Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(8):741-749. doi:10.7150/ijms.20001 This issue Cite
Research Paper
Molecular mechanism of action and safety of 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione - a novel anticonvulsant drug candidate
Barbara Kaproń1, Jarogniew Łuszczki2, 3, Agata Paneth1, Monika Wujec1, Agata Siwek4, Tadeusz Karcz5, Barbara Mordyl4, Monika Głuch-Lutwin4, Anna Gryboś4, Gabriel Nowak4, Karolina Pająk6, Krzysztof Jóźwiak6, Adam Tomczykowski1, Tomasz Plech7 ![]()
1. Department of Organic Chemistry, Medical University of Lublin, Chodzki 4A, 20-093 Lublin, Poland;
2. Department of Pathophysiology, Medical University of Lublin, Jaczewskiego 8, Lublin 20-090, Poland;
3. Isobolographic Analysis Laboratory, Institute of Rural Health, Jaczewskiego 2, Lublin 20-950, Poland;
4. Department of Pharmacobiology, Jagiellonian University Medical College, Medyczna 9, 30-688 Kraków, Poland;
5. Department of Technology and Biotechnology of Drugs, Jagiellonian University Medical College, Medyczna 9, 30-688 Kraków, Poland;
6. Department of Biopharmacy, Medical University of Lublin, Chodzki 4A, 20-093 Lublin, Poland;
7. Department of Pharmacology, Medical University of Lublin, Chodzki 4A, 20-093 Lublin, Poland.
Received 2017-3-9; Accepted 2017-6-6; Published 2017-7-18
Abstract
Previously, it was found that 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (TP-315) effectively protects mice from maximal electroshock-induced seizures. The aim of this study was to determine possible interactions between TP-315 and different molecular targets, i.e. GABAA receptors, voltage-gated sodium channels, and human neuronal α7 and α4β2 nicotinic acetylcholine receptors. The influence of TP-315 on the viability of human hepatic HepG2 cells was also established using PrestoBlue and ToxiLight assays. It was found that the anticonvulsant activity of TP-315 results (at least partially) from its influence on voltage-gated sodium channels (VGSCs). Moreover, the title compound slightly affected the viability of human hepatic cells.
Keywords: Sodium channels, [3H]-batrachotoxin, patch-clamp, cell viability assays.
Introduction
Epilepsy is a neurological disorder in which abnormal excessive and/or hypersynchronous neuronal activity occurs causing seizures or other symptoms, like temporary confusion, loss of consciousness or awareness, unusual sensations, etc. The main aim of treatment of epilepsy is to obtain a seizure-free state or to reduce seizure frequency in patients. The main form of treatment of epilepsy in humans is the administration of anti-epileptic drugs (AEDs). However, about one-third of patients with epilepsy suffer from seizures that cannot be fully controlled with currently available AEDs [1, 2]. Simultaneously, it is well known that dose-dependent and reversible adverse effects of AEDs are common, especially in patients treated with polytherapy [3]. Therefore, there is an immense demand for the development of novel anticonvulsant drugs, especially such ones that offer better tolerability with limited adverse effects.
Recently, the screening tests performed in our laboratory revealed that 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (TP-315; Fig. 1) produced evident anticonvulsant action in the maximal electroshock-induced seizure model in mice (Fig. 2) [4]. Moreover, the triazole derivative TP-315 appeared to have a beneficial protective index.
Chemical structure of TP-315
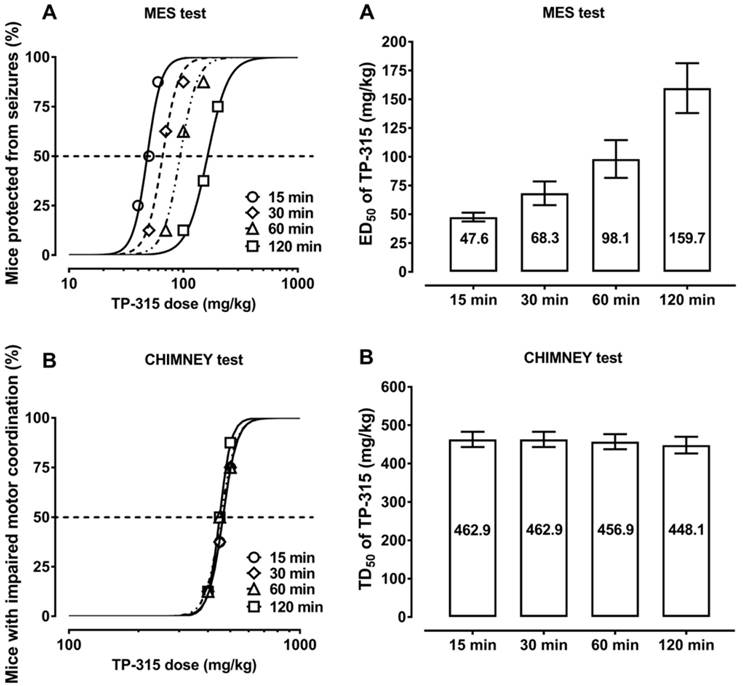
Time-course and dose-response effects of TP-315 in the maximal electroshock-induced seizure (MES) and chimney tests in mice. Left panel: Dose-response function for the anticonvulsant (protective) activity (A) and acute neurotoxic impairment of motor coordination (B) of TP-315 in the MES and chimney tests. Each data point (n=8 mice/data point) represents percent of mice protected from the MES test (A), or percent of mice with impairment of motor coordination in the chimney test (B), at a given dose (in mg/kg) of TP-315 in various pretreatment times (15, 30, 60 and 120 min). Sigmoidal curves are the result of a least squares fit of dose-response function for the tested compound (TP-315). Points of intersections with the dashed line at 50% correspond to approximate ED50 values (A) or TD50 values (B) of TP-315. Right panel: Columns represent median effective doses (ED50 in mg/kg ± S.E.M.) of TP-315 (A), that protect 50% of animals tested against MES-induced seizures in mice, or median toxic doses (TD50 in mg/kg ± S.E.M.) of TP-315 (B), that impair motor coordination in 50% of the mice subjected to the chimney test. The log-probit method was used for calculating the ED50 and TD50 values of TP-315 at 4 various pretreatment times (15, 30, 60 and 120 min).
