International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(5):506-514. doi:10.7150/ijms.18142 This issue Cite
Research Paper
Traditional Chinese Medication Qiliqiangxin Protects Against Cardiac Remodeling and Dysfunction in Spontaneously Hypertensive Rats
Hui Wang1, 2*, Xiaomin Zhang1, 2*, Pujiao Yu3*, Qiulian Zhou4, 5, Jialiang Zhang1, 2, Haifeng Zhang1, Hongsheng Zhu6, Chenlin Zhang6, Wenming Yao1, Lin Che3, Jiahong Xu3, Yihua Bei4, 5 ![]() , Xinli Li1
, Xinli Li1 ![]()
1. Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China;
2. Department of Cardiology, Jiangsu Shengze Hospital, Nanjing Medical University, Suzhou 215228, China;
3. Department of Cardiology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China;
4. Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, Shanghai 200444, China;
5. Innovative Drug Research Center of Shanghai University, Shanghai 200444, China;
6. Department of Anesthesiology, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 200032, China.
* These three authors contributed equally to this work.
Received 2016-10-30; Accepted 2017-3-14; Published 2017-4-9
Citation:
Wang H, Zhang X, Yu P, Zhou Q, Zhang J, Zhang H, Zhu H, Zhang C, Yao W, Che L, Xu J, Bei Y, Li X. Traditional Chinese Medication Qiliqiangxin Protects Against Cardiac Remodeling and Dysfunction in Spontaneously Hypertensive Rats. Int J Med Sci 2017; 14(5):506-514. doi:10.7150/ijms.18142. https://www.medsci.org/v14p0506.htm
Other stylesAbstract
Qiliqiangxin (QLQX), a traditional Chinese herbs medication, exerted protective effect in chronic heart failure patients in a multicenter randomized double-blind study. QLQX has also been found to improve cardiac function and reduce cardiac fibrosis in spontaneously hypertension animal model. However, the effect of longterm treatment with QLQX in such a condition and the related molecular mechanisms remain largely unknown. In the present study, thirteen-week-old spontaneously hypertensive rats (SHRs) were treated by daily intragastric administration of QLQX or saline for one year. Echocardiography, electron microscopy, and Masson's trichrome staining were used to determine cardiac function, mitochondria ultrastructure, and cardiac fibrosis, respectively. Quantitative reverse transcription polymerase chain reactions (qRT-PCRs) and Western blotting were used to determine gene expressions. We found that QLQX significantly improved cardiac function and reduced gene markers of pathological hypertrophy including ANP, BNP, and Myh7. QLQX also attenuated cardiac fibrosis and apoptosis in SHRs as evidenced by downregulation of α-SMA, collagen I, collagen III, and TGF-β expressions and reduction of Bax to Bcl-2 ratio. Moreover, the damage of mitochondrial ultrastructure was greatly improved and the reduction of PPAR-α, PPAR-γ, and PGC-1α expression levels was significantly restored in SHRs by treatment with QLQX. In conclusion, longterm treatment with QLQX protects against cardiac remodeling and dysfunction in hypertension by increasing PPARs and PGC-1α.
Keywords: Qiliqiangxin, cardiac remodeling, fibrosis, spontaneously hypertension, PPAR, PGC-1α.
Introduction
Hypertension represents a common condition that progresses toward severe cardiovascular morbidity. Hypertensive heart disease (HHD), which is characterized by diastolic dysfunction, cardiac remodeling, and cardiac fibrosis, is one of the major clinical phenotypes caused by elevated blood pressure [1]. HHD leads to the worsening of entire cardiac function and results in an increased risk of adverse cardiac events by further damaging systolic function [2]. Preventing cardiac dysfunction and adverse remodeling would be beneficial for patients with hypertension and HHD, though effective therapeutic strategies are still limited [3]. Therefore, additional therapeutic targets and treatment options are highly required [4].
Qiliqiangxin (QLQX), a traditional Chinese herbs medication, is composed of 11 distinct herbs including astragali radix, ginseng radix et rhizoma, aconiti lateralis radix preparata, salvia miltiorrhiza radix et rhizoma, semen descurainiae lepidii, alismatis rhizoma, cinnamomi ramulus, polygonati odorati rhizoma, carthami flos, periploca cortex, and citri reticulatae pericarpium [5, 6]. QLQX has been reported to attenuate the progression of cardiac remodeling and fibrosis after acute myocardial infarction and inhibit the development of cardiac hypertrophy and failure after transverse aorta constriction [7, 8]. It has also been shown that QLQX can prevent cardiac hypertrophy and dysfunction both in vitro and in vivo through several pathways such as activation of mTOR and PPAR-γ, regulation of IL-6, IL-10, and TNF-α, and inhibition of angiotensin II [7-13]. Noteworthy, our group previously reported that QLQX exerted protective effects in chronic heart failure patients in a multicenter randomized double-blind study [14]. These protective effects included the improvement of left ventricle ejection fraction, New York Heart Association (NYHA) functional classification, 6-minute walking distance, and quality of life, as well as the reduction of plasma N-terminal pro-B-type natriuretic peptide (NT-proBNP) level [14]. Interestingly, QLQX has also been demonstrated to improve cardiac function and reduce cardiac fibrosis in spontaneously hypertensive animal model through inhibition of cardiac chymase [15]. However, the effect of longterm treatment with QLQX in such a condition and the related molecular mechanisms remain largely unknown.
