Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(3):246-256. doi:10.7150/ijms.17785 This issue Cite
Research Paper
Functional analysis of a nonsyndromic hearing loss-associated mutation in the transmembrane II domain of the GJC3 gene
Swee-Hee Wong1, 3, Wen-Hung Wang4, Pin-Hua Chen1, Shuan-Yow Li1,2 ![]() , Jiann-Jou Yang1,2
, Jiann-Jou Yang1,2 ![]()
1. Department of BioMedical Sciences, Chung Shan Medical University, Taichung, Taiwan;
2. Department of Medical Research, Chung Shan Medical University Hospital, Taichung, Taiwan;
3. Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan;
4. Department of Otolaryngology, Cathay General Hospital, Taipei, Taiwan.
Received 2016-10-1; Accepted 2016-12-12; Published 2017-2-23
Abstract
In a previous study, we identified a novel missense mutation, p.W77S, in the GJC3 gene encoding connexin30.2/connexin31.3 (CX30.2/CX31.3) from patients with hearing loss. The functional alteration of CX30.2/CX31.3 caused by the p.W77S mutant of GJC3 gene, however, remains unclear. In the current study, our result indicated that the p.W77 is localized at the second membrane-spanning segments (TM2) and near border of the E1 domain of the CX30.2/CX31.3 protein and highly conserved (Conseq score = 8~9) in all species. The p.W77S missense mutation proteins in the intracellular distribution are different CX30.2/CX31.3WT and an accumulation of the mutant protein in the endoplasmic reticulum (ER) of the HeLa cell. Furthermore, co-expression of WT and p.W77S mutant chimerae proteins showed that the heteromeric connexon accumulated in the cytoplasm, thereby impairing the WT proteins' expression in the cell membranes. In addition, we found that CX30.2/CX31.3W77S missense mutant proteins were degraded by lysosomes and proteosomes in the transfected HeLa cell. Based on these findings, we suggest that p.W77S mutant has a dominant negative effect on the formation and function of the gap junction. These results give a novel molecular elucidation for the mutation of GJC3 in the development of hearing loss.
Keywords: CX30.2/CX31.3, GJC3, mutation, hearing loss.
Introduction
The mammalin inner ear comprise the cochlea, which is the hearing organ. The functions of the organ are dependent on tightly controlled ionic environments, in particular for K+ ions, for hearing transduduction [1]. Gap junction system is highly probable pathway for cochlear K+ ions recirculation in the cochlea [2]. CXs genes code for a large and highly homologous family of proteins that form intercellular gap junction chanels. More than 20 CXs have been described in the mammalian. There are twenty-one CXs genes within the human genome. The topological model of CX protein shows that the polypeptide comprise a short cytoplasmic amino-terminal domain (NT), four transmembrane domains (TM1 to TM4) linked by one cytoplasmic loop (CL) and two extracellular loops (E1 and E2), and a most variable carboxyl-terminal cytoplasmic domain (CT) [3].
Mutations in the CXs have been identified as associated with a variety of human inherited disease, such as deafness, epidermal disease, neuropathies, oculoden todigital dysplsia and cataracts. The inheritance of this disease more likely to be autosomal dominant, autosomal recessive, or X-linked [4]. Disease-causing mutations can potentially take place anywhere in the CXs. These mutations may cause disease through a variety of mechanisms, most of which alter intercellular communication by affecting various processes of the CXs life cycle or channel function. The plurality of identified CXs mutations are located within the coding region of protein. These different mutations generate abnormalities at diverse steps in the CX life cycle, including synthesis, assembly, channel function, and degradation [5].
Up to now, six CXs protein (CX26, CX30, CX31, CX30.3, CX30.2/CX31.3 and CX43) are reported expression in the gap junction-rich regions of the cochlear duct and association with human genetic hearing lose [6-12]. The human GJC3 gene, coding for CX30.2/CX31.3, is located on chromosome 7q22.1 and the coding region is localized on both exon 1 and exon 2 and is interrupted by an intron. The CX30.2/CX31.3 contains 279 amino acid residues and has a molecular weight of 31.29 kDa. Human CX30.2/CX31.3, orthologs of the mouse Cx29, was first identified by database analysis in 2002 and has been shown to be highly expressed in the cochlea using cDNA macroarray hybridization [13-15]. Furthermore, previous animal studies also indicate that the Cx29 protein is expressed in the cochlear tissue of mice and rats [16-17]. Previously, we have been identified four heterozygous missense mutations [c.807A>T (E269D), c.43C>G (R15G), c.68T>A (p.L23H) and c.230C>G (W77S)] of the GJC3 gene in Taiwanese patients with nonsyndromic deafness [10-12]. To understand the play role of GJC3 mutation in nonsyndromic hearing loss, it is necessary to investigate the functional alteration of mutant Cx30.2/CX31.3 in intercellular communication. Previously, we have found that p.E269D mutation in the GJC3 gene has a dominant negative effect on the formation and function of the gap junction [18]. In addition, we found that p.R15G and p.L23H mutants do not decrease the trafficking of CX proteins, but the mutations in GJC3 genes result in a loss of function of the CX30.2/CX31.3 protein [19]. However, the functional alternation of CX30.2/CX31.3 caused by the p.W77S mutant remains unclear. This study, therefore, investigates the affecting of the p.W77S mutations on the functional properties and subcellular localization of the mutant CX30.2/CX31.3 protein in tet-on HeLa cells.