However, until now nothing was known about the mechanism(s) involved in the anticonvulsant activity of TP-315. Molecular target identification is one of the most important steps in developing drug candidates [5]. Nonetheless, even such molecules that strongly interact with their biological target(s) can fail to become successful drug because of safety concerns. So, it is important to evaluate the potential toxicity in early stages of drug development. The aim of the present study was to examine the effect of TP-315 on different molecular targets in the central nervous system, i.e. GABAA receptors, voltage-gated sodium channels, and human neuronal α7 and α4β2 nicotinic acetylcholine receptors. Moreover, the cellular toxicity of TP-315 towards human hepatic cells was investigated using two types of viability measuring assays, i.e. PrestoBlue and ToxiLight assays.
Experimental procedures
GABAA electrophysiology
Electrophysiology experiments were carried out on a QPatch16X automatic patch clamp platform (Sophion Biosciences). HEK293 cells, stably expressing the α1β2γ2 subunits of the human GABAA receptor were cultured using standard procedures. On the day of experiment cells were collected from the culture flask using Detachin solution (VWR) and resuspended in serum-free media. Cell suspension was placed in the magnetic stirred tube, located onboard the automated electrophysiology instrument and allowed to recover for 60 minutes at room temperature. In the next step, cells were automatically transferred to a built-in centrifuge, spun down and washed in extracellular Ringer's solution. Cells were applied to the pipetting wells of a disposable 16-channel planar patch chip plates (QPlate 16X, with 10 patch clamp holes per measurement site) and gigaseals were formed upon execution of a combined suction/voltage protocol. Further suction lead to whole-cell configuration. GABAA receptor chloride currents were recorded for 7s after each addition of compound. During whole-cell recording, the holding potential was set to -90 mV. Recordings were performed at room temperature. Extracellular solution consisted of (in mM): 2 CaCl2, 1 MgCl2, 10 HEPES, 4KCl, 145 NaCl, 10 glucose (pH 7.4, 300 mOsm) and intracellular solution contained (in mM):140 CsF, 1 EGTA, 5 CsOH, 10 HEPES, 20 NaCl (pH 7.2, 320 mOsm).
In the agonist assays, the sequential application of: 10 μM γ-aminobutyric acid (GABA, reference agonist at concentration close to half maximal effective concentration); 10 μM tested compound (TP-315) and second addition of 10 μM GABA was set up in the instrument software. The interval between additions of particular compounds was at least 60s. Typically 5 μl of ligand was added to the cells, which was followed after 3s by washout with extracellular solution (two times 5 μl).
In the allosteric modulator/antagonist dose-response mode (simultaneous addition of GABA and tested compound) first 10 μM GABA application was followed by 6 additions of increasing concentrations of tested compound (0.001-100 μM) in combination with 10 μM GABA. In this setup, cells were preincubated with potential allosteric modulator/antagonist alone for at least 50 s, before the addition of combination with agonist. Concentration of reference agonist used in this study (being close to EC50 value) was chosen during method validation process and enabled the detection of both antagonist (e.g. bicuculline) and positive allosteric modulator (e.g. zolpidem) activity in single setup [6].
Data were analyzed using QPatch Assay Software (v5.0, Sophion Biosciences) and representthe mean of at least three experiments carried out on distinct cells. Validation criteria for eachexperiment were: current amplitude evoked by addition of GABA higher than 500 pA anddifference between cells response to both GABA applications in agonist mode not higher than 25%.
