International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907
- Current Issue
- Volume 22; 2025
- Volume 21; 2024
- Volume 20; 2023
- Volume 19; 2022
- Volume 18; 2021
- Archive
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(10):927-936. doi:10.7150/ijms.18812 This issue Cite
Research Paper
Thrombospondin-1 Gene Deficiency Worsens the Neurological Outcomes of Traumatic Brain Injury in Mice
Chongjie Cheng1, 2*, Zhanyang Yu2*, Song Zhao3, Zhengbu Liao1, 2, Changhong Xing2, Yinghua Jiang1, 2, Yong-Guang Yang4, Michael J. Whalen5, Eng H. Lo2, Xiaochuan Sun1 ![]() , Xiaoying Wang2
, Xiaoying Wang2 ![]()
1. Department of Neurosurgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China
2. Neuroprotection Research Laboratory, Departments of Neurology and Radiology, Massachusetts General Hospital, Harvard Medical School, Charlestown, MA, USA;
3. Departments of Orthopedic and Neurosurgery, The First Bethune Hospital of Jilin University, Changchun, Jilin, China;
4. Columbia Center for Translational Immunology, Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY, USA;
5. Department of Pediatrics, Pediatric Critical Care Medicine, Massachusetts General Hospital, Harvard Medical School, Charlestown, MA, USA.
* These authors contributed equally to this work
Received 2016-12-18; Accepted 2017-3-14; Published 2017-7-31
Citation:
Cheng C, Yu Z, Zhao S, Liao Z, Xing C, Jiang Y, Yang YG, Whalen MJ, Lo EH, Sun X, Wang X. Thrombospondin-1 Gene Deficiency Worsens the Neurological Outcomes of Traumatic Brain Injury in Mice. Int J Med Sci 2017; 14(10):927-936. doi:10.7150/ijms.18812. https://www.medsci.org/v14p0927.htm
Other stylesAbstract
Background: Thrombospondin-1 (TSP-1) is an extracellular matrix protein that plays multiple physiological and pathophysiological roles in the brain. Experimental reports suggest that TSP-1 may have an adverse role in neuronal function recovery under certain injury conditions. However, the roles of TSP-1 in traumatic brain injury (TBI) have not been elucidated. In this study we for the first time investigated the roles of TSP-1 in a controlled cortical impact (CCI) model of TBI in TSP-1 knockout (TSP-1 KO) and wild type (WT) mice.
Methods: We examined blood brain-barrier (BBB) damage using at 1 day post-TBI by measuring Evans Blue leakage, and neurological functional recovery at 3 weeks post-TBI by measuring neurological severity score (NSS), wire gripping, corner test and Morris Water Maze (MWM). Mechanistically, we quantified pro-angiogenic biomarkers including cerebral vessel density, vascular endothelial growth factors (VEGF) and angiopoietin-1 (Ang-1) protein expression, synaptic biomarker synaptophysin, and synaptogenesis marker brain-derived neurotrophic factor (BDNF) protein expression in contralateral and ipsilateral (peri-lesion) cortex at 21 days after TBI using immunohistochemistry and Western Blot.
Results: TSP-1 is upregulated at early phase of TBI in WT mice. Compared to WT mice, TSP-1 KO (1) significantly worsened TBI-induced BBB leakage at 1 day after TBI; (2) had similar lesion size as WT mice at 3 weeks after TBI; (3) exhibited a significantly worse neurological deficits in motor and cognitive functions; (4) had no significant difference in cerebral vessel density, but significant increase of VEGF and Ang-1 protein expressions in peri-lesion cortex; (5) significantly increased BDNF but not synaptophysin protein level in peri-lesion cortex compared to sham, but both synaptophysin and BDNF expressions were significantly decreased in contralateral cortex compared to WT.
Conclusion: Our results suggest that TSP-1 may be beneficial for maintaining BBB integrity in the early phase and functional recovery in late phase after TBI. The molecular mechanisms of TSP-1 in early BBB pathophysiology, and long-term neurological function recovery after TBI need to be further investigated.
Keywords: traumatic brain injury, Thromspondin-1 (TSP-1), neurological severity score (NSS), blood-brain-barrier, morris water maze (MWM), angiogenesis, synaptogenesis.
Introduction
Thrombospondin-1 (TSP-1) is a member of the thrombospondin family, an extracellular matrix protein secreted mainly by astrocytes in the brain [1]. It has been known that TSP-1 mediates cell-cell and cell-matrix interactions through communicating with membrane receptors, other extracellular matrix proteins, and cytokines, thus playing important roles in multiple physiological processes including platelet function, vascular remodeling/angiogenesis, synaptogenesis, and wound healing [2, 3]. Multiple adhesion receptors for TSP-1 have been identified, including CD36, integrins, syndecan, and integrin-associated protein (IAP or CD47) [4]. Due to the multi-functions of TSP-1 in association with components of neurovascular unit, TSP-1 has been considered a new target for therapeutic development against traumatic brain injury [5-7]. However, the roles and mechanisms of TSP-1 in TBI remain unknown. Very importantly, a most recently published clinical study showed that TSP-1 was increased in plasma and highly associated with 6-month mortality and unfavorable 6-month outcomes after traumatic brain injury, which further supports the rationale and significance for investigating the role of TSP-1 in TBI [8].
We have previously found exposure to 4N1K, a specific CD47-activating peptide derived from TSP-1, induces neuronal cell death [9]. It also up-regulates vascular endothelial growth factor (VEGF) and matrix metalloproteinase-9 (MMP-9) in brain endothelial cell and astrocytes cultures, suggesting a potential role of TSP-1 in alteration of blood-brain barrier (BBB) homeostasis [10]. Moreover, besides regulating BBB integrity, emerging experimental reports have demonstrated that TSP-1 is mainly produced by astrocytes in the brain, which functions as an anti-angiogenic [11, 12], but pro- synaptogenesis factor [13].
