Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(5):425-433. doi:10.7150/ijms.18037 This issue Cite
Research Paper
Isoflurane preconditioning inhibits the effects of tissue-type plasminogen activator on brain endothelial cell in an in vitro model of ischemic stroke
So Yeong Cheon1, 2*, So Yeon Kim1, 2*, Eun Hee Kam1, 2, Jae Hoon Lee1, 2, Jeong Min Kim1, 2, Eun Jung Kim1, 2, Tae Whan Kim1, Bon-Nyeo Koo1, 2 ![]()
1. Department of Anesthesiology and Pain Medicine, Yonsei University College of Medicine, Seoul, Republic of Korea;
2. Anesthesia and Pain Research Institute, Yonsei University College of Medicine, Seoul, Republic of Korea.
* These authors contributed equally to this work.
Received 2016-10-23; Accepted 2017-1-30; Published 2017-4-8
Abstract
Tissue-type plasminogen activator (tPA) is the only treatment for ischemic stroke. However, tPA could induce the intracranial hemorrhage (ICH), which is the main cause of death in ischemic stroke patient after tPA treatment. At present, there is no treatment strategy to ameliorate tPA-induced brain injury after ischemia. Therefore, we investigated the effect of pre-treated isoflurane, which is a volatile anesthetic and has beneficial effects on neurological dysfunction, brain edema and infarct volume in ischemic stroke model. In this study, we used oxygen/glucose deprivation and reperfusion (OGD/R) condition to mimic an ischemic stroke in vitro. Matrix metalloproteinases (MMP) activity was measured in endothelial cell media. Also, neuronal cell culture was performed to investigate the effect of pretreated isoflurane on the neuronal cell survival after tPA-induced injury during OGD/R. Isoflurane pretreatment prevented tPA-induced MMP-2 and MMP-9 activity and suppressed tPA-triggered LRP/NF-κB/Cox-2 signaling after OGD/R. Neuronal cells, incubated with endothelial cell conditioned medium (EC-CM) after tPA + OGD/R, showed upregulation of pro-apoptotic molecules. However, neurons incubated with isoflurane-pretreated EC-CM showed increased anti-apoptotic molecules. Our findings suggest that isoflurane pretreatment could attenuate tPA-exaggerated brain ischemic injury, by reducing tPA-induced LRP/NF-κB/Cox-2 in endothelial cells, endothelial MMP-2 and MMP-9 activation, and subsequent pro-apoptotic molecule in neurons after OGD/R.
Keywords: Tissue-type plasminogen activator, isoflurane, oxygen/glucose deprivation, endothelial cell, matrix metalloproteinase, neuronal cell.
Introduction
Tissue-type plasminogen activator (tPA) is a thrombolytic agent used for the treatment of ischemic stroke within 3 hours of onset [1, 2]. It is useful to dissolve the blood clot but this effect also leads to intracranial hemorrhage (ICH) in stroke patients [3]. Many clinical and experimental studies have proven that ICH after tPA treatment closely correlates with blood brain barrier (BBB) breakdown and matrix metalloproteinases (MMPs) activation in the brain, which is involved in aggravation of infarction, morbidity, and mortality [4-6]. MMPs play an essential role in tissue remodeling in normal conditions; however, in pathological conditions, MMPs are related to the degradation of the BBB [4]. In addition, dysfunction of MMPs results in interruption of cell to cell or cell to matrix homeostasis for cell survival through the degradation of major neurovascular substrates [7]. Especially, it is reported that MMP-9 induces neuronal cell death directly by disturbing cell-to-matrix communication [7, 8].
Isoflurane is a volatile anesthetic used clinically [1]. Many experimental studies have demonstrated that isoflurane provides protective effects in many organs after ischemic reperfusion injury [8-10]. Isoflurane pretreatment reduced neurological deficits, brain edema, and infarction volume in ischemic animal model [11]. Furthermore, isoflurane preconditioning attenuates inflammation and injury by decreasing pro-apoptotic factors and increasing anti-apoptotic factors [12]. Although many experimental evidences have proved that isoflurane exerts the prevention of ischemic brain injury, cerebral vascular protection and suppression of brain inflammation [13], it is unknown whether isoflurane treatment may have protective effects in tPA-induced brain injury in cerebral ischemia. In this study, we investigated the effect of isoflurane pretreatment on tPA-induced cerebral endothelial cell damage and activation of MMPs from endothelial cells after oxygen and glucose deprivation/reperfusion (OGD/R). We revealed that isoflurane pretreatment could affect neuronal cell survival against tPA-induced MMPs activation after OGD/R.
Materials and Methods
The bEnd.3 Cell culture
Murine brain endothelial cell line, bEnd.3, was purchased from American Type Culture Collection (Manassas, VA, USA). The bEnd.3 cells were cultured with Dulbecco's Modified Eagle's Medium high glucose (DMEM, Hyclone™, GE Healthcare Life Sciences, Logan, UT, USA), adding 10% fetal bovine serum (FBS, GE Healthcare Life Sciences) and 1% penicillin-streptomycin solution (Thermo Scientific). The cells were incubated at 37°C in a humid atmosphere in the presence of 5% CO2.