Radioligand binding assay for Na+ channel - site 2 using [3H]batrachotoxin
Radioligand binding assay was performed according to the method of Callaway et al. [7] with slight modifications. Rat cerebral cortex was weighed and homogenised at concentration of 10% (w/v) in ice-cold 0.32 M sucrose, 10 mM phosphate buffer (pH 7.4) using an ULTRA TURRAX homogeniser. The homogenate was centrifuged at 1000 x g for 10 min (0-4oC). Collected supernatant was centrifuged at 40 000 x g for 45 min (0-4oC) to obtain membrane fraction. The resulting pellet was resuspended in incubation buffer containing (mM): 50 Tris base, 50 HEPES, 130 choline chloride, 5.4 KCl, 0.8 MgSO4 and 5.5 glucose (pH 7.4). 300 µl of the tissue suspension (300 µg/well) containing LQ scorpion venom (10 µg/well), 50 µl of [3H]-BTX solution (final conc. 10 nM, spec. act. 38.6 Ci/mmol, PerkinElmer) and 50 µl of the tested compounds were incubated at 37ºC for 1 h. The incubation was terminated by rapid filtration over glass fiber filters (FilterMate B, PerkinElmer) using 96-well harvester (PerkinElmer). Eight rapid washes with 500 µl of ice-cold 50 mM Tris-HCl buffer, pH 7.4, were performed. Veratridine (300 μM) was used to define nonspecific binding.
Filter mates were dried at 37°C in forced-air incubator and solid scintillator (MeltiLex, PerkinElmer) was then melted on them at 100°C for 5 minutes. The radioactivity on the filters was measured in MicroBetaTriLux 1450 scintillation counter (PerkinElmer).Each compound was tested in a screening assay at final concentration of 100 µM. Results were expressed as percent inhibition of [3H]-batrachotoxin binding. Then compound TP-315, qualified for dose-response binding experiments, was tested in 8 concentrations ranging from 300 to 0.1 µM.
Interaction of TP-315 with orthosteric sites of human α7 and human α4β2 nicotinic acetylcholine receptors (nAChRs)
Reagents
See Table A.
Reagents
| [3H]Epibatidine (62.2 Ci/mmol) | Perkin Elmer |
| (±)-Epibatidine dihydrochloride hydrate | Sigma-Aldrich |
| Polyethylenimine (PEI) | |
| Protease inhibitors cocktail | |
| Sodium azide (NaN3) | |
| Trizma base | |
| Trizma hydrochloride | |
| Potassium chloride (KCl) | Baxter (Lublin, Poland) |
| Sodium chloride (NaCl) | |
| Di-sodium hydrogen phosphate (NaH2PO4) | POCH (Gliwice, Poland) |
| Postassium phosphate monobasic (KH2PO4) | |
| CaCl2 | |
| MgCl2 | |
| Fetal bovine serum (FBS) | Life Technologies |
| Trypsin/EDTA | |
| Dulbecco's Modified Eagle Medium (DMEM) | |
| Ham's F-12 nutrient mixture | |
| Hygromycin B | |
| Geneticin (G-418 sulfate) |
Membrane preparation
nAChRs binding experiments were performed on membranes from HEK-293 and SHSY5Y cells stably transfected with genes encoding hα4β2 and hα7 nAChRs, respectively. Method of Arias et al. was used to obtain membranes and perform binding assay [8].
HEK293-hα4β2 cells were maintained in 1:1 mixture of Dulbecco's Modified Eagle Medium (DMEM) and Ham's F‑12 nutrient mixture supplemented with 10% fetal bovine serum (FBS), 200 µg/ml G-418 and 200 µg/ml hygromycine B. SHSY5Y-hα7 were cultured in DMEM supplemented with 10% FBS and 200 µg/ml G-418. Cells were cultured for approximately 3 weeks in suspension on nontreated Petri dishes, then gently scraped and centrifuged at 500 x g for 5 min. Cells were then homogenized in binding saline buffer (50 mM Tris-HCl, pH 7.4 containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2) suplemented with a cocktail of protease inhibitors and 0.025% sodium azide and centrifuged at 10 000 rpm for 30 min at 4°C. The pellet was finally resuspended in binding saline buffer containing 20% sucrose. Aliquotes of AChR membranes were stored at -80°C.
Radioligand binding assay
Radioligand binding assay was performed in 96-well plate format. AChR membranes (1 mg/ml), were suspended in binding saline buffer containing 10 nM [3H]epibatidine and preincubated for 10 min at room temperature following the addition of TP-315. Nonspecific binding was determined in the presence of 1 µM (±)-epibatidine. After 90 min incubation at room temperature, the AChR-bound radioligand was separated from the free [3H]epibatidine by a filtration assay using a 96-sample harvester system with GF/B Whatman filters, previously soaked with 0.5% polyethylenimine for 30 min. The bound radioactivity was determined using the MicroBetaTriLux scintillation counter (Perkin Elmer).