In the context of TBI pathophysiology, a complex cascade of processes is initiated following traumatic brain injury (TBI). Among them, three pathological events or mechanisms are closely linked to the TSP-1 functions, including BBB integrity disruption at early acute injury phase, vascular remodeling/angiogenesis and synaptogenesis at late recovery phase, which in coordination control overall functional outcomes after TBI [14, 15]. Therefore, in this exploratory study, we investigated the roles of TSP-1 in neurological outcomes up to 3 weeks in a TBI model, performed with a controlled cortical impact device (CCI), in TSP-1 gene knockout (TSP-1 KO) and matching WT mice. Potential mechanisms involving BBB integrity disruption at early acute injury phase (1 day), and vascular remodeling/angiogenesis and synaptogenesis at late recovery phase (3 weeks) are also examined.
Materials and Methods
Animals and CCI model
Experimental protocols were approved by the Massachusetts General Hospital Animal Care and Use Committee in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. 12-week-old mice of WT (C57BL/6J, Jackson Laboratory) and TSP-1 deletion (KO) (B6.129S2-Thbs1tmHyn/J, Jackson Laboratory) were used. Totally 142 mice (71 WT, 71 TSP 1-KO) were used in this study. TBI was conducted as previously reported [16, 17]. Briefly, the mice were anesthetized with 4% isoflurane and positioned in a stereotaxic frame. Anesthesia was maintained using 2-3% isoflurane. A midline longitudinal incision was then performed and the skin retracted and skull exposed. A 5.0 mm-diameter craniotomy was made in the left parietal bone midway between bregma and lambda with the medial edge 1 mm lateral to the midline. Mice were impacted at 5.0 m/s with a 40 ms dwell time and 0.6 mm depth using a 3 mm diameter convex tip, mimicking a moderate TBI based on literature [17, 18]. The bone flap was discarded, and the scalp was sutured closed, surgical knots being used to secure the suture. The mice were then returned to their cages to recover from anesthesia.
BBB leakage assessment
The integrity of BBB was investigated by measuring the extravasation of evans blue at 24h after injury following our previously published protocol [19]. Briefly, evans blue dye (2% wt/vol in saline) in a volume of 4ml/kg was given by tail vein injection and allowed to circulate for 1 hour before being sacrificed. After cardio-perfusion with 0.1 mol/l phosphate-buffered saline, the mice were decapitated and brains were removed, weighed, and homogenized in 1.0 ml of trichloroacetic acid (50% in pure water), and centrifuged at 10,000 rpm for 20 min. Then 0.1 ml of the resultant supernatant was added to 0.3 ml of ethanol (100%). The fluorescence were analyzed at 630nm for excitation and 680nm for emission using a spectrophotometer (SpectraMax M5, Molecular devices). The amount of Evans blue was quantified according to Evans blue external standard curve (25-2000 ng/ml) in 50% TCA /ethanol (1:3), and expressed as nanograms of Evans blue per gram of brain tissue.
Behavioral tests
For the timeline of behavioral assay, previous studies have revealed that the injury effect for moderate CCI in mice generally lasts about 3-4 weeks, as summarized in a comprehensive review article by Fujimoto et al [20], and practiced by a large number of experimental studies [21-23], we therefore examined the behavioral deficit and brain lesion volume of the mice up to 21 days after CCI in this study. Before and after CCI (Day -1, 1, 3, 5, 7, 10, 14, 21), the behavioral function of the mice was evaluated according to a set of neurobehavioral tasks (neurological severity score, NSS), corner test and wire gripping test. A 10-point NSS was used for assessment of posttraumatic neurological impairment, as previous described [24, 25]. The NSS was assessed at 1, 3, 5, 7, 10, 14 and 21 days after TBI. All mice were trained and pre-tested prior to injury. Vestibulomotor function was assessed using a standard wire-grip test [26], and performed in triplicate and an average value calculated for each mouse on each day of testing. Furthermore, Morris Water Maze was applied to evaluate spatial memory performance after TBI as previously described [27]. Briefly, from 15 days post-CCI, five consecutive daily training sessions were performed to learn the locational quadrant of the slight underwater platform. For probe trial, the stay time and entry times into the platform area and target quadrant was recorded at day 21. To exclude the potential difference of visual ability between groups, extra visible trial was performed using a labeled platform above the surface of the water. The assessment process was carried out by an investigator who was blinded to the animal groups.
Lesion volume
Lesion volume was measured as we previously described [16]. Briefly, at day 21 after TBI, the animals were perfused with 0.1 mol/l phosphate-buffered saline under deep anesthesia. Brains were frozen-sectioned at the thickness of 10μm. Brain slices 500μm apart were stained with hematoxylin and eosin (H&E) and photographed. The volume of injured tissue was measured with image J software. Damaged tissue volume = contralateral hemisphere volume- ipsilateral hemisphere volume.
Immunohistochemistry
Brain slices were air dried and fixed in 4% paraformaldehyde, then blocked in 5% fetal bovine serum for 80 minutes. After incubation overnight at 4°C with rat anti-mouse CD31 (1:100, BD science), slides were analyzed using fluorescence microscope (ECLIPSE Ti-s, Nikon). For quantification of vessel density, the optical area fraction of CD31 positive cells per 20x field in the peri-lesion area was calculated in 4 randomized areas (2 in cortex, and 2 in sub-cortex) in each animal.
Western blot
Western blot was performed following the protocols as we previously described [28]. Briefly, brain tissue dissected from contused cortex was homogenized in lysis buffer (cell signaling, Cambridge, MA) on ice, and centrifuged at 14,000 RPM for 15mins at 4°C. Equal amount of protein were separated in a 4-20% Tris-glycine gel (Invitrogen) (40 μg/lane) and then transferred onto PVDF Membranes. Membranes were blocked for 1h in 5% non-Fat milk in Tris-buffered saline (pH 7.4) containing 0.1% Tween 20, then incubated overnight at 4°C with mouse anti-actin (Sigma Aldrich), mouse anti-synaptophysin (Millipore), rabbit anti-Ang-1 (Abcam), rabbit anti-VEGF (Santa cruz) and rabbit anti-BDNF (Santa Cruz). After washing with PBST for three times, 20 min each, the membranes were then incubated for 1h with an appropriate horseradish peroxidase-conjugated secondary antibody at room temperature and developed by enhanced chemiluminescent (Pierce, Rockford, IL, USA). Densitometric analysis was performed for quantitation with Image J software.