In this study, we investigated the effect of QLQX in spontaneously hypertensive rats (SHRs) and demonstrated that QLQX could attenuate cardiac remodeling and dysfunction likely by inducing peroxisome proliferator-activated receptors (PPARs) and PPAR-γ coactivator-1α (PGC-1α).
Materials and Methods
Animals
All rats were purchased and raised at the Experimental Animal Center of Nanjing Medical University (Nanjing, China). All procedures with rats were in accordance with the guidelines on the use and care of laboratory animals for biomedical research published by National Institutes of Health (No. 85-23, revised 1996), and the experimental protocol was reviewed and approved by the ethical committees of Nanjing Medical University.
Thirteen-week-old spontaneously hypertensive rats (SHRs) were randomly divided into two groups: the SHR control group with intragastric administration of saline (SHR+Saline, n=3) and the SHR treatment group with intragastric administration of 1 g/kg/d QLQX (SHR+QLQX, n=3). Moreover, four Wistar-Kyoto (WKY) rats were treated with saline as the normal control group (Wistar+Saline, n=4). The three groups of rats were housed in one room and fed with a standard diet and water. QLQX was provided by Shijiazhuang Yiling Pharmaceutical Co., Ltd. (Shijiazhuang, Hebei, China). To guarantee the quality and consistency of QLQX, the raw medicinal materials were of a certain variety, and their areas of origin, medicinal parts, and processing methods were kept consistent. The stability of the product was verified by analyzing 10 batches of QLQX and fingerprints. The three groups of rats were treated by daily intragastric administration of QLQX or saline for one year.
Echocardiography
In order to assess the cardiac structure and function, all rats were fixed in hypsokinesis position after anesthesia with 1.5-2.0% isoflurane and echocardiography was performed blinded using Vevo 2100 (VisualSonics Inc, Toronto, Ontario, Canada) with a 30 MHz central frequency scan head. Evaluation indexes include left ventricular internal dimension-diastole (LVIDd), left ventricular internal dimension-systole (LVIDs), left ventricular fractional shortening (FS), and left ventricular ejection fraction (EF). These parameters were measured from M-mode images taken from the parasternal short-axis view at papillary muscle level.
Electron microscopy
Heart samples were fixed in 2.5% glutaraldehyde for 1 h, treated with 1% osmium tetroxide, and then dehydrated and embedded in Durcupan. Heart samples were sectioned into 60 nm and mounted on Cu-grids, then stained with uranyl acetate and lead citrate, and finally examined under an electron microscope (JEM-1010). Images were taken at 15,000× magnification. A total of 3 sections/specimen from 3 rats per group were used for analyses of mitochondria ultrastructure.
Histological analysis
After intragastric administration of QLQX or saline for one year, hearts were excised after being perfused with phosphate-buffered saline and then fixed with 4% paraformaldehyde. These sections were processed in paraffin using standard techniques and the degree of cardiac fibrosis was evaluated with Masson's trichrome staining. Images were acquired from at least 20 fields of each heart section randomly. The degree of cardiac fibrosis was assessed by the ratio of fibrotic area (blue) to total myocardial area using Image J software.
Western blotting
Heart tissues were homogenized in RIPA lysis buffer and then centrifuged to acquire protein supernatants. The equal amounts of total proteins were subjected to SDS-PAGE gels and transferred onto PVDF membranes. The membranes were incubated with primary antibodies, including transforming growth factor-β (TGF-β, 1:1000 dilution; Cell Signaling Technology, Boston, Massachusetts, USA), α-smooth muscle actin (α-SMA, 1:1000 dilution; Cell Signaling Technology), B-cell lymphoma 2 (Bcl-2, 1:1000 dilution; Cell Signaling Technology), Bcl-2-associated X protein (Bax, 1:1000 dilution; Cell Signaling Technology), PPAR-α (1:1000 dilution; Abcam, Cambridge, UK), PPAR-γ (1:400 dilution; Abcam), PGC-1α (1:1000 dilution; NOVUS, Littleton, COLO, USA), Protein kinase B (Akt, 1:1000 dilution; Cell Signaling Technology), p-Akt (Ser473, 1:1000 dilution; Cell Signaling Technology), p-Akt (Thr308, 1:1000 dilution; Cell Signaling Technology), LC3 (Light chain 3, 1:1000 dilution; Cell Signaling Technology), and P62 (EBI 3 associated protein 62, 1:1000 dilution; Cell Signaling Technology). Glyceraldehyde 3-phosphate dehydrogenase antibody (GAPDH, 1:2000 dilution; Kangchen, Shanghai, China) was used as a loading control. After this, the membranes were incubated with secondary antibodies for 2 h at room temperature. Then the signals were visualized using the ECL Plus Western blotting detection reagents (Bio-Rad, Hercules, CA, USA) and the ChemiDoc XRS Plus luminescent image analyzer (Bio-Rad). The densitometry of each protein band was analyzed by Imagelab software (Bio-Rad).