Cell viability and cytotoxicity assays
Cell culture
The human hepatocellular carcinoma cells (HepG2) were obtained from the ATCC (HB-8065). The cells were thawing according to the manufacturer's protocol form ATCC. HepG2 cells are cultivated in Eagle's Minimum Essential Medium (EMEM, ATCC) supplemented with 10% heat inactivated fetal bovine serum (ATCC), with added 100 IU/ml penicillin (ATCC) and 100 µg/ml streptomycin (ATCC). The cells were passaged three times a week, until they reached confluence. HepG2 cells culture were carried out in accordance with basic techniques of cell culture in sterile conditions. Cells were cultured in flasks with an area of 175 cm2, and incubated at 37°C, 5% CO2. For the test of compounds with the HepG2 cells line, hepatocytes were seeded on 96-well culture plate at a density of 2x104 cells per well in fresh medium. Cells grew for 24 hours in the incubator (37°C, 5% CO2) before performing experiments.
Sample preparation
TP-315 was dissolved in dimethyl sulfoxide (DMSO) with stock concentrations of 10 mM. The compounds were incubated for 5 minutes with ultrasound in a water bath. From the stock, dilutions were prepared in phosphate buffered saline (PBS). All experiments were performed in triplicates, in two independent experiments.
PrestoBlue assay
Cell viability was measured using the PrestoBlue reagent (Invitrogen). PrestoBlue reagent is a resazurin-based solution that functions as a cell viability indicator. Metabolically active cells are capable of reducing the PrestoBlue reagent, with the colorimetric changes used as an indicator to quantify the viability of cells in culture. This change can be determined by measuring the fluorescence. After 24 hours of incubation with the compounds the PrestoBlue reagent was added to wells of a microplate in an amount equals to one tenth of the remaining medium volume. After 15 minutes of incubation at 37°C, the fluorescence intensity (EX 530 EM 580 nm) was measured in a plate reader (POLARstar Omega, BMG Labtech). Viability values were calculated as a percentage of live cells with respect to the control sample (DMSO). The negative control was medium without cells.
ToxiLight assay
The bioluminescent ToxiLight bioassay (Lonza) is a cytotoxicity highly sensitive assay designed to measure cell membrane damage. It quantitatively measures the release of Adenylate Kinase (AK) from the membranes of damaged cells. AK is a protein presented in all eukaryotic cells, which is released into the culture medium when cells die. The enzyme actively phosphorylates ADP and the resultant ATP is then measured using the bioluminescent firefly luciferase reaction with the ToxiLight reagent. The emitted light intensity expressed as a RLU value is linearly related to the adenylate kinase activity. After 24h of treatments, 5 µl of the clear fluid above sediment was transferred into 384-well plate (Perkin Elmer). Then 20 μl of the Adenylate Kinase Detection Reagent (AKDR) was added. As a positive control for lysis 10% Triton X- 100 (Sigma-Aldrich) in growth medium is used, the negative control is growth medium alone. The luminescence was measured in a plate reader (POLARstar Omega, BMG Labtech) after 5 minutes of incubation. The results were expressed as a percentage of positive control, which corresponded to the percentage of dead cells with respect to the control sample.
Results
Influence of 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (TP-315) on the GABAA receptor chloride currents
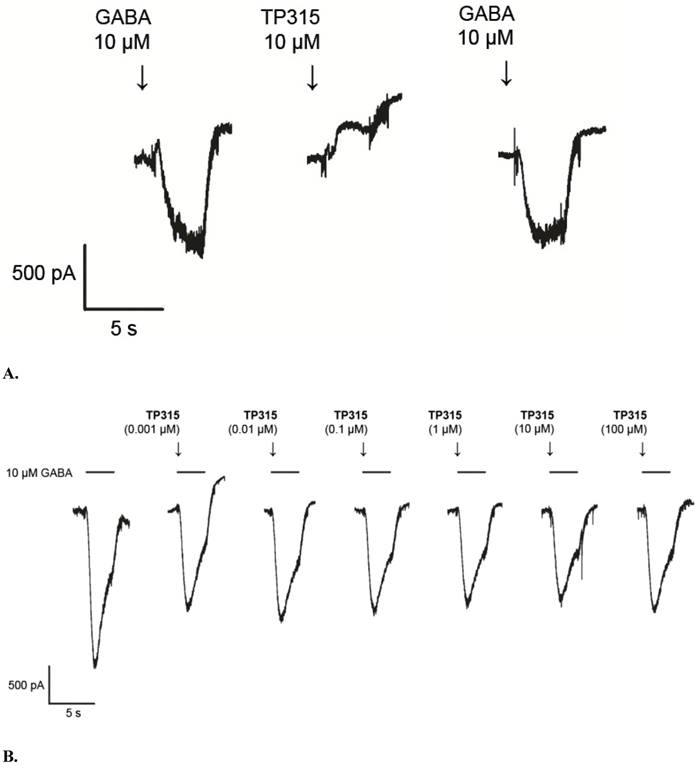
5-(3-Chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (TP-315) in 10 µM concentration was not able to evoke GABAA receptor chloride current, when added directly to QPlate measurement site containing recombinant HEK-293 cells in whole-cell configuration (Fig. 3A). Thus, it can be stated that TP-315 lacks significant efficacy at GABAA receptors. Moreover, no potentiation, neither blockade of GABA-evoked currents was observed when GABA (10 µM) was used in combination with different concentrations of TP-315 (i.e. 0.001 µM - 100 µM) (Fig. 3B). Therefore, it can be concluded that TP-315 does not possess allosteric modulatory nor (ant)agonistic properties at GABAA receptors.