Oxygen and glucose deprivation (OGD)
Mouse Endothelial cells were seeded in 100 mm cell culture dish and incubated in an incubator which conditioned with an atmosphere of 5% CO2 and 95% N2. Before oxygen and glucose deprivation, the culture medium was discarded, washed with phosphate buffer saline (PBS). Endothelial cells were placed in an anaerobic chamber (tension = 0.1%) (Forma Scientific, Inc., Marietta, GA, USA) and PBS was washed away and changed to deoxygenated glucose-free balanced salt solution (BSS). Cells were exposed to OGD conditionduring6 hours. After OGD, BSS solution was discarded and changed into DMEM cultured media and cells were transferred to a CO2 incubator for 3 hours. After reperfusion, cells and supernatants were collected and stored at - 80°C. The bEnd.3 cells were treated with the concentration of tPA (20µg/mL, Actilyse®; BoehringerIngelheim, Ingelheim am Rhein, Germany) during one hour after OGD. Isoflurane (Hana Pharm Co., Ltd, Hwa-Sung, South Korea) (10mM) stock solution was made 130ml isoflurane into 103ml of DMEM high glucose serum free media as following previous study [14]. The concentration of isoflurane stock solution was confirmed by gas chromatography. One hour before OGD, Endothelial cells were treated isoflurane (0.1mM). The receptor associated protein (RAP) (200 nM), an antagonist for ligand interaction with LRP, and MG-132 (10 µM), a proteasome inhibitor and NF-κB inhibitor, were treated for 1 hour before OGD/R injury (Figure 1A).
Experimental procedure and measurement of cell viability. (A) It presents the diagram of experimental procedure. (B) Endothelial cells, after OGD/R injury, showed a reduction in cell viability (74.4%), compared with that of the control (100%); moreover, tPA treatment resulted in lower cell viability (51.8%) compared to that of only OGD/R-injured cells. After testing different concentrations of isoflurane, 0.1mM isoflurane treatment was shown to restore decreased cell viability up to 72.5%. (C) Under normoxic conditions, there were no significant differences in cell viability except for a modest reduction at 1mM isoflurane (81.4%).*p< 0.05,**p< 0.01, ***p< 0.001, the one-way ANOVA (means ± SEM, n = 5).
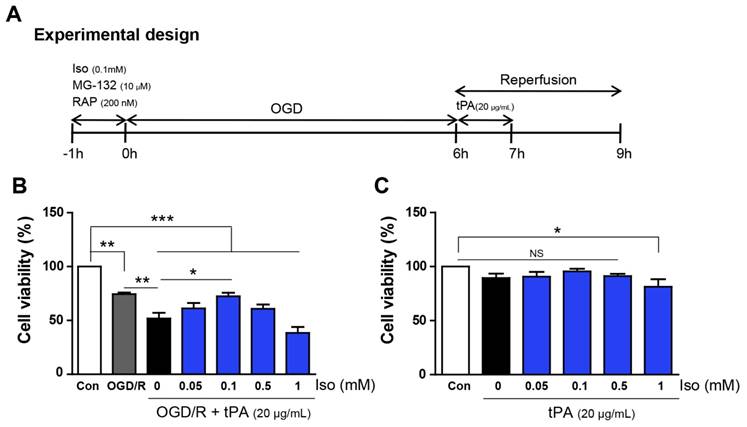
Cell viability assay
Cell viability was assessed by a WST assay (EZ-CYTOX, DAEILLAB SERVICE, Seoul, South Korea). The bEnd3 endothelial cells were seeded in 96-well plates at a density 2×105cells in 100 µl of DMEM culture medium per well. After pretreatment isoflurane and OGD/R, WST was added 10 µl per well. After 1 hour 30 min incubation at 37°C in the CO2 incubator, it was analyzed using VERSASA max microplate reader (Moelcular Devices, Sunnyvale, CA, USA) at a wavelength 450nm.
The Neuro-2A Cell culture
Murine neuronal cell line, Neuro-2A, was cultured in DMEM high glucose cultured media, supplemented with 10% fetal bovine serum (GE Healthcare Life Sciences) and 1% penicillin-streptomycin solution (Thermo Scientific). Neuro-2A cells were incubated at 37°C in a humid atmosphere in the presence of 5% CO2. To examine effects of endothelial cell-released factor on neuronal cells, neuronal cells were incubated with endothelial cell conditioned medium (EC-CM) which collected from endothelial cell culture after OGD/R.
Western blot analysis
The endothelial cells were lysate with a T-PER® Tissue Protein Extraction Reagent (Thermo Scientific) adding HaltTM protease & Phosphatase inhibitor cocktail (1:100, Thermo Scientific). Cells were centrifuged at 13000rpm at 4°C for 20 min. Pierce® BCA protein assay kit was used for detecting concentration of protein as followed manufacture's protocol (Thermo Scientific). Samples were mixed with 5× sample buffer (BIOSESANG, INC. Seongnam, South Korea) and boiled at 95°C for 5 min. Proteins were transferred to polyvinylidene difluoride membranes (PVDF, Millipore, Bedford, MA, USA). After blocking with 5% bovine serum albumin (BSA), membranes were incubated with the anti-LRP (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-NF-κB p65 (1:500, Santa Cruz Biotechnology), anti-Cox-2 (1:500, Santa Cruz Biotechnology), anti-Bax (1:1000, Merck Millipore, Bedford, MA, USA), anti-Bcl-2 (1:1000, Abcam, Cambridge, UK) primary antibodies respectively. Horseradish peroxidase-conjugated anti-goat, anti-mouse, or anti-rabbit IgG reagents (1:5000) were used as secondary antibodies. The β-actin (1:5000, Santa Cruz Biotechnology) was used as an internal control. The bands were visualized with enhanced chemiluminescence reagents (ECL Plus; Amersham Biosciences, Piscataway, NJ, USA) under the LAS 4000 program (GE Healthcare, Pittsburgh, PA, USA).