Quantitative reverse transcription polymerase chain reactions (qRT-PCRs)
Total RNA was extracted from left ventricular tissues of rats with miRNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse transcribed into cDNA using Bio-Rad iScriptTM cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer's instruction. The cDNA was then subjected to 40 cycles of quantitative PCR in the 7900HT Fast Real-Time PCR System with SYBR green (Takara, Tokyo, Japan). The relative expression levels of mRNA were calculated with GAPDH as the control gene. The primer sequences are as follows (forward and reverse, 5'-3').
GAPDH, GGCACAGTCAAGGCTGAGAATG and ATGGTGGTGAAGACGCCAGTA.
ANP, CTCCCAGGCCATATTGGAG and TCCAGGTGGTCTAGCAGGTT.
BNP, TGGGAAGTCCTAGCCAGTCTC and TCTGAGCCATTTCCTCTGAC.
MyH7, GGATGACGTTACCTCCAACA and GTGTCTCCTCAGCCTTGCTC.
α-SMA, GTCCCAGACATCAGGGAGTAA and TCGGATACTTCAGCGTCAGGA.
Collagen I, GAGCGGAGAGTACTGGATCGA and CTGACCTGTCTCCATGTTGCA.
Collagen III, TGCCATTGCTGGAGTTGGA and GAAGACATGATCTCCTCAGTGTTGA.
Statistical analysis
All data were shown as mean±SEM. The one-way ANOVA followed by Bonferroni's post-hoc test was used to compare the one-way layout data. A P value less than 0.05 was considered to be statistically significant. All analyses were performed using SPSS and presented with GraphPad Prism 5.
Results
QLQX improves cardiac function in SHRs
Cardiac function was measured by echocardiography in SHRs after one year treatment with QLQX. As shown in Figure 1A, QLQX significantly improved cardiac function, including EF, FS, and LVIDs. Moreover, QLQX prevented cardiac hypertrophy as evidenced by the reduction of the ratio of left ventricle weight to body weight and the ratio of left ventricle weight to tibia length (Figure 1B). In addition, the gene markers of pathological hypertrophy including ANP, BNP, and Myh7 were also reduced by treatment with QLQX (Figure 1C).
QLQX attenuates cardiac fibrosis and apoptosis in SHRs
Based on histological analysis of cardiac tissue sections, we found that the degree of cardiac fibrosis in the SHR+QLQX group was significantly alleviated when compared with the SHR+Saline group (Figure 2A), which was further supported by the decreased expression levels of α-SMA, collagen I, and collagen III as determined by qRT-PCRs (Figure 2B) and Western blotting (Figure 2C). Besides, the expression level of TGF-β, a key mediator responsible for cardiac fibrosis and remodeling [19, 20], was also reduced by treatment with QLQX (Figure 2C). Meanwhile, the ratio of proapoptotic protein Bax to antiapoptotic protein Bcl-2 was increased in SHRs, while reduced by QLQX treatment, indicating that cardiac apoptosis was also attenuated (Figure 3A). However, the autophagy was not altered in SHRs regardless of QLQX treatment, as indicated by no alteration of the expression levels of P62 and LC3 (Figure 3B).