Influence of TP-315 on GABAA-receptor mediated currents; A. current recordings for electrophysiological agonist-mode screening experiments; B. dose-dependent influence of TP-315 on GABA-gated chloride current in HEK-293 cells expressing recombinant GABAA receptor (TP-315 used in combination with 10 µM GABA).
Radioligand binding assay for Na+ channel (site 2) using [3H]batrachotoxin
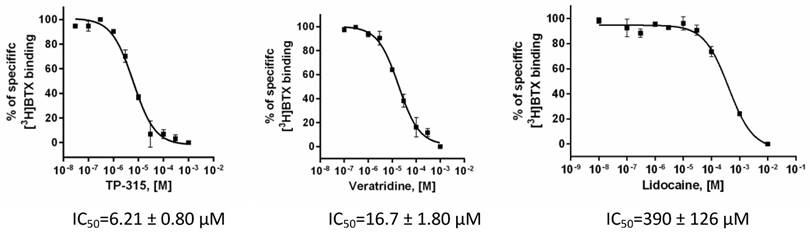
Affinity of TP-315 to sodium channels - site 2 was expressed as a percentage of inhibition of the binding of [3H]batrachotoxin. Initially, TP-315 was tested in a screening assay at final concentration of 100 µM and it was found to be very effective sodium channel blocker with %inhibition amounted to 92 ± 4% (Table 1). Interestingly, carbamazepine, known as anti-epileptic drug acting on sodium channels, revealed much weaker inhibition of [3H]batrachotoxin binding (i.e. 17.4%). In the next step, radioligand binding assay was performed with varying concentrations of TP-315 in order to obtain dose-response curve (Fig. 4). Median inhibitory concentration (IC50) for TP-315 turned out to be 6.21 ± 0.80 µM whereas IC50 for carbamazepine equalled 131 µM. The other two reference substances, veratridine and lidocaine, inhibited the binding of [3H]batrachotoxin to a receptor site of sodium channel with IC50 of 16.7 ± 1.8 µM and 390 ± 126 µM, respectively.
Radioligand binding assay for Na+ channel (site 2) using [3H]batrachotoxin.
| Sodium channel - site 2 (rat brain cortical membranes) | ||
|---|---|---|
| Compounds | % inhibition ± SEM at 100 µM | IC50 ± SEM [µM] |
| TP-315 | 92 ± 4 | 6.21 ± 0.80 |
| Veratridine | 100 ± 2 | 16.7 ± 1.8 |
| Lidocaine | 26 ± 2 | 390 ± 126 |
| Carbamazepine | 17.41 | 1312 |
Dose - response binding curves for TP-315, veratridine and lidocaine obtained in [3H]batrachotoxin binding assay for Na+ channels - site 2. Graphs from two independent experiments performed in duplicates with mean IC50values ± SEM are shown.
Interaction of TP-315 with orthosteric sites of human α7 and human α4β2 nicotinic acetylcholine receptors (nAChRs)
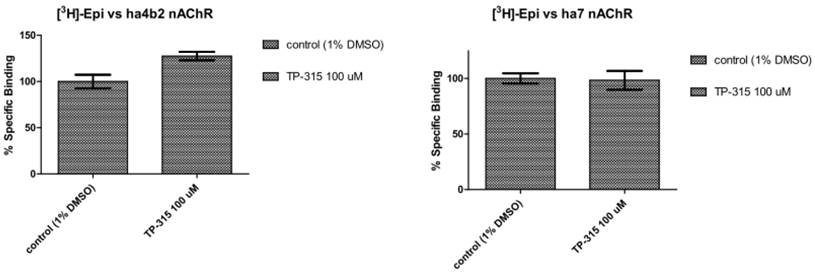
Binding experiments were performed on membranes derived from HEK-293 and SH-SY5Y cells stably transfected with genes encoding human α4β2 and human α7 nicotinic acetylcholine receptors (nAChRs). TP-315 at 100 µM concentration does not displace [3H]epibatidine from its complexes neither with hα4β2 nor hα7 nAChRs (Fig. 5).
Displacement of [3H]epibatidine from its complexes with human α7 and human α4β2 nicotinic acetylcholine receptors (nAChRs).
Influence of TP-315 on the viability of human HepG2 cells
Human liver-derived cell lines are often used in drug development process for assessing safety of drug candidates. Toxicity of TP-315 was tested using immortalized hepatocellular carcinoma cells HepG2. The mentioned cell line constitutes one of the most frequently used model to study toxicity of drugs and chemicals [11-13]. In order to evaluate the viability of HepG2 cell after exposure to TP-315 PrestoBlue and ToxiLight assays were used. While PrestoBlue assay measures metabolic activity of cells (precisely, it measures cellular reducing activity), the ToxiLight assay was designed to measure cell membrane damage (i.e. it quantitatively measures the release of adenylate kinase from the membranes of damaged cells). The reference drug, astemizole, inhibited the viability of HepG2 cells in a dose-dependent manner. As its concentration increased from 1 µM to 100 µM the viability of cells decreased from 113% to 2% and from 93% to 14% in PrestoBlue and Toxilight assays, respectively (Table 2). At the same experimental conditions TP-315 used in the highest concentration of 100 µM inhibited the growth of cells only by 6% (as measured in ToxiLight assay) and 15% (as measured in PrestoBlue assay).