Immunocytochemistry
Brain endothelial cells were fixed with 4% paraformaldehyde and washed with PBS for three times. Samples were blocked with 5% BSA for 1 hour at room temperature. Immunolabelling was performed with anti-NF-κB p65 (1:100, Santa Cruz Biotechnology) overnight at 4°C. After washing with PBS for 3 times, cells were incubated with Rhodamine-conjugated anti-rabbit IgG (1:1000, Jackson ImmunoResearch) at room temperature for 1 hour. After washing with PBS for 3 times, samples were mounted with Vectashield with 4', 6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Inc., Burlingame, CA, USA). Endothelial cells were observed under LSM700 confocal microscope (Carl Zeiss, Thornwood, NY, USA).
MMP-2 and -9 activity assay
Active form of MMP-2 and MMP-9 in endothelial cell media was measured by using Biotrak Activity Assay system from Amersham Biosciences (Piscataway, NJ, USA). All procedures were followed by the manufacturer's protocol. Each sample was added into wells and incubated overnight for immunoreactivity at 4°C. For detection active- form of MMP-2 or MMP-9, detection enzyme was added and incubated for 1 hour at 37°C according to the protocol. The parameter was estimated in each well using an automatic ELISA reader at 450 nm.
Statistical analysis
Data are expressed as the means± standard error of the mean (SEMs). Statistical comparisons among groups were assessed with a one way ANOVA followed by a Turkey post hoc test using Prism version 6.0. Statistical significance between groups was considered to be present at *p< 0.05, **p< 0.01, *** p< 0.001
Results
Preconditioning with 0.1mM isoflurane increased cell viability after tPA-induced injury during OGD/R
To examine the cell viability of endothelial cells after tPA treatment under conditions of OGD/R, cell viability assays were performed under different concentration of isoflurane (Figure 1B, n=5). Cell viability decreased after OGD/R injury; however, tPA treatment resulted in significantly lower cell viability compared to that of the experimental control (OGD/R group). Isoflurane pre-treatment increased the viability of tPA-treated endothelial cells after OGD/R injury. After preconditioning with 0.1 mM isoflurane, the cell viability was higher than that in the tPA-treated group after OGD/R injury. Among four concentrations of isoflurane, preconditioning with 0.1 mM isoflurane resulted in the highest cell viability; therefore, we conducted subsequent experiments using 0.1mM isoflurane. In normoxic conditions, cell viabilities after preconditioning with 0.05-0.5 mM isoflurane were not changed, however, that with 1 mM isoflurane pretreatment decreased compared to that of the control (Figure 1C, n=5).
MMP-2 and MMP-9 activities were reduced after isoflurane pretreatment
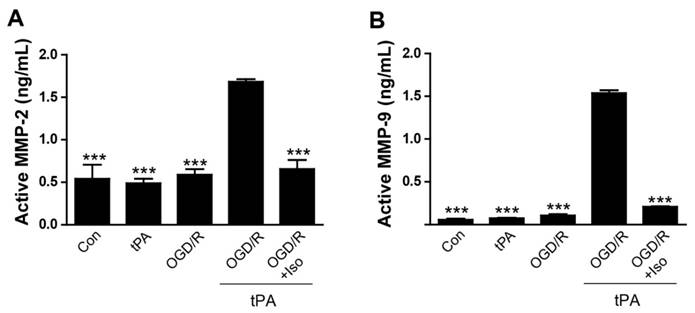
To demonstrate that treatment of tPA induces MMP-2 and MMP-9 activation after OGD/R injury, we measured MMP-2 and MMP-9 activities in endothelial cell conditioned medium (EC-CM) (Figure 2). Our results demonstrated that a significant increase of MMP-2 activity in OGD/R + tPA-treated EC-CM was observed, however, isoflurane pretreatment efficiently inhibited MMP-2 activation in EC-CM despite tPA and OGD/R injuries (Figure 2A, n=3, *** p< 0.001 vs OGD/R + tPA group). MMP-9 activation was also measured by the same method. Activated MMP-9 level was significantly enhanced after tPA treatment and OGD/R insult, whereas MMP-9 activation was attenuated by isoflurane pretreatment in EC-CM (Figure 2B, n=6, *** p< 0.001 vs OGD/R + tPA group). Our results revealed that isoflurane inhibited the MMP-2 and MMP-9 activities after tPA treatment under conditions of OGD/R. These results indicated that isoflurane has an important role in the suppression of MMP activation after tPA-induced injury under OGD/R.
Reduction of tPA-induced MMP-2 and MMP-9 activations by isoflurane pretreatment (A) The level of active MMP-2 was evaluated using an activity assay kit. Isoflurane pretreatment blocked MMP-2 activation by tPA during OGD/R conditions. (B) Increased MMP-9 activation in the tPA-treated group after OGD/R, was significantly reduced in the isoflurane-pretreated group. ***p< 0.001 vs. OGD/R + tPA group, the one-way ANOVA (means ± SEM, active MMP-2 (ng/mL) (n=3),active MMP-9 (ng/mL) (n=6))
LRP/NF-κB/Cox-2 signaling pathway was inhibited by isoflurane pretreatment
To examine the protective mechanism of isoflurane pretreatment against tPA-induced injury in endothelial cells during OGD/R, we performed western blot analysis and immunofluorescent staining (Figure 3). It is known that ischemic stress increases LRP signaling [4], therefore, we first assessed LRP protein levels by western blotting. Based on our results, LRP expression was slightly increased after OGD/R injury, but not by tPA itself (Figure 3B, n=3). Preconditioning with isoflurane considerably reduced LRP levels, compared to those of the OGD/R group and OGD/R + tPA group (Figure 3B).