Statistical analysis
Data are presented as Mean+SEM. Lesion volume, immunoblot and immunohistochemistry were analyzed by Student t test. Neurobehavioral assessments were analyzed by repeated measures ANOVA. Differences with P<0.05 were considered statistically significant.
Results
TSP-1 expression in peri-lesion cortex is transiently upregulated within 3 days after TBI
TSP-1 gene deficiency was verified by genotyping with PCR using genomic DNA from TSP-1-KO mice. The size of PCR product from TSP-1-KO mice is as expected and clearly different from WT, confirming TSP-1 gene deficiency (supplemental Figure S1).
To examine the expression of TSP-1 in response to TBI, we measured the TSP-1 protein level by Western blot at different time points following TBI. Three different antibodies for TSP-1 (mouse anti-TSP-1, NeoMarkers; mouse anti-TSP-1, Santa Cruz; rabbit anti-TSP-1, Abcam) were used to ensure the validity of Western blot. Our results showed that TSP-1 protein was not detectable in the mouse brain cortex before TBI. After TBI, TSP-1 expression in the peri-lesion cortex was significantly upregulated at 6 h and lasted for 3 days, then returned to basal level (Figure 1A), showing a transient upregulation of TSP-1 expression during acute and sub-acute phases within about 3 days after TBI.
TSP-1 knockout worsens CCI-induced BBB permeability increase
To test the effect of TSP-1 knockout on BBB permeability after CCI, we examined Evans blue extravasation into the brain at 24h after TBI (n=4/group). We noticed there was a very low level of Evans blue extravasation in contralateral hemisphere, no difference between TSP-1 KO and WT mice, indicating a similar baseline of BBB permeability at least to large molecules in both group mice. As expected, TBI significantly increased Evans blue extravasation in ipsilateral hemisphere of both TSP-1 KO and WT mice, however, this increase of Evans blue extravasation was significantly potentiated in TSP-1 KO mice compared to WT mice, (Figure 1B), demonstrating that TSP-1 gene knockout exacerbates TBI-induced BBB permeability.
Figure 1
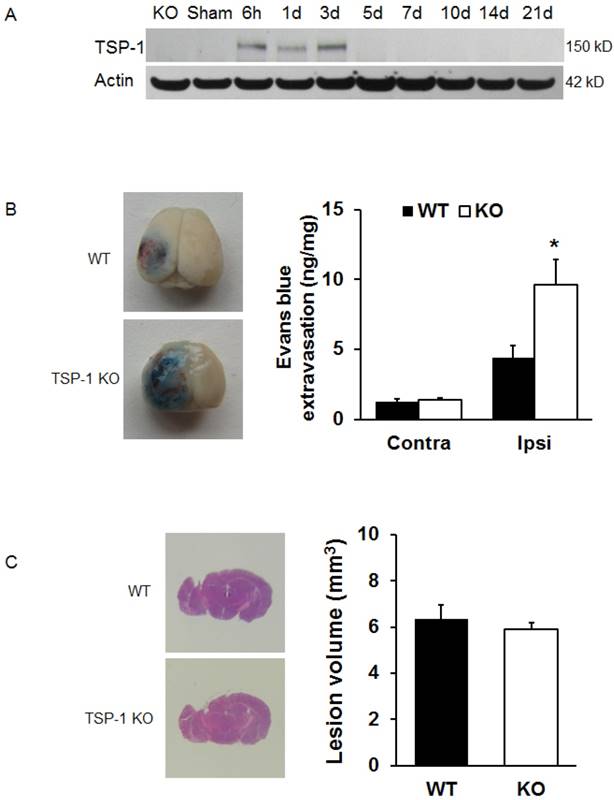
Effects of TSP-1 knockout on BBB leakage and brain lesion in mice after TBI. To examine the contribution of TSP-1 in the outcomes of TBI, we first measured the TSP-1 protein expression changes in response to TBI, then measured the BBB leakage and brain lesion volumes in TSP-1 KO mice and WT mice. (A) Representative Western Blot image of TSP-1 protein levels in the ipsilateral hemisphere at 6 h, 1d, 3d, 5d, 7d, 10d, 14d, 21d following TBI. Samples from TSP-1 KO mice was used as negative control (n=4 for each time point). (B) BBB leakage was measured and quantified at 24 hrs after TBI by testing Evans Blue extravasation (n=4, *p<0.05 vs WT group). (C) Brain lesion volume was measured and quantified at 28 days after TBI (n=16).
TSP-1 knockout does not alter brain lesion volumes after TBI
To examine if TSP-1 contributes to the brain tissue damage progression after TBI, we assessed brain lesion volumes of WT and TSP-1 KO mice at 28 days post-TBI (n=16/group). Our results show that there were no significant differences in lesion volumes between WT and TSP-1 KO mice after CCI (Figure 1C), suggesting that TSP-1 gene knockout during the first 3 weeks does not affect brain tissue damage progression.
TSP-1 knockout potentiates neurological function deficits after TBI
To determine the roles of TSP-1 in neurological functional recovery after TBI, we assessed and compared the motor-sensor behavior outcomes, including neurological severity score (NSS), wire grip and corner test, in WT and TSP-1 KO mice at 1 day before TBI (day -1) and day 1, 3, 5, 7, 10, 14, 21 after TBI (n=16/group). As expected, the motor-sensor functions were significantly impaired by TBI. For NSS, TSP-1 KO mice had significantly worse recovery compared to WT mice (Figure 2A). While both wire grip and corner test of WT and TSP-1 KO mice recovered to baseline at 10 days post-TBI, no significant differences were detected between the two groups (Figure 2B, 2C). These data indicate a significantly worsened motor-sensor function recovery by the gene knockout of TSP-1 after TBI.