Figure 1
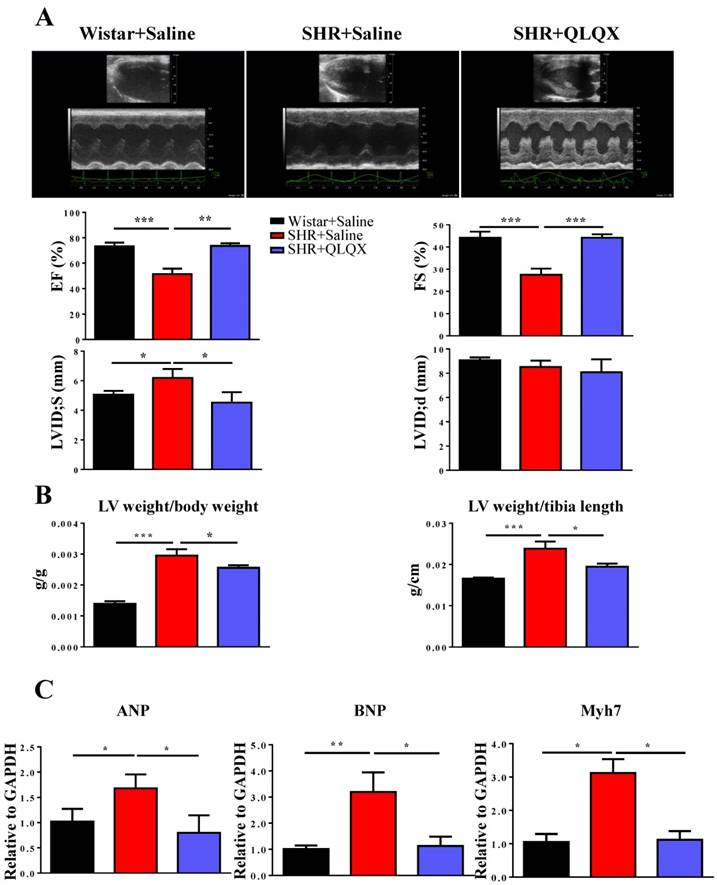
QLQX improves cardiac function in SHRs (A) QLQX significantly improved cardiac function in SHRs, by preserving ejection fraction (EF), fractional shortening (FS), and left ventricular internal dimension-systole (LVIDs). (B) QLQX reduced the ratio of left ventricle weight to body weight and the ratio of left ventricle weight to tibia length in SHRs. (C) QLQX reduced the gene markers of pathological hypertrophy in SHRs. N=3-4 per group. *, P<0.05; **, P<0.01; ***, P <0.001.
QLQX attenuates mitochondrial ultrastructure damage and restores downregulation of PPAR-α and PPAR-γ in SHRs
Using transmission electron microscopy (TEM), we demonstrated that mitochondria ultrastructure of cardiac myocytes showed more swelling and vacuolization in SHRs, which could be greatly improved by QLQX treatment (Figure 4A). The peroxisome proliferator-activated receptors (PPARs) are involved in various aspects of cardiac energy metabolism and we previously reported that QLQX could increase PPAR-α, PPAR-γ, and PGC-1α in acute myocardial infarction [7]. Here we also found that PPAR-α, PPAR-γ, and PGC-1α were decreased in SHRs and QLQX could elevate all of them (Figure 4B), which is consistent with our previous report [7]. Similarly, the Akt signaling pathway was not changed in SHRs regardless of QLQX treatment (Figure 4C). Thus, we speculate that QLQX might probably improve cardiac energy metabolism in SHRs via increasing PPARs and PGC-1α.
Figure 2
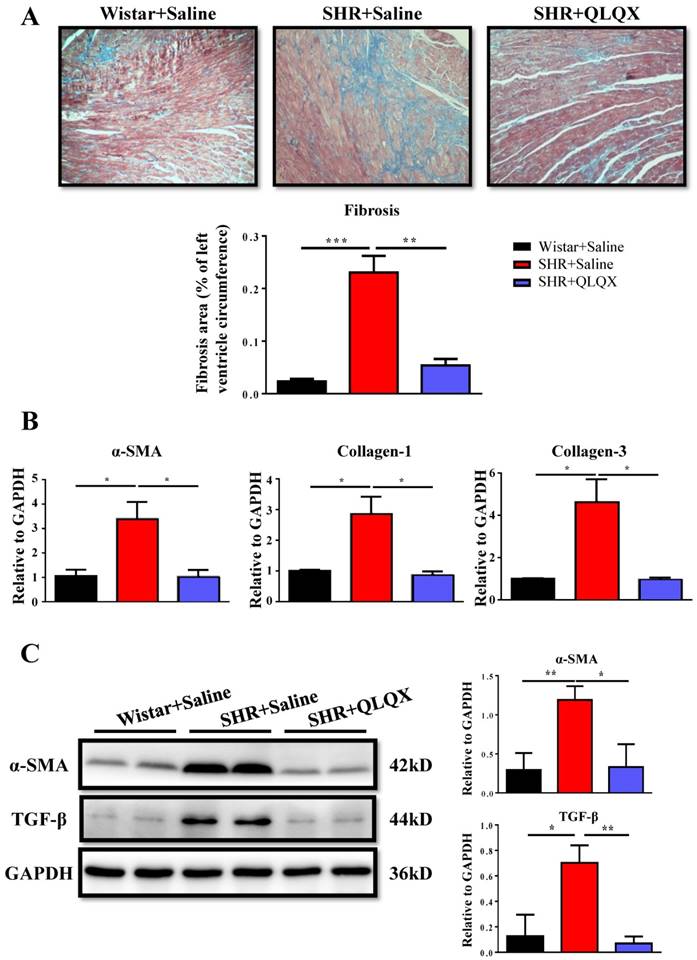
QLQX attenuates cardiac fibrosis in SHRs (A) QLQX decreased cardiac fibrosis in SHRs as determined by Masson's trichrome staining. (B) QLQX downregulated mRNA levels of α-SMA, collagen I, and collagen III in SHRs. (C) QLQX decreased protein levels of α-SMA and TGF-β in SHRs. N=3-4 per group. *, P<0.05; **, P<0.01; ***, P <0.001.
