heart defects. Hum Mutat. 2016;37:308-14
82. Priest JR, Osoegawa K, Mohammed N, Nanda V, Kundu R, Schultz K, Lammer EJ, Girirajan S, Scheetz T, Waggott D, Haddad F, Reddy S, Bernstein D, Burns T, Steimle JD, Yang XH, Moskowitz IP, Hurles M, Lifton RP, Nickerson D, Bamshad M, Eichler EE, Mital S, Sheffield V, Quertermous T, Gelb BD, Portman M, Ashley EA. De novo and rare variants at multiple loci support the oligogenic origins of atrioventricular septal heart defects. PLoS Genet. 2016;12:e1005963
83. Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, Prigmore E, Rajan D, Abdul-Khaliq H, Banka S, Bauer UM, Bentham J, Berger F, Bhattacharya S, Bu'Lock F, Canham N, Colgiu IG, Cosgrove C, Cox H, Daehnert I, Daly A, Danesh J, Fryer A, Gewillig M, Hobson E, Hoff K, Homfray T; INTERVAL Study, Kahlert AK, Ketley A, Kramer HH, Lachlan K, Lampe AK, Louw JJ, Manickara AK, Manase D, McCarthy KP, Metcalfe K, Moore C, Newbury-Ecob R, Omer SO, Ouwehand WH, Park SM, Parker MJ, Pickardt T, Pollard MO, Robert L, Roberts DJ, Sambrook J, Setchfield K, Stiller B, Thornborough C, Toka O, Watkins H, Williams D, Wright M, Mital S, Daubeney PE, Keavney B, Goodship J; UK10K Consortium, Abu-Sulaiman RM, Klaassen S, Wright CF, Firth HV, Barrett JC, Devriendt K, FitzPatrick DR, Brook JD; Deciphering Developmental Disorders Study, Hurles ME. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060-5
84. Wang X, Charng WL, Chen CA, Rosenfeld JA, Al Shamsi A, Al-Gazali L, McGuire M, Mew NA, Arnold GL, Qu C, Ding Y, Muzny DM, Gibbs RA, Eng CM, Walkiewicz M, Xia F, Plon SE, Lupski JR, Schaaf CP, Yang Y. Germline mutations in ABL1 cause an autosomal dominant syndrome characterized by congenital heart defects and skeletal malformations. Nat Genet. 2017;49:613-7
85. Ebrahim MA, Williams MR, Shepard S, Perry JC. Genotype positive long QT syndrome in patients with coexisting congenital heart disease. Am J Cardiol. 2017;120:256-61
86. Guo C, Wang Q, Wang Y, Yang L, Luo H, Cao XF, An L, Qiu Y, Du M, Ma X, Li H, Lu C. Exome sequencing reveals novel IRXI mutation in congenital heart disease. Mol Med Rep. 2017;15:3193-7
87. Liu S, Su Z, Tan S, Ni B, Pan H, Liu B, Wang J, Xiao J, Chen Q. Functional analyses of a novel CITED2 nonsynonymous mutation in Chinese Tibetan patients with congenital heart disease. Pediatr Cardiol. 2017;38:1226-31
88. Cristo F, Inácio JM, de Almeida S, Mendes P, Martins DS, Maio J, Anjos R, Belo JA. Functional study of DAND5 variant in patients with congenital heart disease and laterality defects. BMC Med Genet. 2017;18:77
89. Li YJ, Yang YQ. An update on the molecular diagnosis of congenital heart disease: focus on loss-of-function mutations. Expert Rev Mol Diagn. 2017;17:393-401
90. McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol. 2012;100:253-77
91. Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404-7
92. Barnes RM, Harris IS, Jaehnig EJ, Sauls K, Sinha T, Rojas A, Schachterle W, McCulley DJ, Norris RA, Black BL. MEF2C regulates outflow tract alignment and transcriptional control of Tdgf1. Development. 2016;143:774-9
93. Dong C, Yang XZ, Zhang CY, Liu YY, Zhou RB, Cheng QD, Yan EK, Yin DC. Myocyte enhancer factor 2C and its directly-interacting proteins: A review. Prog Biophys Mol Biol. 2017;126:22-30
94. Morin S, Charron F, Robitaille L, Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000;19:2046-55
95. Lilly B, Zhao B, Ranganayakulu G, Paterson BM, Schulz RA, Olson EN. Requirement of MADS domain transcription factor D-MEF2 for muscle formation in Drosophila. Science. 1995;267:688-93
96. Hinits Y, Pan L, Walker C, Dowd J, Moens CB, Hughes SM. Zebrafish Mef2ca and Mef2cb are essential for both first and second heart field cardiomyocyte differentiation. Dev Biol. 2012;369:199-210
97. Guo Y, Kühl SJ, Pfister AS, Cizelsky W, Denk S, Beer-Molz L, Kühl M. Comparative analysis reveals distinct and overlapping functions of Mef2c and Mef2d during cardiogenesis in Xenopus laevis. PLoS One. 2014;9:e87294
98. Edmondson DG, Lyons GE, Martin JF, Olson EN. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994;120:1251-63
99. Iida K, Hidaka K, Takeuchi M, Nakayama M, Yutani C, Mukai T, Morisaki T. Expression of MEF2 genes during human cardiac development. Tohoku J Exp Med. 1999;187:15-23
100. Kodo K, Nishizawa T, Furutani M, Arai S, Ishihara K, Oda M, Makino S, Fukuda K, Takahashi T, Matsuoka R, Nakanishi T, Yamagishi H. Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ J. 2012;76:1703-11
101. Rocha H, Sampaio M, Rocha R, Fernandes S, Leão M. MEF2C haploinsufficiency syndrome: Report of a new MEF2C mutation and review. Eur J Med Genet. 2016;59:478-82
Author contact
![]() Corresponding authors: Dr. Xing-Yuan Liu, Department of Pediatrics, Tongji Hospital, Tongji University School of Medicine, 389 Xincun Road, Shanghai 200065, China. E-mail: liuxingyuan402com; Prof. Yi-Qing Yang, Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. E-mail: dryyqedu.cn.
Corresponding authors: Dr. Xing-Yuan Liu, Department of Pediatrics, Tongji Hospital, Tongji University School of Medicine, 389 Xincun Road, Shanghai 200065, China. E-mail: liuxingyuan402com; Prof. Yi-Qing Yang, Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. E-mail: dryyqedu.cn.