Inhibition of tPA-induced activation of LRP/NF-κB/Cox-2 signaling pathways in endothelial cells after isoflurane pretreatment.(A) To measure protein expression of LRP, NF-κB p65, and Cox-2, we performed western blot analysis; representative data are shown. (B) LRP protein expression in the OGD/R and OGD/R + tPA-treated group was strongly increased compared to that of the control. LRP levels were attenuated after pretreatment with isoflurane after tPA and OGD/R injury. (C) The relative protein expression of NF-κB p65 in the OGD/R and OGD/R + tPA-treated group showed a significant increase; however, isoflurane pretreatment efficiently reduced NF-κB p65 protein levels. (D) NF-κB p65 expression in the nucleus of endothelial cells were highly detected after tPA and OGD/R insults, however, isoflurane pretreatment efficiently reduced the expression of NF-κB p65. (E) Cox-2 protein levels in the isoflurane-pretreated group were slightly decreased compared with those of the OGD/R + tPA-treated group. *p< 0.05, **p< 0.01, ***p< 0.001, the one-way ANOVA (means ± SEM, Relative optical density (OD) of LRP, NF-κB p65, and Cox-2 (n=3))
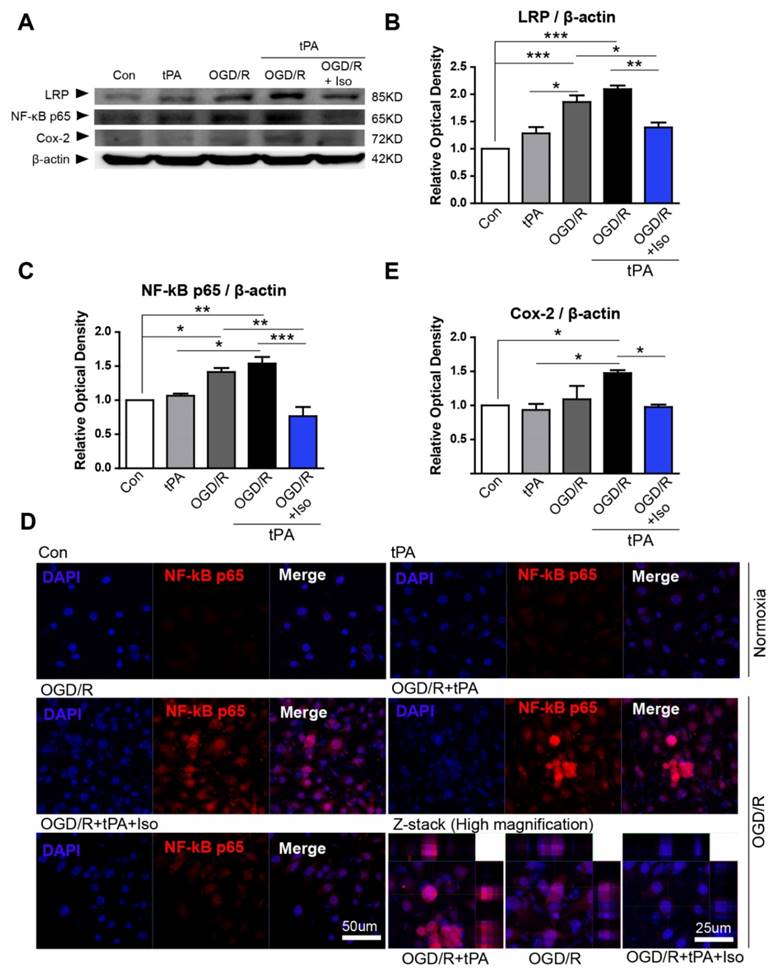
To study the role of isoflurane in OGD/R + tPA-induced nuclear factor-kappa B (NF-κB) signaling, we measured NF-κB p65, a marker of NF-κB activation. We observed that tPA alone did not increase NF-κB p65 protein levels; however, combining tPA treatment with OGD/R injury upregulated NF-κB p65. Isoflurane pretreatment significantly abolished tPA-activated NF-κBp65 with conditions of OGD/R (Figure 3A, 3C, n=3). In addition, we assessed NF-κB p65 expression level in the nucleus by immunofluorescent staining. In the control group, NF-κB p65 expressions were not easily detected in the nucleus of endothelial cells, however, OGD/Rand OGD/R and tPA treatment increased NF-κB p65 level in the nucleus. These apparent expression of NF-κB p65 was abolished after preconditioning of isoflurane (Figure 3D).
We next investigated the level of cyclooxygenase-2 (Cox-2), which is known to be controlled by NF-κB and associated with mediating the inflammatory processes [15]. We found that Cox-2 protein level was upregulated after tPA treatment in endothelial cells with OGD/R; however, isoflurane pretreatment suppressed expression of Cox-2 (Figure 3A, 3E, n=3).
These findings suggested that OGD/R and tPA-induced LRP/NF-κB/Cox-2 pathway was inhibited by isoflurane pretreatment.