Figure 3
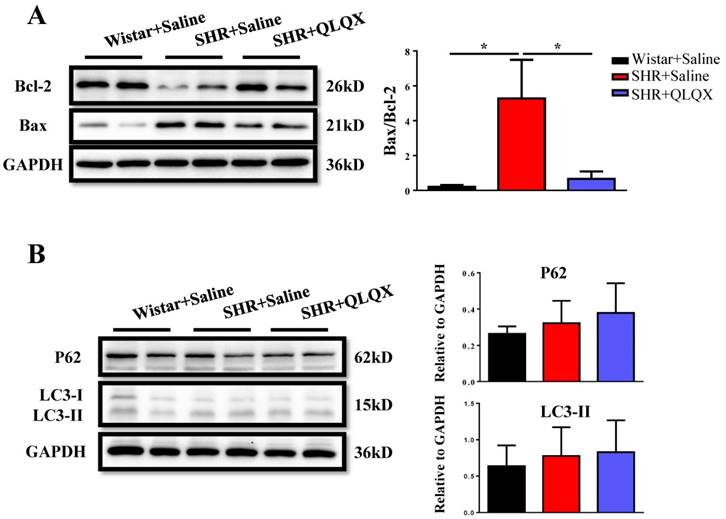
QLQX decreases cardiac apoptosis without affecting autophagy in SHRs QLQX reduced the ratio of Bax to Bcl-2 in SHRs. (B) QLQX did not affect the autophagic proteins including P62 and LC3. N=3-4 per group. *, P<0.05.
Discussion
Hypertension, which presents vascular sclerosis and left ventricle hypertrophy, will finally develop to cardiac decompensation if not effectively interfered [16]. To slow the progress of cardiac remodeling and heart failure induced by hypertension, the present therapeutic strategies lay emphasis on inhibiting β-adrenergic receptor, angiotensin II, and aldosterone. However the effects of these treatments are still limited for chronic phase of hypertension. Traditional Chinese medication has a certain foundation in clinic with a long history and may set a new direction for therapeutic strategies. Previous study has indicated that QLQX, a traditional Chinese medication, had beneficial effect on chronic heart failure patients and could prevent the heart from adverse remodeling [14, 17]. Here, in this study, we focused on the protective effect of QLQX in a rat model of genetic hypertension and found that QLQX could improve cardiac function and attenuate cardiac remodeling and fibrosis after one-year treatment.
The major findings of the present study are as follows. Firstly, QLQX could significantly improve cardiac function by preserving EF and FS in SHRs. In addition, the gene markers of pathological hypertrophy including ANP, BNP, and Myh7 were reduced by QLQX treatment. Secondly, QLQX alleviated cardiac fibrosis and apoptosis, and improved mitochondrial ultrastructure of cardiomyocytes in SHRs. Finally, QLQX restored the reduction of PPAR-α, PPAR-γ, and PGC-1α expressions in SHRs. Taken together, these data demonstrated that QLQX could protect against cardiac remodeling and dysfunction in hypertensive rats by increasing PPARs and PGC-1α.
Sustained hypertension progressively leads to left ventricular hypertrophy, cardiac fibrosis, and ultimately heart failure [18]. It was previously reported that QLQX was effective to improve cardiac function and reduce cardiac fibrosis in SHRs [15]. However, the effect of long-term treatment with QLQX in hypertension and the related molecular mechanisms remain largely unknown. Here, we demonstrated that treatment with QLQX for one year was effective to preserve EF and FS in SHRs, and prevent ventricular hypertrophy as well. Moreover, hypertension-induced cardiac fibrosis was greatly alleviated by QLQX, suggesting a potential protective effect of long-term treatment with QLQX on cardiac remodeling and dysfunction in hypertension.