Figure 2
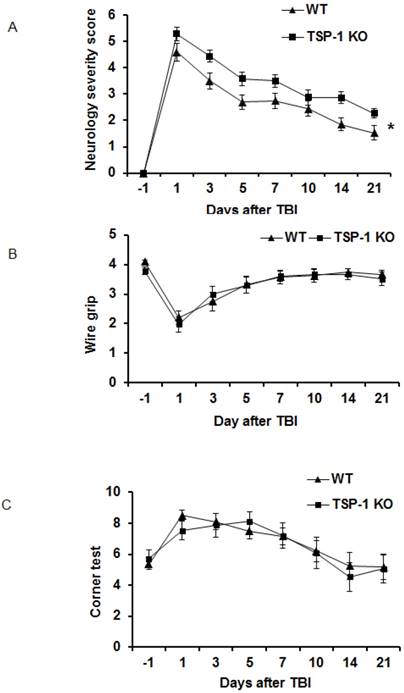
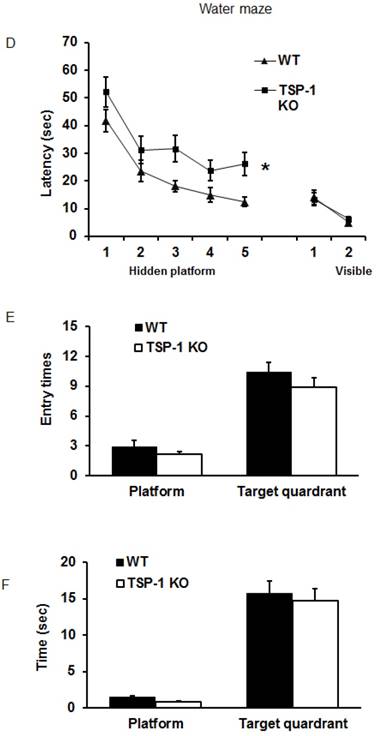
Effect of TSP-1 knockout on behavior outcomes in mice after TBI. Neurological function outcomes were measured after TBI in TSP-1 KO and WT mice. Motor-sensor functions including NSS, wire gripping and corner test were assessed before and 1d, 3d, 5d, 7d, 10d, 14d, 21d after TBI. (A) NSS test; (B) wire gripping test; (C) corner test. Moreover, spatial memory ability was assessed by Morris Water Maze starting from 14d post-TBI. (D) latency training; (E) probe trials measuring the entry times to platform; (F) probe trials measuring the stay time in targeted quadrant. (n=16, * p<0.05 vs WT group).
To examine the effect of TSP-1 knockout on post-injury cognitive function, we performed Morris water maze (MWM) assay starting from 14 days after TBI for 5 consecutive days. Both WT and TSP-1 KO groups showed a time-dependent improvement in latency to hidden platform tests. WT group showed significantly better improvement than TSP-1 KO group in latency to find the hidden platform (Figure 2D), but no differences were detected in probe tests between the two groups measuring entry times or time stay in platform and target quadrant (Figure 2E, 2F), suggesting that TSP-1 knockout might potentiate the TBI-induced memory loss.
TSP-1 knockout does not change vessel density, but elevates pro-angiogenic factors VEGF and Ang-1 expression after TBI
To investigate whether TSP-1 knockout affects vascular remodeling/angiogenesis after TBI, we tested brain vessel density in WT and TSP-1 KO mice by immunostaining with anti-CD31 antibody at 21 days post-TBI (n=4/group). In the sham control cortex, there was no significant difference between TSP-1 KO and WT mice. TBI did not increase the vessel density in contralateral peri-lesion cortex of both groups. However, TBI significantly increased the vessel density in ipsilateral peri-lesion cortex, but no difference was detected between the WT and TSP-1 KO groups (Figure. 3A, 3B). These data suggest that TSP-1 knockout may not alter brain vessel density during development and after TBI.
Figure 3
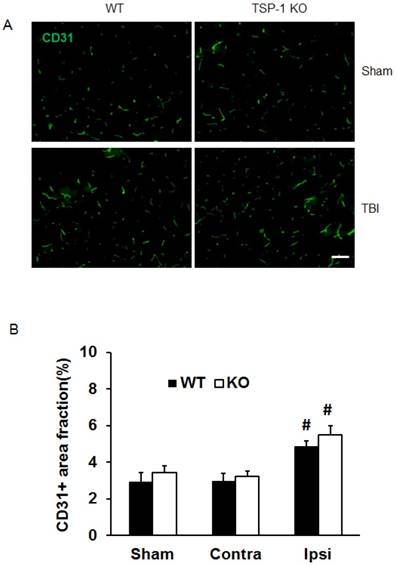
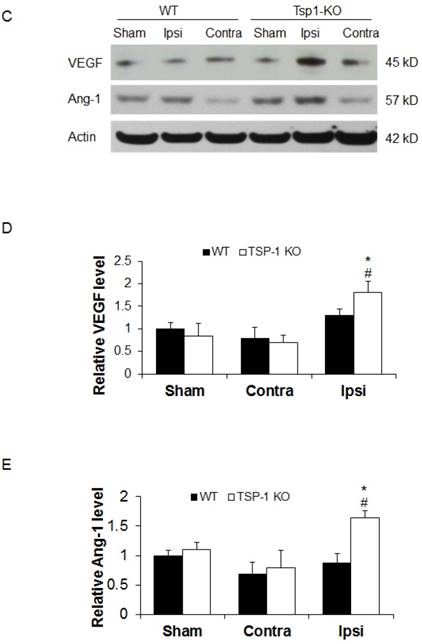
Effect of TSP-1 knockout on vessel density and vascular response in mouse brains after CCI. At 21 days after TBI, the brain vessel density in the peri-lesion cortex area was measured by immunostaining with anti-CD31 antibody. Vascular responses was measured by testing the protein expression of angiogenic factors VEGF and Ang-1. (A) Representative images of CD31-positive vessels in the ipsilateral peri-lesion cortex at 21 days after TBI or sham operation (Scale bar =50um). (B) Quantitative analysis of vessel density defined as the area fraction of positive signal. (n=4/group). (C) Representative images of Western blot for VEGF and Ang-1 protein levels in sham and injured hemispheres at 21 days after TBI (n=4/group). (D) Quantification of VEGF protein level showed no difference between WT and TSP-1 KO groups. (E) Quantification of Ang-1 protein level showed significant increase in the ipsilateral hemisphere of TSP-1 KO mice compared with WT mice, after TBI (n=4/group). (# p<0.05 vs sham, *p<0.05 vs WT group).