Influence of different concentrations of TP-315 on the viability of human liver HepG2 cells using PrestoBlue and ToxiLight assays
| Type of assay | PrestoBlue | ToxiLight | ||||||
|---|---|---|---|---|---|---|---|---|
| Viability (% of control) | Viability (% of control) | |||||||
| Concentration of compounds | 100µM | 50µM | 10µM | 1µM | 100µM | 50µM | 10µM | 1µM |
| TP-315 | 75% | 92% | 103% | 87% | 94% | 95% | 95% | 95% |
| Astemizole | 2% | 20% | 68% | 113% | 14% | 20% | 85% | 93% |
Discussion
Molecular mechanisms of action of antiepileptic drugs (AEDs) are varied. Three main groups of mechanisms include: (i) modulation of ionchannels (sodium, calcium, potassium), (ii) enhancement of GABA-ergic (inhibitory) neurotransmission, and (iii) diminishing of glutamatergic (excitatory) neurotransmission. In case of manyAEDs, their activity is decided by more than one molecular mechanism. The chemical structure of TP-315, containing a five-membered 1,2,4-triazole ring, made it possible for the compound to have effect on GABAA receptors. When researching the antiepileptic effect of loreclezole Wingrove and co-workers proved that 1,2,4-triazole ring of loreclezole interact with the amide group of Asn-289, which is situated on the β2 subunit of the GABAA receptor [14]. Our previously conducted radioligand binding experiments with the use of radiolabeled ligands of GABAA receptor, i.e. [3H]muscimol and [3H]flunitrazepam, showed that 4,5-disubstituted 1,2,4-triazole-3-thione derivatives did not possess the affinity to their respective active sites [15, 16]. The mentioned lack of affinity concerned both aromatic and aliphatic derivatives of 1,2,4-triazole-3-thione. However, GABAA receptor complex contains many orthosteric and allosteric binding sites [17], and therefore, finding that there is no interaction between 4,5-disubstituted 1,2,4-triazole-3-thiones and muscimol- and benzodiazepine-binding sites does not exclude the possibility of other effects on GABAA receptor. Therefore, in the course of searching for the mechanism of anticonvulsant activity of TP-315 we applied the patch clamp technique, that allows to investigate the influence of the compound on the chloride current through the GABAA receptor. It has been determined, during the electrophysiological experiments, that TP-315 did not cause any changes inchloride ion current. Moreover, it neither intensify nor inhibit GABA-evoked currents.Therefore, the obtained results justify the statement that TP-315 does not show the (ant)agonistic nor allosteric effect on GABAA receptor.
The anticonvulsant activity of TP-315 was identified in the mouse maximal electroshock-induced seizure (MES) test, which is an experimental model of human generalized tonic-clonic seizures [18]. The MES-induced seizure test still remains one of the basic screening models used for the identification of new compounds showing anticonvulsant effects [19]. Numerous researches have proven that the anticonvulsant activity observed in the MES-induced seizure test is a result of interaction between the tested compound and voltage-gated sodium channels (VGSCs) [20]. In spite of such premises, the results presented herein provide the first experimental proof that TP-315 shows strong affinity towards the VGSCs. TP-315 displaced [3H]batrachotoxin from its binding site much stronger than veratridine and lidocaine. Moreover, TP-315 turned out to possess stronger affinity towards sodium channels than carbamazepine, the first generation AED acting on VGSCs.
Sodium channels blockers, i.e. compounds disturbing the conduction of sodium ions through sodium channels, serve as local anaesthetics as well as antiarrhythmic and antiepileptic drugs. The quite new application of sodium channel blockers results from the fact that they are thought to inhibit excitotoxic damage of neurons [7]. Therefore they may show neuroprotective effects [21, 22]. Certainly, the hypothesis assuming that TP-315, beside the anticonvulsant activity, also shows neuroprotective effects shall require confirmation in experimental conditions. Nevertheless, in case of other AEDs acting on sodium channels, e.g. carbamazepine and phenytoin, the neuroprotective effects have been proven at the same time [23].