Figure 4
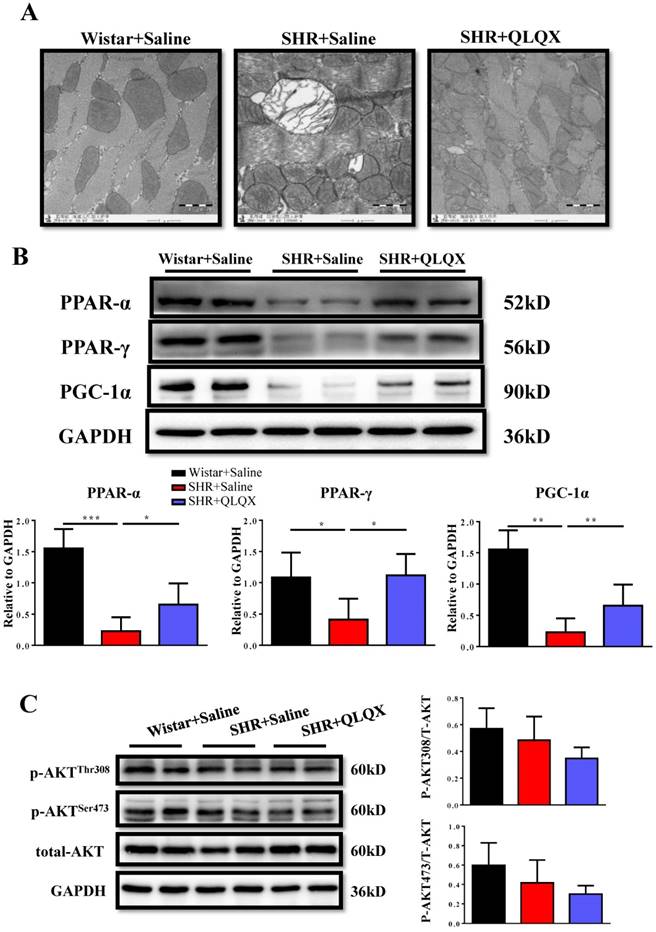
QLQX attenuates the damage of mitochondrial ultrastructure and restores the expression levels of PPAR-α, PPAR-γ, and PGC-1α in SHRs (A) QLQX preserved the mitochondria ultrastructure in SHRs as determined by transmission electron microscopy. (B) QLQX increased PPAR-α, PPAR-γ, and PGC-1α expression levels in SHRs. (C) QLQX did not affect the Akt signaling in SHRs. N=3-4 per group. *, P<0.05; **, P<0.01; ***, P <0.001.
Cardiomyocyte apoptosis is a hallmark induced by a variety of cardiac injuries including hypertension. Here we showed that treatment with QLQX for one year was effective to change the apoptotic proteins in SHRs. Additionally, QLQX was previously reported to reduce cardiomyocyte autophagy during 4 weeks of pressure overload in mice [8]. However, in the present study, the autophagic proteins were not modified in SHRs regardless of QLQX treatment. The differences between this study and ours are that we used different animal models of hypertension and treated animals with QLQX for much longer period. Thus, the effect of QLQX in cellular autophagy during hypertension may be related to both disease stage and length of treatment.
Cardiac energy metabolism defect is an important mediator of remodeling progression and it also contributes to the development of myocardial diseases [19-21]. The peroxisome proliferator-activated receptor (PPAR) family has been confirmed to regulate cardiac energy metabolism at the gene expression level [22-25]. The PPAR family consists of PPAR-α, PPAR-δ, and PPAR-γ. It has been reported that activation of PPAR-γ prevented lipopolysaccharide-mediated cardiac dysfunction and that PGC-1α was required for QLQX-associated enhancement in cardiomyocyte mitochondrial energy metabolism [26, 27]. In our previous study, we also demonstrated that QLQX could improve cardiac function and attenuate cardiac remodeling after AMI by activating PPAR-γ [7]. Moreover, the regulation of PPAR family was associated with cardiac fibrosis and ventricular remodeling [28, 29]. In line with these studies, the present study found that QLQX treatment significantly attenuated the damage of myocardium mitochondrial ultrastructure in SHRs. Moreover, QLQX treatment upregulated the expression levels of PPAR-α, PPAR-γ, and PGC-1α in SHRs, suggesting that QLQX might deliver the protective effect on cardiac remodeling and dysfunction in SHRs through activation of PPARs and PGC-1α.
Several limitations of the present study should be highlighted. Firstly, it has been reported that activations of PPAR-α and PPAR-γ have beneficial effects on cardiac hypertrophy and fibrosis by regulating fibroblast proliferation and differentiation, however, the direct evidence for the role of PGC-1α in regulating cardiac fibrosis is still lacking [29, 30]. Secondly, though we confirmed the elevation of PPAR-α, PPAR-γ, and PGC-1α with QLQX treatment, it deserves to determine if the activation of PPAR pathway is necessary for the protective effects of QLQX in preventing cardiac remodeling and dysfunction in hypertension. Thirdly, as QLQX contains 11 distinct active components, which compound(s) in QLQX should be responsible for these protective effects observed in this study warrants further investigation.