To delineate the vascular signaling response to brain trauma, we measured the typical pro-angiogenic factors VEGF and Ang-1 expression at 21 days after TBI (n=4/group). No significant differences were detected in sham level of VEGF and Ang-1 between WT and TSP-1 KO mice. There was slight decrease (but no significance) of VEGF and Ang-1 protein expression in contralateral peri-lesion cortex compared to the sham controls and between WT and TSP-1 KO mice after TBI. However, the expression of both VEGF and Ang-1 were significantly increased in the ipsilateral peri-lesion cortex of TSP-1 KO mice after TBI compared with sham, or WT control group (Figure. 3C, 3D, 3E). These data indicate that Tsp-1 gene knockout significantly elevated the two pro-angiogenic factors VEGF and Ang-1 protein expression, but might not significantly alter vascular histological response, at least in vessel density.
TSP-1 Knockout diminishes synaptogenic responses in contralateral but not ipsilateral cortex after TBI
To determine the effect of TSP-1 knockout on synaptogenic response following TBI, a biomarker for synapse quantification [29], synaptophysin, was examined in the brain cortex by Western blot at 21 days after TBI (n=4/group). In shame control animals, we detected a similar baseline level of synaptophysin expression in cerebral cortex of both WT and TSP-1 knockout mice (Figure 4A, 4B), suggesting the quantity of synapse is not altered by TSP-1 gene depletion at pre-TBI baseline. After TBI, we found a slight and similar increase (~20%) of synaptophysin expression in both contralateral and ipsilateral cortex of WT mice, and ipsilateral cortex of TSP-1 KO mice, compared to the sham controls. Interestingly, there was a significant decrease of synaptophysin expression in the contralateral hemisphere cortex of TSP-1 KO mice compared to its sham (~20% decrease), contralateral cortex of WT (~33% decrease) and ipsilateral (~30% decrease) hemisphere cortex (Figure 4A, 4B) of TSP-1 KO mice, indicating TSP-1 gene depletion diminishes synaptogenic response of contralateral cortex after TBI. However, the similar synaptophysin level in the ipsilateral cortex of the two groups indicates a different response in the injured cortex that may compensate for TSP-1 deficiency-associated synaptophysin decrease in the contralateral cortex after TBI.
Since BDNF is an important neurotrophic factor that promotes both vascular remodeling and synaptic plasticity during recovery phase after TBI [30], we therefore examined BDNF protein expression at 21 days after TBI (n=4/group). We found there was no significant difference of BDNF protein expression at cortex between WT and TSP-1 KO mice (Figure 4A, 4C). TBI significantly increased BDNF expression in ipsilateral cortex of both WT (~70% increase) and TSP-1 KO (~105% increase) mice compared to the sham controls, but there was no significance difference between the two groups. However, in the contralateral cortex, BDNF expression was not altered by TBI in TSP-1 KO mice; in contrast, TBI significantly increased BDNF expression in the contralateral cortex compared to its sham control (~60% increase), and TSP-1 KO mice (~45% increase) (Figure 4A, 4C). These data indicate that similarly as synaptophysin, there is a different response in the injured cortex that may compensate the TSP-1 gene depletion-associated resistance of BDNF expression in the contralateral cortex after TBI.
Figure 4
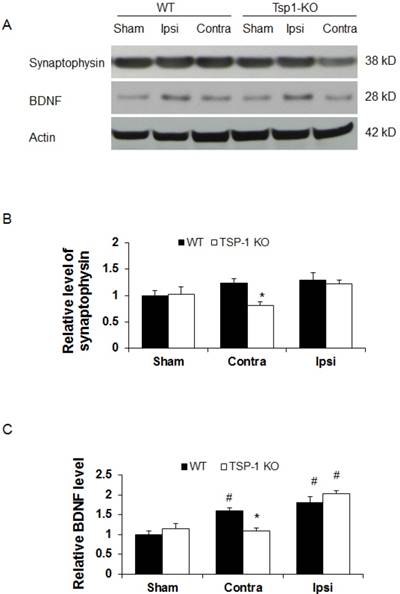
Effect of TSP-1 knockout on synaptogenic responses in mouse brains after TBI. Synaptogenic response was examined by testing synaptophysin and BDNF protein levels in bilateral hemispheres by Western blot at 21 days after TBI. (A) Representative Western blot images for synaptophysin and BDNF in sham and injured brain tissue (n=4/group). (B) Quantification of synaptophysin protein levels showed significant decrease in the contralateral hemisphere of TSP-1 KO mice after TBI, compared with WT. (C) Quantification of BDNF protein level showed significant decrease in the contralateral hemisphere of TSP-1 KO mice compared to WT after TBI, and significant elevation in the ipsilateral hemisphere of both groups compared to sham after TBI. (# p<0.05 vs sham, *p<0.05 vs WT group).