The NF-κB/Cox-2 signaling pathway was downregulated by inhibitors, RAP or MG-132
To demonstrate if inhibition of LRP and NF-κB signaling pathway using RAP or MG-132 could exert the protective effects as same as isoflurane treatment on tPA-induced cell injury during OGD/R, RAP and MG-132 were applied 1 hour before OGD. We used RAP to investigate LRP role in OGD/R + tPA treatment. Our results showed that upregulated NF-κB p65 by OGD/R + tPA was efficiently downregulated by RAP (Figure 4A, n=3). In addition, a semi-quantitative graph indicated that co-treatment with isoflurane and RAP also inhibited the expression of NF-κB p65 induced by tPA and OGD/R, but did not have any synergistic effects (Figure 4A, 4B, n=3).
Isoflurane, RAP, or MG-132 treatment inhibited LRP/NF-κB/Cox-2 pathway and MMP-2 and MMP-9 activities. (A and B) Isoflurane pretreatment significantly reduced NF-κB expression in the tPA and OGD/R condition; RAP and isoflurane + RAP treatment also reduced NF-κB p65 in the tPA and OGD/R condition. (C, D, and E) Isoflurane pretreatment significantly reduced NF-κB and Cox-2 expression in the tPA and OGD/R condition; MG-132 and isoflurane + MG-132 treatment reduced NF-κB p65 and Cox-2 in the tPA and OGD/R condition. To measure the activity of MMP-2 and MMP-9 after treatment with RAP or MG-132, we performed an activity assay. (F and G) Isoflurane pretreatment significantly reduced the activities of MMP-2 and MMP-9 under tPA and OGD/R condition. RAP and isoflurane + RAP treatment reduced the activities of MMP-2 and MMP-9 in the tPA and OGD/R condition. (H and I) MG-132and isoflurane + MG-132 treatment reduced the activities of MMP-2 and MMP-9 in the tPA and OGD/R condition. *p< 0.05, **p< 0.01, ***p< 0.001, the one-way ANOVA (means ± SEM, OD of NF-κB, and Cox-2 (n=3), Active MMP-2 (ng/mL) (n=3), Active MMP-9 (ng/mL) (n=3-4))
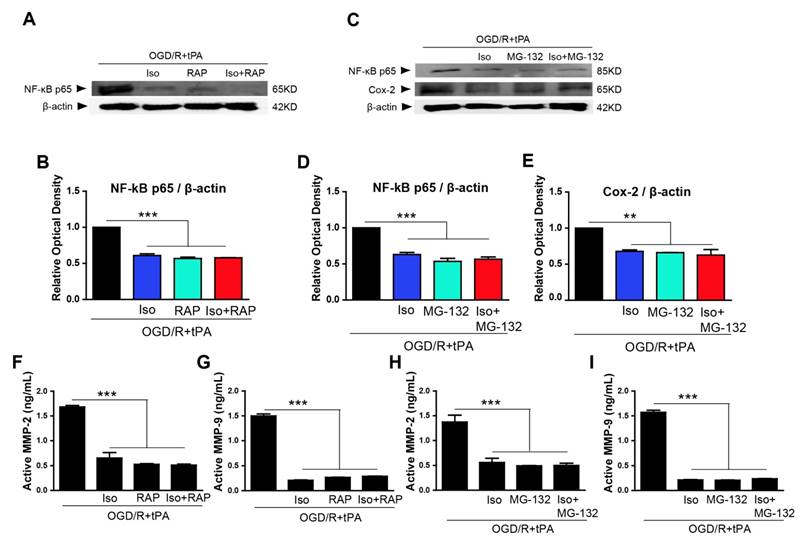
To elucidate the involvement of NF-κB p65, we treated MG-132, which is a proteasome inhibitor that blocks NF-κB translocation to the nucleus [4, 16], and examined NF-κB p65 and Cox-2 levels by western blotting. Based on our results, NF-κB p65 expression was significantly blocked after treated with MG-132 and co-treatment with isoflurane and MG-132 (Figure 4C, 4D, n=3). Furthermore, MG-132 treatment and combination of isoflurane and MG-132 significantly abolished Cox-2 expression in tPA-treated endothelial cells after OGD/R without any synergistic effect (Figure 4C, 4E, n=3).
Based on these findings, OGD/R + tPA-induced NF-κB/Cox-2 signals were hampered by isoflurane, as well as by the inhibition of LRP or NF-κB, however, isoflurane did not have any synergistic effect with the inhibition of LRP or NF-κB.
tPA-induced MMP-2 and MMP-9 activities were reduced by RAP or MG-132
The activities of MMP-2 and MMP-9 also were measured after treatment with RAP or isoflurane/RAP co-treatment. We observed that MMP-2 and MMP-9 activities were efficiently decreased when RAP or isoflurane + RAP was treated in endothelial cells (Figure 4F, n=3 and 4G, n=3-4). Next, we measured active MMP-2 and MMP-9 levels after treatment with MG-132 or isoflurane + MG-132 co-treatment, and showed that these enzymes were considerably inhibited, compared with activities of the tPA and OGD/R-treated group (Figure 4H, n=3 and 4I, n=3-4). These results suggested that OGD/R + tPA-induced MMP-2 and MMP-9 activities were inhibited by isoflurane, as well as by the inhibition of LRP or NF-κB in endothelial cells after OGD/R. However, isoflurane did not have any synergistic effect with the inhibition of LRP or NF-κB.