Conclusion
In conclusion, our study demonstrates that QLQX protects against cardiac remodeling and dysfunction in spontaneously hypertensive rats by activation of PPARs and PGC-1α.
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (81370332 and 81170201 to XL Li; 81400647 to Y Bei), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD20102013 to XL Li), the National Basic Research Program of China (973 Program, Grant no.2012CB518606), Natural Science Foundation of Jiangsu Provincial (BK2012648 to Hui Wang), and “Six talent peaks” project in Jiangsu Province (2015-WSN-033). Dr. XL Li is an Associate Fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine.
Competing Interests
Dr. Xinli Li received research grants from Shijiazhuang Yiling Pharmaceutical Co., Ltd. All other authors have reported that they have no relationships to disclose.
References
1. Zile MR, Brutsaert DL. New concepts in diastolic dysfunction and diastolic heart failure: Part I: diagnosis, prognosis, and measurements of diastolic function. Circulation. 2002;105:1387-93
2. Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD. et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247-59
3. Zile MR, Brutsaert DL. New concepts in diastolic dysfunction and diastolic heart failure: Part II: causal mechanisms and treatment. Circulation. 2002;105:1503-8
4. Bernardo BC, Ooi JY, Lin RC, McMullen JR. miRNA therapeutics: a new class of drugs with potential therapeutic applications in the heart. Future Med Chem. 2015;7:1771-92
5. Chen F, Wu JL, Fu GS, Mou Y, Hu SJ. Chronic treatment with qiliqiangxin ameliorates aortic endothelial cell dysfunction in diabetic rats. J Cardiovasc Pharmacol Ther. 2015;20:230-40
6. Tao L, Shen S, Li X. Future prospects of Qiliqiangxin on heart failure: epigenetic regulation of regeneration. Front Genet. 2013;4:221
7. Tao L, Shen S, Fu S, Fang H, Wang X, Das S. et al. Traditional Chinese Medication Qiliqiangxin attenuates cardiac remodeling after acute myocardial infarction in mice. Sci Rep. 2015;5:8374
8. Zou Y, Lin L, Ye Y, Wei J, Zhou N, Liang Y. et al. Qiliqiangxin inhibits the development of cardiac hypertrophy, remodeling, and dysfunction during 4 weeks of pressure overload in mice. J Cardiovasc Pharmacol. 2012;59:268-80
9. Zhou J, Jiang K, Ding X, Fu M, Wang S, Zhu L. et al. Qiliqiangxin inhibits angiotensin II-induced transdifferentiation of rat cardiac fibroblasts through suppressing interleukin-6. J Cell Mol Med. 2015;19:1114-21
10. Wei Y, Liu X, Hou L, Che W, The E, Jhummon MV. Qiliqiangxin affects L type ca(2+) current in the normal and hypertrophied rat heart. Evid Based Complement Alternat Med. 2012;2012:131830
11. Zhou Y, Fang H, Lin S, Shen S, Tao L, Xiao J. et al. Qiliqiangxin Protects Against Cardiac Ischemia-Reperfusion Injury via Activation of the mTOR Pathway. Cell Physiol Biochem. 2015;37:454-64
12. Xiao H, Song Y, Li Y, Liao YH, Chen J. Qiliqiangxin regulates the balance between tumor necrosis factor-alpha and interleukin-10 and improves cardiac function in rats with myocardial infarction. Cell Immunol. 2009;260:51-5
13. Zhang J, Wei C, Wang H, Tang S, Jia Z, Wang L. et al. Protective effect of qiliqiangxin capsule on energy metabolism and myocardial mitochondria in pressure overload heart failure rats. Evid Based Complement Alternat Med. 2013;2013:378298
14. Li X, Zhang J, Huang J, Ma A, Yang J, Li W. et al. A multicenter, randomized, double-blind, parallel-group, placebo-controlled study of the effects of qili qiangxin capsules in patients with chronic heart failure. J Am Coll Cardiol. 2013;62:1065-72
15. Liu W, Chen J, Xu T, Tian W, Li Y, Zhang Z. et al. Qiliqiangxin improves cardiac function in spontaneously hypertensive rats through the inhibition of cardiac chymase. Am J Hypertens. 2012;25:250-60
16. Dent MR, Das S, Dhalla NS. Alterations in both death and survival signals for apoptosis in heart failure due to volume overload. J Mol Cell Cardiol. 2007;43:726-32
17. Ye Y, Gong H, Wang X, WU J, Wang S, Yuan J. et al. Combination Treatment With Antihypertensive Agents Enhances the Effect of Qiliqiangxin on Chronic pressure Overload-induced Cardiac Hypertrophy and Remodeling in Male Mice. J Cardiovasc Pharmacol. 2015;65:628-39
18. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021-40
19. van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovasc Res. 2004;61:218-26
20. Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. 2008;44:968-75
21. Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM. et al. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930-9
22. Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JR, Belke DD. et al. A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002;277:4098-103
23. Panagia M, Gibbons GF, Radda GK, Clarke K. PPAR-alpha activation required for decreased glucose uptake and increased susceptibility to injury during ischemia. Am J Physiol Heart Circ Physiol. 2005;288:H2677-83
24. Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ. et al. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92:518-24
25. Cheng L, Ding G, Qin Q, Xiao Y, Woods D, Chen YE. et al. Peroxisome proliferator-activated receptor delta activates fatty acid oxidation in cultured neonatal and adult cardiomyocytes. Biochem Biophys Res Commun. 2004;313:277-86
26. Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS. et al. Peroxisome proliferator-activated receptor-gamma activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550-62
27. Lin S, Wu X, Tao L, Bei Y, Zhang H, Zhou Y. et al. The Metabolic Effects of Traditional Chinese Medication Qiliqiangxin on H9C2 Cardiomyocytes. Cell Physiol Biochem. 2015;37:2246-56
28. Liu HJ, Liao HH, Yang Z, Tang QZ. Peroxisome Proliferator-Activated Receptor-gamma Is Critical to Cardiac Fibrosis. PPAR Res. 2016;2016:2198645
29. Ogata T, Miyauchi T, Sakai S, Irukayama-Tomobe Y, Goto K, Yamaguchi I. Stimulation of peroxisome-proliferator-activated receptor alpha (PPAR alpha) attenuates cardiac fibrosis and endothelin-1 production in pressure-overloaded rat hearts. Clin Sci (Lond). 2002;103(Suppl 48):284S-8S
30. Chen M, Li H, Wang G, Shen X, Zhao S, Su W. Atorvastatin prevents advanced glycation end products (AGEs)-induced cardiac fibrosis via activating peroxisome proliferator-activated receptor gamma (PPAR-gamma). Metabolism. 2016;65:441-53
Author contact
![]() Corresponding author: Dr. Xinli Li, Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, China Tel: 0086-25-84352775; Fax: 0086-25-84352775 E-mail: xinli3267_njcom Dr. Yihua Bei, Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, 333 Nan Chen Road, Shanghai 200444, China Tel: 0086-21-66138131; Fax: 0086-21-66138131 E-mail: beiyh36edu.cn
Corresponding author: Dr. Xinli Li, Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, China Tel: 0086-25-84352775; Fax: 0086-25-84352775 E-mail: xinli3267_njcom Dr. Yihua Bei, Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, 333 Nan Chen Road, Shanghai 200444, China Tel: 0086-21-66138131; Fax: 0086-21-66138131 E-mail: beiyh36edu.cn
Citation styles
APA
Wang, H., Zhang, X., Yu, P., Zhou, Q., Zhang, J., Zhang, H., Zhu, H., Zhang, C., Yao, W., Che, L., Xu, J., Bei, Y., Li, X. (2017). Traditional Chinese Medication Qiliqiangxin Protects Against Cardiac Remodeling and Dysfunction in Spontaneously Hypertensive Rats. International Journal of Medical Sciences, 14(5), 506-514. https://doi.org/10.7150/ijms.18142.
ACS
Wang, H.; Zhang, X.; Yu, P.; Zhou, Q.; Zhang, J.; Zhang, H.; Zhu, H.; Zhang, C.; Yao, W.; Che, L.; Xu, J.; Bei, Y.; Li, X. Traditional Chinese Medication Qiliqiangxin Protects Against Cardiac Remodeling and Dysfunction in Spontaneously Hypertensive Rats. Int. J. Med. Sci. 2017, 14 (5), 506-514. DOI: 10.7150/ijms.18142.
NLM
Wang H, Zhang X, Yu P, Zhou Q, Zhang J, Zhang H, Zhu H, Zhang C, Yao W, Che L, Xu J, Bei Y, Li X. Traditional Chinese Medication Qiliqiangxin Protects Against Cardiac Remodeling and Dysfunction in Spontaneously Hypertensive Rats. Int J Med Sci 2017; 14(5):506-514. doi:10.7150/ijms.18142. https://www.medsci.org/v14p0506.htm
CSE
Wang H, Zhang X, Yu P, Zhou Q, Zhang J, Zhang H, Zhu H, Zhang C, Yao W, Che L, Xu J, Bei Y, Li X. 2017. Traditional Chinese Medication Qiliqiangxin Protects Against Cardiac Remodeling and Dysfunction in Spontaneously Hypertensive Rats. Int J Med Sci. 14(5):506-514.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.