Discussion
Compared to WT control mice, our experimental results showed that TSP-1 KO (1) significantly worsened TBI-induced BBB leakage at 1 day after TBI; (2) had similar lesion size as WT mice at 3 weeks after TBI; (3) exhibited a significantly worse neurological deficits in motor function, and cognitive function; (4) had no significant difference in cerebral vessel density, but a significant increase of VEGF and Ang-1 protein expression in peri-lesion cortex; (5) significantly increased BDNF but not synaptophysin protein level in peri-lesion cortex compared to sham, but both synaptophysion and BDNF expressions were significantly decreased in contralateral cortex compared to WT.
In this study we found TSP-1 KO mice exhibited significantly worse motor sensory function deficit and significantly more impairment in learning ability compared to WT controls at 3 weeks after TBI (Figure 2). However, no difference in lesion size was detected between the two groups (Figure 1). The roles of TSP-1 in BBB permeability in the acute phase after brain injury have not been fully examined in vivo. Only limited knowledge was learnt from in vitro endothelial cell culture studies, but the results were controversial. One study suggested that increased TSP-1 expression was associated with the roles of astrocytes under reoxygenation acting as a significant driving force for BBB maturation [31], but many other studies imply that TSP-1 is a destabilizing factor of endothelial barrier [32, 33]. In this study we for the first time show that TSP-1 KO potentiated TBI-mediated BBB permeability disruption. We did not find any increase of Evans blue BBB leakage in the contralateral hemisphere of TSP-1 KO mice, indicating TSP-1 KO might not alter physiological resting baseline of BBB permeability, at least to larger size molecules. Although earlier in vitro experimental reports have suggested TSP-1 is an inflammatory mediator which can cause endothelial cell death and permeability increase via activating TGFβ, binding CD47 and upregulating MMP-9 [34, 35], our results for the first time reveals that TSP-1 may play an important role in maintaining, but not disrupting, BBB integrity after brain injury. Moreover, it raises an important notion that more preclinical investigations are needed to define the roles of TSP-1 in brain injury-induced BBB damage and associated pathophysiology.
Among many physiological functions of TSP-1, anti-angiogenesis and pro-synaptogenesis are two major functions closely related to post TBI functional recovery process [36, 37]. After TBI, angiogenesis may provide the critical neurovascular microenvironment for neuronal remodeling [38]. Two mechanistic biomarkers, VEGF and Ang-1, which can promote angiogenesis and vascular stability after TBI [39], were examined and compared in this study. Synaptogenesis forms new connections between existing neurons [40]; synaptophysin is a commonly used biomarker [29]. Additionally, BDNF was also selected as a biomarker for synaptogenesis in this study, since BDNF can cause neural regeneration, reconnection, and dendritic sprouting, and can improve synaptic efficacy [30].
In this study we observed transient upregulation of TSP-1 protein expression within 3 days after TBI, suggesting that TSP-1 actively responds to TBI, and might play a more important role at acute and sub-acute phases. A previous study also detected a peak elevation of TSP-1 expression in the first three days after focal cerebral ischemia of rats, however, TSP-2 was elevated peaking at two weeks after ischemia [41]. Both TSP-1 and TSP-2 are mostly produced by astrocytes, belonging to a family of extracellular glycoproteins with very similar angiostatic and synaptogenic properties [12, 13]. Very interestingly, a previous study reported that TSP-1/2 double KO mice exhibited significantly impaired motor function recovery after stroke. The TSP-1/2 double KO mice had significant deficits in synapse density and axonal sprouting, but had not difference in brain vessel density compared to WT control [42].
In the present study, at late three weeks after TBI, there were no differences between the two group mice on VEGF-A and Ang-1 expressions in contralateral hemisphere, but both were significantly increased in ipsilateral hemisphere of TSP-1 KO mice, suggesting vascular remodeling response may be potentiated from the TSP-1 KO mice after TBI. However we found brain vessel density was significantly increased after TBI, but no difference between the two group mice (Figure 3), which might be speculated that possible involvements of other anti- or pro- vascular remodeling factors in the late vascular response. Furthermore, we found a significant decline of synaptogenesis markers synaptophysin and BDNF expression in the contralateral cortex, but not in the sham and ipsilateral cortex of TSP-1 KO mice compared to the WT controls. We speculate that TSP-1 depletion might lead to a latent response for synaptogenesis in the contralateral hemisphere at late phase after TBI, while other signaling molecules such as TSP-2 in the injured hemisphere might actively respond to TBI and compensate the latency caused by the TSP-1 deficiency [42].
There are a few caveats in this study. Firstly, although previous studies suggest that astrocytes is the major source of TSP-1 in the brain [43], due to the lack of reliable anti-TSP-1 antibodies for immunohistochemistry, we were unable to dissect cellular localization of TSP-1 protein expression in this study, further investigations on the cellular sources of TSP-1 will be conducted in the future. Secondly, vascular remodeling and synaptogenesis may take several months after TBI, but we only examined one single time point, i.e, 21 days after CCI. Examination for these important endogenous recovery mechanisms for longer time at multiple time points would facilitate better understanding of the roles and mechanisms of TSP-1. Third, in this study we observed worsened neurological outcome in TSP-1 KO mice, associated with decreased synaptophysin and BDNF expressions, but increased pro-angiogenesis markers VEGF and Ang-1. However, the causality between these phenomena has not been established, which warrants further investigation in the future. Lastly, the different responses of TSP-1 KO to TBI versus WT mice might be partially due to TSP-1 gene deficiency-associated developmental deficits that result in alterations in key signaling pathways of pre-TBI baseline. The baseline changes of molecular signaling needs to be carefully characterized for better result interpretation. TSP-1 conditional and inducible gene KO mice would therefore be more powerful tools for further evaluating the roles and mechanisms of TSP-1 in TBI.
In summary, in this study we found TSP-1 protein expression was transiently up-regulated within the first 3 days after TBI. TSP-1 may be beneficial for maintaining BBB integrity in the early phase, but its role for functional recovery in the late phase of TBI remains unclear. The pathological roles and molecular mechanisms of TSP-1 in early BBB pathophysiology, and long-term neurological function recovery after TBI need to be further investigated.
Supplementary Material
Figure S1.