Pro-apoptotic Bax was suppressed after incubation with isoflurane-pretreated endothelial cell conditioned medium
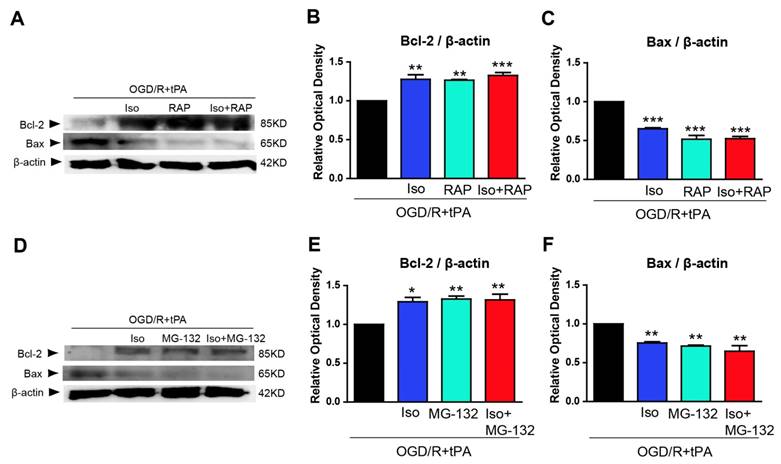
To examine whether pretreated isoflurane in endothelial cells could affect neuronal cell survival, we conducted western blot analysis of anti-apoptosis marker and pro-apoptosis marker in neuronal cells after incubation with EC-CM. We probed for Bcl-2, an anti-apoptosis marker, and Bax, a pro-apoptosis marker, in neurons after incubation with EC-CM for 24 hours (Figure 5). Neuro2A cells, incubated with tPA and OGD/R-treated EC-CM, showed decreased Bcl-2 and increased Bax protein expression, whereas isoflurane pretreated OGD/R + tPA-EC-CM abolished these changes in neuronal cells. Neuro2A cells incubated with RAP-treated EC-CM, or isoflurane + RAP co-treated EC-CM showed significant upregulation of Bcl-2, however, Bax protein expression was reduced (Figure 5B and 5C, n=3,*p< 0.05, **p< 0.01, *** p< 0.001 vs OGD/R + tPA group). In addition, Neuro2A cells, incubated with MG-132-treated EC-CM or isoflurane + MG-132 co-treated EC-CM, showed an obvious increase in Bcl-2 and suppression of Bax, similar to the pattern observed with RAP treatment (Figure 5E and 5F, n=3,*p< 0.05, **p< 0.01, *** p< 0.001 vs OGD/R + tPA group). These results indicated that neuronal cell death was attenuated by isoflurane preconditioned EC-CM despite tPA and OGD/R injury. Therefore, we suggest that isoflurane could lead to an inhibition in tPA-induced neuronal cell death after OGD/R.
Inhibition of endothelial cell conditioned medium (EC-CM) induced pro-apoptotic molecule expression by isoflurane. To examine the effect of pretreated isoflurane on neuronal cell survival and death under culture with EC-CM, we performed western blot analysis using Bcl-2 and Bax. (B) The Bcl-2 protein was significantly upregulated in neuronal cells incubated with EC-CM of isoflurane, RAP, or isoflurane RAP treated groups in the tPA and OGD/R condition. (C) In contrast to Bcl-2, the protein levels of Bax was significantly downregulated in neuronal cells incubated with EC-CM of isoflurane, RAP or isoflurane + RAP-treated groups in the tPA and OGD/R condition. (E) Bcl-2 protein levels were significantly increased in neuronal cells incubated with EC-CM of isoflurane, MG-132, or isoflurane + MG-132-treated groups in the tPA and OGD/R condition. (F) Bax was significantly decreased in neuronal cells incubated with EC-CM of isoflurane, MG-132 and isoflurane + MG-132-treated groups in the tPA and OGD/R condition. *p< 0.05, **p< 0.01, ***p< 0.001 vs. OGD/R + tPA group, the one-way ANOVA (means ± SEM, OD of Bcl-2 and Bax(n=3))
Discussion
In this study, we investigated the protective effects of isoflurane pretreatment on tPA-induced endothelial cell damage and neuronal cell death, during conditions of OGD/R. Based on our results, endothelial cell viability was significantly decreased after tPA treatment during conditions of OGD/R, compared with only OGD/R injury. Secretion of MMPs was activated by tPA treatment and OGD/R insult. This process is likely to upregulate the LRP/NF-κB/Cox-2 signaling pathway. In addition, EC-CM from endothelial cell culture treated with tPA and OGD/R had negative effects on neuronal cell survival by upregulating pro-apoptotic Bax. However, pretreatment with isoflurane enhanced endothelial cell viability despite tPA and OGD/R injury. Combination of tPA- and OGD/R-induced LRP/NF-κB/Cox-2 signaling pathway was significantly reduced by isoflurane, RAP, or MG-132. Taken together, isoflurane resulted in decreased activation of MMP-2 and MMP-9 in endothelial cell conditioned media, which might lead to decrease neuronal Bax levels even after treatment with tPA and OGD/R. Our findings demonstrated that isoflurane has preventive effects against tPA- and OGD/R-induced cell damage and neuronal apoptotic-molecule through inhibition of MMPs activation.