The research aiming at the understanding of molecular mechanism of action of TP-315 also included human α7 and human α4β2 nicotinic acetylcholine receptors (nAChRs). Although they do not belong to the main molecular targets of AEDs, the literature reports suggest that these receptors may play a significant role in pathophysiology of many neurological diseases, including epilepsy [24]. Significantly altered activity of nAChRs was observed in some types of epilepsy. This mainly concerns juvenile myoclonic epilepsy and autosomal dominant nocturnal frontal lobe epilepsy - ADNFLE [24]. Moreover, it has been found out that AEDs affect the function of nAChRs and that some of nAChRs antagonists show strong anticonvulsant effects in animal models of epilepsy [25]. For example, amino-alkyl-cyclohexanes efficiently protected mice and rats from seizures induced by electric shock (MES test) or nicotine. Median effective doses (ED50) for these derivatives reached 3.6 mg/kg in the MES test [26], and 2.46 mg/kg [25] in the nicotine-induced seizure model of epilepsy.
During the present research, the affinity of TP-315 towards some of the nAChRs was tested using radioligand binding technique. During the experiments, the level of displacement of [3H]epibatidine from its complexes with α4β2 and α7 receptors was measured. The mentioned receptors constitute the two main types of nicotinic acetylcholine receptors in the brain [27]. They differ in their sensibility to nicotine. Nicotine's affinity for human α4β2 nAChRs is nearly 1000 times higher than that for α7 receptor. It is also thought that nicotine-induced seizures are mediated mainly by α7 nAChRs [28]. In the course of our experiments it has been found that TP-315 does not possess affinity to epibatidine-binding sites on human α4β2 and α7 nAChRs.
An important stage of the pre-clinical trials, the drug-candidates are subject to, is to check the effect of those compounds on human cells and tissues. Therefore, it is recommended that, at the stage of cytotoxicity testing, there are applied methods allowing determination of the tested compound's effect on different phases of the cell cycle [29]. First of the two applied assays, i.e. PrestoBlue assay, measures the mitochondrial activity of cells, and therefore it is an early indicator of cellular toxicity. In turn, ToxiLight assay, whose principle consists in the measurement of adenylate kinase released from damaged cell membranes, is an indicator of cellular necrosis. Based on the results obtained from both PrestoBlue and ToxiLight assays it has been demonstrated that TP-315 is characterized by low toxicity for human hepatic HepG2 cells. While assessing the possible toxicity of drug-candidates, one should also observe the restrictions associated with in vitro methods. First of all, such methods measure the effect of chemical compounds on isolated cells, and therefore no interactions occurring in a living organism between different tissues and organs are taken into account.Therefore, it is worth mentioning that TP-315 was also tested in in-vivo model of neurotoxicity, i.e. in the chimney test [4]. The median toxic dose (TD50) for TP-315 administered 15min before the test was 462.9 ± 20.0 mg/kg, and the respective protective index (PI = TD50/ED50) value amounted to 9.7.
Conclusions
The presented study reports that the anticonvulsant activity of 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (TP-315) is at least the effect of its influence on the voltage-gated sodium channels (VGSCs). At the same time there has been excluded the possibility of interactions of TP-315 with orthosteric and allosteric binding sites on GABAA receptors.Moreover, it has been found that TP-315 does not interact with epibatidine-binding sites on human α4β2 and α7 nAChRs. We also found that TP-315 affected the viability of human hepatic HepG2 cells to a slight degree only. The results obtained enabled to identify the plausible mechanism of action for TP-315. Moreover, the potent anticonvulsant activity together with low (neuro)toxicity, justify the further preclinical and hopefully clinical development of TP-315 as potential candidate for new antiepileptic drug.
Acknowledgements
The research was supported by grant from the National Science Centre, Poland (Sonata funding, UMO-2013/11/D/NZ7/01170).
The authors thank to Novartis for kind donation of HEK-293 and SH-SY5Y cells transfected with genes encoding hα4β2 and hα7 nAChRs, respectively.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schmidt D, Löscher W. Drug resistance in epilepsy: putative neurobiologic and clinical mechanisms. Epilepsy. 2005;46:858-877
2. Löscher W. How to explain multidrug resistance in epilepsy? Epilepsy Curr. 2005;5:107-112
3. Perucca E, Meador KJ. Adverse effects of antiepileptic drugs. Acta Neurol Scand Suppl. 2005;181:30-35
4. Plech T, Łuszczki JJ, Wujec M, Flieger J, Pizoń M. Synthesis, characterization and preliminary anticonvulsant evaluation of some 4-alkyl-1,2,4-triazoles. Eur J Med Chem. 2013;60:208-215
5. Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162:1239-1249
6. Marcinkowska M, Kołaczkowski M, Kamiński K, Bucki A, Pawłowski M, Siwek A, Karcz T, Mordyl B, Starowicz G, Kubowicz P, Pękala E, Wesołowska A, Samochowiec J, Mierzejewski P, Bienkowski P. Design, synthesis, and biological evaluation of fluorinated imidazo[1,2-a]pyridine derivatives with potential antipsychotic activity. Eur J Med Chem. 2016;124:456-467
7. Callaway JK, Castillo-Melendez M, Giardina SF, Krstew EK, Beart PM, Jarrot B. Sodium channel blocking activity of AM-36 and sipatrigine (BW619C89): in vitro and in vivo evidence. Neuropharmacology. 2004;47:146-155
8. Arias HR, Gu RX, Feuerbach D, Guo BB, Ye Y, Wei DQ. Novel positive allosteric modulators of the human α7 nicotinic acetylcholine receptor. Biochemistry. 2011;50:5263-5278
9. Kamiński K, Zagaja M, Łuszczki JJ, Rapacz A, Andres-Mach M, Latacz G, Kieć-Kononowicz K. Design, synthesis, and anticonvulsant activity of new hybrid compounds derived from 2-(2,5-dioxopyrrolidin-1-yl)propanamides and 2-(2,5-dioxopyrrolidin-1-yl)butanamides. J Med Chem. 2015;58:5274-5286
10. Willow M, Catterall WA. Inhibition of binding of [3H]batrachotoxinin A 20-alpha-benzoate to sodium channels by the anticonvulsant drugs diphenylhydantoin and carbamazepine. Mol Pharmacol. 1982;22:627-635
11. Westerink WM, Schoonen WG. Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol In Vitro. 2007;21:1581-1591
12. Knasmüller S, Mersh-Sundermann V, Kevekordes S, Darroudi F, Huber WW, Hoelzl C, Bichler J, Majer BJ. Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants: current state of knowledge. Toxicology. 2004;198:315-328
13. Gasnier C, Dumont C, Benachour N, Clair E, Chagnon M-C, Séralini G-E. Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines. Toxicology. 2009;262:184-191
14. Wingrove PB, Wafford KA, Bain C, Whiting PJ. The modulatory action of loreclezole at the γ-aminobutyric acid type A receptor is determined by a single amino acid in the β2 subunit. Proc Natl Acad Sci USA. 1994;91:4569-4573
15. Plech T, Kaproń B, Łuszczki JJ, Paneth A, Siwek A, Kołaczkowski M, Żołnierek M, Nowak G. Studies on the anticonvulsant activity of 4-alkyl-1,2,4-triazole-3-thiones and their effect on gabaergic system. Eur J Med Chem. 2014;86:690-699
16. Plech T, Kaproń B, Łuszczki JJ, Wujec M, Paneth A, Siwek A, Kołaczkowski M, Żołnierek M, Nowak G. Studies on the anticonvulsant activity and influence on GABA-ergic neurotransmission of 1,2,4-triazole-3-thione based compounds. Molecules. 2014;19:11279-11299
17. Sigel E, Steinmann ME. Structure, function, and modulation of GABAA receptors. J Biol Chem. 2012;287:40224-40231
18. Löscher W, Fassbender CP, Nolting B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. II. Maximal electroshock seizure models. Epilepsy Res. 1991;8:79-94
19. Rogawski MA, Tofighy A, White HS, Matagne A, Wolff C. Current understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res. 2015;110:189-205
20. Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18-61
21. Taylor CP, Meldrum BS. Na+ channels as targets for neuroprotective drugs. Trends Pharmacol Sci. 1995;16:309-316
22. Waxman SG. Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis - current status. Nature Clin Pract Neurol. 2008;4:159-169
23. Cunha AO, Mortari MR, Liberato JL, dos Santos WF. Neuroprotective effects of diazepam, carbamazepine, phenytoin and ketamine after pilocarpine-induced status epilepticus. Basic Clin Pharmacol Toxicol. 2009;104:470-477
24. Ghasemi M, Hadipour-Niktarash A. Pathologic role of neuronal nicotinic acetylcholine receptors in epileptic disorders: implication for pharmacological interventions. Rev Neuroscience. 2015;26:199-223
25. Löscher W, Potschka H, Wlaź P, Danysz W, Parsons CG. Are neuronal nicotinic receptors a target for antiepileptic drug development? Studies in different seizure models in mice and rats. Eur J Pharmacol. 2003;466:99-111
26. Parsons CG, Danysz W, Bartmann A, Spielmanns P, Frankiewicz T, Hesselink M, Eilbacher B, Quack G. Amino-alkyl-cyclohexanes are novel uncompetitive NMDA receptor antagonists with strong voltage-dependency and fast blocking kinetics: in vitro and in vivo characterization. Neuropharmacology. 1999;38:85-108
27. Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555-561
28. Damaj MI, Glassco W, Dukat M, Martin BR. Pharmacological characterization of nicotine-induced seizures in mice. J Pharmacol Exp Ther. 1999;291:1284-1291
29. Miret S, de Groene EM, Klaffke W. Comparison of in vitro assays of cellular toxicity in the human hepatic cell line HepG2. J Biomol Screen. 2006;11:184-193
Author contact
![]() Corresponding author: Tomasz Plech, Department of Pharmacology, Faculty of Health Sciences, Medical University of Lublin, Chodźki 4a, 20-093 Lublin, Poland. Phone/Fax: +48 (81) 448 6770 email: tomasz.plechpl
Corresponding author: Tomasz Plech, Department of Pharmacology, Faculty of Health Sciences, Medical University of Lublin, Chodźki 4a, 20-093 Lublin, Poland. Phone/Fax: +48 (81) 448 6770 email: tomasz.plechpl