Abbreviations
TSP-1, Thrombospondin-1; KO, knockout; WT, wild type; BBB, blood-brain-barrier; TBI, traumatic brain injury; CCI, controlled cortical impact; NSS, neurological severity score; MWM, morris water maze; VEGF, vascular endothelial growth factors; Ang-1, angiopoietin-1; BDNF, brain-derived neurotrophic factor; IAP, integrin-associated protein; MMP-9, matrix metalloproteinase-9.
Acknowledgements
This work was supported in part by NIH grant R01AI-064569 (X.Wang) and the National Natural Science Foundation of China (81301035 to S.Zhao, 81571159 to X.Sun).
Authors' contributions
CC, ZY, SZ, and ZL performed the study and analyzed the data. YJ and JK helped in the neurological behavior study. YY, JL, MW, EL helped analyze the data. ZY, XS and XW designed the study and wrote the paper. All authors have read and approved the manuscript.
Compliance with Ethics Requirements
All animal experiments were performed following protocols approved by the Massachusetts General Hospital Animal Care and Use Committee in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tran MD, Furones-Alonso O, Sanchez-Molano J, Bramlett HM. Trauma-induced expression of astrocytic thrombospondin-1 is regulated by P2 receptors coupled to protein kinase cascades. Neuroreport. 2012;23(12):721-726
2. Esemuede N, Lee T, Pierre-Paul D, Sumpio BE, Gahtan V. The role of thrombospondin-1 in human disease. The Journal of surgical research. 2004;122(1):135-142
3. Resovi A, Pinessi D, Chiorino G, Taraboletti G. Current understanding of the thrombospondin-1 interactome. Matrix biology: journal of the International Society for Matrix Biology. 2014;37:83-91
4. Chen H, Herndon ME, Lawler J. The cell biology of thrombospondin-1. Matrix biology: journal of the International Society for Matrix Biology. 2000;19(7):597-614
5. Lok J, Wang XS, Xing CH, Maki TK, Wu LM, Guo SZ, Noviski N, Arai K, Whalen MJ, Lo EH. et al. Targeting the neurovascular unit in brain trauma. CNS neuroscience & therapeutics. 2015;21(4):304-308
6. Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Translational stroke research. 2011;2(4):492-516
7. Cho S. CD36 as a therapeutic target for endothelial dysfunction in stroke. Current pharmaceutical design. 2012;18(25):3721-3730
8. Wang JL, Jin GL, Yuan ZG, Yu XB, Li JQ, Qiu TL, Dai RX. Plasma thrombospondin-1 and clinical outcomes in traumatic brain injury. Acta neurologica Scandinavica. 2015
9. Xing C, Lee S, Kim WJ, Jin G, Yang YG, Ji X, Wang X, Lo EH. Role of oxidative stress and caspase 3 in CD47-mediated neuronal cell death. Journal of neurochemistry. 2009;108(2):430-436
10. Xing C, Arai K, Park KP, Lo EH. Induction of vascular endothelial growth factor and matrix metalloproteinase-9 via CD47 signaling in neurovascular cells. Neurochemical research. 2010;35(7):1092-1097
11. Xing C, Lee S, Kim WJ, Wang H, Yang YG, Ning M, Wang X, Lo EH. Neurovascular effects of CD47 signaling: promotion of cell death, inflammation, and suppression of angiogenesis in brain endothelial cells in vitro. Journal of neuroscience research. 2009;87(11):2571-2577
12. Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harbor perspectives in medicine. 2012;2(5):a006627
13. Risher WC, Eroglu C. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix biology: journal of the International Society for Matrix Biology. 2012;31(3):170-177
14. Sharp DJ, Scott G, Leech R. Network dysfunction after traumatic brain injury. Nature reviews Neurology. 2014;10(3):156-166
15. Kabadi SV, Faden AI. Neuroprotective strategies for traumatic brain injury: improving clinical translation. International journal of molecular sciences. 2014;15(1):1216-1236
16. Zhao S, Yu Z, Zhao G, Xing C, Hayakawa K, Whalen MJ, Lok JM, Lo EH, Wang X. Neuroglobin-overexpression reduces traumatic brain lesion size in mice. BMC neuroscience. 2012;13:67
17. Yang J, You Z, Kim HH, Hwang SK, Khuman J, Guo S, Lo EH, Whalen MJ. Genetic analysis of the role of tumor necrosis factor receptors in functional outcome after traumatic brain injury in mice. J Neurotrauma. 2010;27(6):1037-1046
18. Sierra-Mercado D, McAllister LM, Lee CC, Milad MR, Eskandar EN, Whalen MJ. Controlled cortical impact before or after fear conditioning does not affect fear extinction in mice. Brain research. 2015;1606:133-141
19. Aoki T, Sumii T, Mori T, Wang X, Lo EH. Blood-brain barrier disruption and matrix metalloproteinase-9 expression during reperfusion injury: mechanical versus embolic focal ischemia in spontaneously hypertensive rats. Stroke; a journal of cerebral circulation. 2002;33(11):2711-2717
20. Fujimoto ST, Longhi L, Saatman KE, Conte V, Stocchetti N, McIntosh TK. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 2004;28(4):365-378
21. Yager PH, You Z, Qin T, Kim HH, Takahashi K, Ezekowitz AB, Stahl GL, Carroll MC, Whalen MJ. Mannose binding lectin gene deficiency increases susceptibility to traumatic brain injury in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28(5):1030-1039
22. Washington PM, Forcelli PA, Wilkins T, Zapple DN, Parsadanian M, Burns MP. The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J Neurotrauma. 2012;29(13):2283-2296
23. You Z, Yang J, Takahashi K, Yager PH, Kim HH, Qin T, Stahl GL, Ezekowitz RA, Carroll MC, Whalen MJ. Reduced tissue damage and improved recovery of motor function after traumatic brain injury in mice deficient in complement component C4. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2007;27(12):1954-1964