MMPs are a family of zinc-dependent endopeptidases associated with the pathogenesis of stroke. MMPs degrade extracellular matrix proteins for tissue remodeling; however, MMPs play a pivotal role in the breakdown of blood vessel barriers which leads to hemorrhagic transformation and edema formation [17, 18]. These processes have been associated with cerebral infarction by activation of apoptotic cell death [19]. Specifically, after hypoxia-reoxygenation, both MMP-2 and MMP-9 are vastly produced [7]. Both MMP-2 and MMP-9 are also increased in ischemic brain and in human endothelial cells [7, 20, 21]. Intravenously administrated tPA-induced ICH might be involved in MMP activation. It was reported that tPA plays an important role in MMP-2 and MMP-9 activation [17, 22] and stimulation of MMPs play important roles in the process of excitotoxic anoikis-like cell death for neurodegeneration [8, 17]. Also, MMP-9-induced breakdown of cell-matrix interaction triggers neuronal cell death [8, 17]. As shown by our data, after OGD/R injury, which mimics ischemic stroke, tPA treatment enhanced MMP-2 and MMP-9 activations in endothelial cells. Upregulated active MMP-2 and MMP-9 in EC-CM might initiate upregulating apoptotic-signal in neuronal cells.
During OGD/R, tPA-induced MMP activation in endothelial cell-related intracellular signaling pathways may be connected with low-density lipoprotein (LDL) receptor-related protein (LRP), which consists of a transmembrane and cytoplasmic domain, and interacts with various ligands including tPA [21, 23]. The previous study demonstrated that ischemic stress easily upregulates LRP expression in vitro and in vivo studies and increases sensitivity of endothelial cells to tPA [4]. Blockade of LRP with RAP or anti-LRP and genetic deletion of endogenous tPA leads to a reduction of LRP expression in vascular structures [24]. Interaction of tPA and LRP regulates cerebrovascular tone and permeability of the neurovascular units under non-ischemic or ischemic condition, and synthesizes MMP-9 [17, 24]. LRP downstream signals in endothelial cells can be activated by intravascular tPA, which can promote activation of NF-κB signaling as shown by our results. Several studies also proved that tPA activates NF-κB signaling in endothelial cells and NF-κB regulates MMP-9 activation [4, 7, 17]. An increase in NF-κB activation participates in the induction of an inflammatory response and inducible nitric-oxide synthase (iNOS). tPA also plays essential roles in induction of inflammation and iNOS production after cerebral ischemia [25]. OGD/R + tPA-induced Cox-2 up-regulation was correlated with NF-κB activation and these changes could be reversed by inhibition of NF-κB translocation to the nucleus using MG-132. Therefore, it is speculated that Cox-2 induction might be through NF-κB signaling. The previous study supported that hypoxia-induced Cox-2 up-regulation is mediated by NF-κB p65 in vascular endothelial cells, with the mechanisms likely involving binding of p65 to the NF-κB -3ʹ site in the Cox-2 upstream promoter region [15].
Studies have demonstrated that isoflurane promotes ischemic tolerance, decreases ischemic brain injury, and preserves the BBB. Moreover, isoflurane may transiently hamper the progression and delay the onset of cerebral infarct [26]. In addition, isoflurane pretreatment reduces neurological deficits, brain edema, and cerebral infarct size after ischemia/reperfusion [11]. We determined in the previous study that isoflurane post-treatment has a protective effect on tPA-exaggerated brain injury [1]; however, the protective mechanisms of isoflurane against tPA-induced injury after ischemic insult were not elucidated. Based on our results, we proved that isoflurane preconditioning ameliorates tPA-induced injury after OGD/R. Downregulation of OGD/R + tPA-triggered LRP/NF-κB/Cox-2 signaling and MMP-2 and MMP-9 activation by isoflurane pretreatment might be involved in this mechanism, which leads to neuronal anti-apoptotic molecule, Bcl-2 upregulation and pro-apoptotic molecule, Bax downregulation. Although we showed changes of protein expression in LRP, Cox-2, and NF-κB by isoflurane pretreatment, it is not clear that preventive effects of isoflurane is from only reduction of these signals or blunted endothelial sensitivity against tPA. However, these results implicate isoflurane preconditioning as having a critical role in vascular protection with respect to tPA-exaggerated injury after OGD/R. These preventive effects of isoflurane preconditioning against tPA-induced injury might contribute to the development of a new treatment strategy for ischemic stroke.
Acknowledgements
This research was supported by special Research Grant funded by a Korean Society of Neuroscience in Anesthesiology and Critical Care (KSNACC-2015).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kim EJ, Kim SY, Lee JH, Kim JM, Kim JS, Byun JI. et al. Effect of isoflurane post-treatment on tPA-exaggerated brain injury in a rat ischemic stroke model. Korean journal of anesthesiology. 2015;68:281-6
2. Clark WM, Wissman S, Albers GW, Jhamandas JH, Madden KP, Hamilton S. Recombinant tissue-type plasminogen activator (Alteplase) for ischemic stroke 3 to 5 hours after symptom onset. The ATLANTIS Study: a randomized controlled trial. Alteplase Thrombolysis for Acute Noninterventional Therapy in Ischemic Stroke. JAMA. 1999;282:2019-26
3. Suzuki Y, Nagai N, Yamakawa K, Muranaka Y, Hokamura K, Umemura K. Recombinant tissue-type plasminogen activator transiently enhances blood-brain barrier permeability during cerebral ischemia through vascular endothelial growth factor-mediated endothelial endocytosis in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:2021-31
4. Suzuki Y, Nagai N, Yamakawa K, Kawakami J, Lijnen HR, Umemura K. Tissue-type plasminogen activator (t-PA) induces stromelysin-1 (MMP-3) in endothelial cells through activation of lipoprotein receptor-related protein. Blood. 2009;114:3352-8
5. Kastrup A, Groschel K, Ringer TM, Redecker C, Cordesmeyer R, Witte OW. et al. Early disruption of the blood-brain barrier after thrombolytic therapy predicts hemorrhage in patients with acute stroke. Stroke; a journal of cerebral circulation. 2008;39:2385-7
6. Kassner A, Roberts TP, Moran B, Silver FL, Mikulis DJ. Recombinant tissue plasminogen activator increases blood-brain barrier disruption in acute ischemic stroke: an MR imaging permeability study. AJNR American journal of neuroradiology. 2009;30:1864-9
7. Lee SR, Lo EH. Induction of caspase-mediated cell death by matrix metalloproteinases in cerebral endothelial cells after hypoxia-reoxygenation. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24:720-7
8. Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A. et al. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186-90
9. Liang Y, Li Z, Mo N, Li M, Zhuang Z, Wang J. et al. Isoflurane preconditioning ameliorates renal ischemia-reperfusion injury through antiinflammatory and antiapoptotic actions in rats. Biological & pharmaceutical bulletin. 2014;37:1599-605
10. Agnic I, Filipovic N, Vukojevic K, Saraga-Babic M, Vrdoljak M, Grkovic I. Effects of isoflurane postconditioning on chronic phase of ischemia-reperfusion heart injury in rats. Cardiovascular pathology: the official journal of the Society for Cardiovascular Pathology. 2015;24:94-101
11. Xiao Z, Ren P, Chao Y, Wang Q, Kuai J, Lv M. et al. Protective role of isoflurane pretreatment in rats with focal cerebral ischemia and the underlying molecular mechanism. Molecular medicine reports. 2015;12:675-83
12. Bedirli N, Bagriacik EU, Emmez H, Yilmaz G, Unal Y, Ozkose Z. Sevoflurane and isoflurane preconditioning provides neuroprotection by inhibition of apoptosis-related mRNA expression in a rat model of focal cerebral ischemia. Journal of neurosurgical anesthesiology. 2012;24:336-44
13. Altay O, Suzuki H, Hasegawa Y, Ostrowski RP, Tang J, Zhang JH. Isoflurane on brain inflammation. Neurobiol Dis. 2014;62:365-71
14. Kudo M, Aono M, Lee Y, Massey G, Pearlstein RD, Warner DS. Absence of direct antioxidant effects from volatile anesthetics in primary mixed neuronal-glial cultures. Anesthesiology. 2001;94:303-212
15. Schmedtje JF Jr, Ji YS, Liu WL, DuBois RN, Runge MS. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. The Journal of biological chemistry. 1997;272:601-8
16. Xie H, Ray PE, Short BL. NF-kappaB activation plays a role in superoxide-mediated cerebral endothelial dysfunction after hypoxia/reoxygenation. Stroke; a journal of cerebral circulation. 2005;36:1047-52
17. Wang X, Lee SR, Arai K, Lee SR, Tsuji K, Rebeck GW. et al. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nature medicine. 2003;9:1313-7
18. Copin JC, Bengualid DJ, Da Silva RF, Kargiotis O, Schaller K, Gasche Y. Recombinant tissue plasminogen activator induces blood-brain barrier breakdown by a matrix metalloproteinase-9-independent pathway after transient focal cerebral ischemia in mouse. The European journal of neuroscience. 2011;34:1085-92
19. Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke; a journal of cerebral circulation. 2003;34:2165-70
20. Lo EH, Wang X, Cuzner ML. Extracellular proteolysis in brain injury and inflammation: role for plasminogen activators and matrix metalloproteinases. J Neurosci Res. 2002;69:1-9
21. Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279-91
22. del Zoppo GJ, von Kummer R, Hamann GF. Ischaemic damage of brain microvessels: inherent risks for thrombolytic treatment in stroke. J Neurol Neurosurg Psychiatry. 1998;65:1-9
23. An J, Zhang C, Polavarapu R, Zhang X, Zhang X, Yepes M. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein induce Akt phosphorylation in the ischemic brain. Blood. 2008;112:2787-94
24. Polavarapu R, Gongora MC, Yi H, Ranganthan S, Lawrence DA, Strickland D. et al. Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood. 2007;109:3270-8
25. Zhang X, Polavarapu R, She H, Mao Z, Yepes M. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein mediate cerebral ischemia-induced nuclear factor-kappaB pathway activation. Am J Pathol. 2007;171:1281-90
26. Taheri S, Shunmugavel A, Clark D, Shi H. Isoflurane reduces the ischemia reperfusion injury surge: a longitudinal study with MRI. Brain research. 2014;1586:173-83
Author contact
![]() Corresponding author: Dr. Bon-Nyeo Koo, Department of Anesthesiology and Pain Medicine, Yonsei University College of Medicine 50, Yonsei-ro, Seodaemun-gu, Seoul120-752, Republic of Korea, E-mail: koobnac
Corresponding author: Dr. Bon-Nyeo Koo, Department of Anesthesiology and Pain Medicine, Yonsei University College of Medicine 50, Yonsei-ro, Seodaemun-gu, Seoul120-752, Republic of Korea, E-mail: koobnac