24. Tsenter J, Beni-Adani L, Assaf Y, Alexandrovich AG, Trembovler V, Shohami E. Dynamic changes in the recovery after traumatic brain injury in mice: effect of injury severity on T2-weighted MRI abnormalities, and motor and cognitive functions. J Neurotrauma. 2008;25(4):324-333
25. Beni-Adani L, Gozes I, Cohen Y, Assaf Y, Steingart RA, Brenneman DE, Eizenberg O, Trembolver V, Shohami E. A peptide derived from activity-dependent neuroprotective protein (ADNP) ameliorates injury response in closed head injury in mice. J Pharmacol Exp Ther. 2001;296(1):57-63
26. Bermpohl D, You Z, Korsmeyer SJ, Moskowitz MA, Whalen MJ. Traumatic brain injury in mice deficient in Bid: effects on histopathology and functional outcome. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2006;26(5):625-633
27. Bermpohl D, You Z, Lo EH, Kim HH, Whalen MJ. TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2007;27(11):1806-1818
28. Wang X, Liu J, Zhu H, Tejima E, Tsuji K, Murata Y, Atochin DN, Huang PL, Zhang C, Lo EH. Effects of neuroglobin overexpression on acute brain injury and long-term outcomes after focal cerebral ischemia. Stroke; a journal of cerebral circulation. 2008;39(6):1869-1874
29. Lu D, Goussev A, Chen J, Pannu P, Li Y, Mahmood A, Chopp M. Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma. 2004;21(1):21-32
30. Kaplan GB, Vasterling JJ, Vedak PC. Brain-derived neurotrophic factor in traumatic brain injury, post-traumatic stress disorder, and their comorbid conditions: role in pathogenesis and treatment. Behavioural pharmacology. 2010;21(5-6):427-437
31. Song HS, Son MJ, Lee YM, Kim WJ, Lee SW, Kim CW, Kim KW. Oxygen tension regulates the maturation of the blood-brain barrier. Biochem Biophys Res Commun. 2002;290(1):325-331
32. Jerkic M, Letarte M. Increased endothelial cell permeability in endoglin-deficient cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2015;29(9):3678-3688
33. Garg P, Yang S, Liu A, Pallero MA, Buchsbaum DJ, Mosher DF, Murphy-Ullrich JE, Goldblum SE. Thrombospondin-1 opens the paracellular pathway in pulmonary microvascular endothelia through EGFR/ErbB2 activation. Am J Physiol Lung Cell Mol Physiol. 2011;301(1):L79-90
34. Nor JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. Journal of vascular research. 2000;37(3):209-218
35. Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93(7):1159-1170
36. Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100(13):1423-1431
37. Xu J, Xiao N, Xia J. Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nature neuroscience. 2010;13(1):22-24
38. Xiong Y, Zhang Y, Mahmood A, Meng Y, Qu C, Chopp M. Erythropoietin mediates neurobehavioral recovery and neurovascular remodeling following traumatic brain injury in rats by increasing expression of vascular endothelial growth factor. Translational stroke research. 2011;2(4):619-632
39. Gong D, Zhang S, Liu L, Dong J, Guo X, Hao M, Tu Y, Diao Y, Zhang J. Dynamic changes of vascular endothelial growth factor and angiopoietin-1 in association with circulating endothelial progenitor cells after severe traumatic brain injury. The Journal of trauma. 2011;70(6):1480-1484
40. Xuan W, Agrawal T, Huang L, Gupta GK, Hamblin MR. Low-level laser therapy for traumatic brain injury in mice increases brain derived neurotrophic factor (BDNF) and synaptogenesis. Journal of biophotonics. 2015;8(6):502-511
41. Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke; a journal of cerebral circulation. 2003;34(1):177-186
42. Liauw J, Hoang S, Choi M, Eroglu C, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA. et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28(10):1722-1732
43. Lu Z, Kipnis J. Thrombospondin 1-a key astrocyte-derived neurogenic factor. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24(6):1925-1934
Author contact
![]() Corresponding authors: Xiaochuan Sun, MD, sunxch1445com or Xiaoying Wang, MD, PhD, wangximgh.harvard.edu
Corresponding authors: Xiaochuan Sun, MD, sunxch1445com or Xiaoying Wang, MD, PhD, wangximgh.harvard.edu
Citation styles
APA
Cheng, C., Yu, Z., Zhao, S., Liao, Z., Xing, C., Jiang, Y., Yang, Y.G., Whalen, M.J., Lo, E.H., Sun, X., Wang, X. (2017). Thrombospondin-1 Gene Deficiency Worsens the Neurological Outcomes of Traumatic Brain Injury in Mice. International Journal of Medical Sciences, 14(10), 927-936. https://doi.org/10.7150/ijms.18812.
ACS
Cheng, C.; Yu, Z.; Zhao, S.; Liao, Z.; Xing, C.; Jiang, Y.; Yang, Y.G.; Whalen, M.J.; Lo, E.H.; Sun, X.; Wang, X. Thrombospondin-1 Gene Deficiency Worsens the Neurological Outcomes of Traumatic Brain Injury in Mice. Int. J. Med. Sci. 2017, 14 (10), 927-936. DOI: 10.7150/ijms.18812.
NLM
Cheng C, Yu Z, Zhao S, Liao Z, Xing C, Jiang Y, Yang YG, Whalen MJ, Lo EH, Sun X, Wang X. Thrombospondin-1 Gene Deficiency Worsens the Neurological Outcomes of Traumatic Brain Injury in Mice. Int J Med Sci 2017; 14(10):927-936. doi:10.7150/ijms.18812. https://www.medsci.org/v14p0927.htm
CSE
Cheng C, Yu Z, Zhao S, Liao Z, Xing C, Jiang Y, Yang YG, Whalen MJ, Lo EH, Sun X, Wang X. 2017. Thrombospondin-1 Gene Deficiency Worsens the Neurological Outcomes of Traumatic Brain Injury in Mice. Int J Med Sci. 14(10):927-936.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.