Materials and Methods
Molecular cloning and construction of the plasmids expressing wild-type or mutants CX30.2/CX31.3
The wild-type CX30.2/CX31.3 expressing plasmids was constructed as previously describe [18]. Mutant GJC3 genes were generated by performing oligonucleotide-directed mutagenesis using the Stratagene Quickchange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). The following oligonucleotide primers (mutated nucleotide is underlined) were used to prepare the mutant GJC gene: CX30.2/31.3 W77S sense 5'-CCgCTgCgTTTCTCggTCTTCCAggTCATC-3' and CX30.2/31.3 W77S antisense 5'-gATgACCTggAAgACCgAgAAACgCAgC gg-3'. The cDNA sequences of the autofluorescent reporter proteins EGFP (pEGFPN1 vector; Clontech, Palo Alto, CA, USA) were fused in-frame to the C terminus of wild type and mutants for fusion protein generation. The coding region of CX30.2/31.3WT and that of mutant CX30.2/31.3W77S were amplified from plasmids containing the CX30.2/31.3 cDNA (CX30.2/31.3wt-EGFP or CX30.2/31.3W77S-DsRed) using two pair primers containing recognition sequences 5'- SalI and 3'- NotI or 5'-NheI and 3'-EcoRV, respectively, and Platinum Pfx DNA polymerase (Invitrogen, Carisbad, CA). Purified products were subcloned into the corresponding site of the bi-directional expression vector pBI (Clontech, Palo Alto, CA). The dideoxy DNA sequencing method, using a DNA Sequencing kit (Applied Biosystems, Foster City, CA, USA) with an ABI PRISM 3730 automated sequencer, were used to confirm the DNA sequence of all constructs.
Transfection and expression of CX30.2/31.3WT, CX30.2/31.3W77S, and CX30.2/31.3WT/ CX30.2/31.3W77S chimerae protein in tet-on HeLa cell line
The tet-on HeLa cell line deficient in the GJIC gene was purchased from BD Biosciences Clontech (Palo Alto, CA, USA) and maintained in Dulbecco's modified Eagle's medium, supplemented with 10% FBS (Gibco BRL, Gaithersburg, USA), 100 μg/ml G418, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a moist atmosphere containing 5% CO2. Transfection was carried out using LipofectAMINE reagent (Invitrogen, Carlsbad, USA) according to the manufacturer's instructions. A ratio of 1 μg DNA vs. 2 μl LipofectAMINE 2000 was used for the tet-on HeLa cells. Cells were harvested at 24 h post-transfection and grown on a coverslip for 24 h at 37℃ in a humidified 5% CO2 incubator. Then, tet-on HeLa cells were treated with 1 μg/ml doxycyclin (Dox) (Sigma-Aldrich Corporation, St. Louis, Mo) in cell culture medium to induce CX30.2/31.3WT or CX30.2/31.3W77S mutant protein expression. Cells were exposed to Dox for 5 h prior to immunofluorescence staining. Tet-on HeLa cells were fixed with 4% paraformaldehyde in 0.1 M PBS for 20 min, rinsed three times in PBS, stained with DAPI for 5 min, and then washed three times with PBS. Mounted slides were visualized and photographed using a fluorescence microscope (Zeiss Axioplam, Oberkochen, Germany).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from wild type or mutant CX30.2/CX31.3 expression cell lines using the Total RNA Extraction Miniprep System according to the manufacturer's directions (VIOGENE, Sunnyvale). cDNA was synthesized according to the manufacturer's directions in a reaction volume of 20 μl, containing 2-5 μg RNA, random hexamer primer, and 200 units Improm-IITM Reverse Transcriptase (Promega, San Luis Obispo). With primers specific for the coding region of the GJC3 gene (forward 5'- ATGTGCGGCAGGTTCCTGAG -3' and reverse 5'- CATGTTTGGGATCAGCGG-3'), PCR was performed (94 oC 30 sec, 58 oC 35 sec, 72 oC 1 min) for 35 cycles in a volume of 25μl containing 1 mM Tris-HCl (pH 9.0), 5 mM KCl, 150 μM MgCl2, 200 μM dNTP, 1 units proTaq DNA polymerase (Promega, San Luis Obispo), 100 ng of cDNA, and 200 μM forward and reverse primers. A fragment of approximately 700 bp was amplified from cDNA of the GJC3 gene. The PCR products were subjected to electrophoresis in an agarose gel (2 w/v %) stained with ethidium bromide. The signals were detected by an Alpha Image 2200 system (Alpha Image 2200 analysis software).
Immunofluorescence staining of post-transfection HeLa cells
Wild-type or mutant CX30.2/CX31.3 protein expression in tet-on HeLa cells was analyzed by a direct fluorescent protein fusion method involving fusion of EGFP or DsRed to the C-terminal ends of the CX30.2/CX31.3 proteins. Briefly, post-transfection tet-on HeLa cells grown on coverslips were fixed with 4% paraformaldehyde in 0.1 M PBS for 20 min and then rinsed three times in PBS. Then, the coverslips were immersed in 10% normal goat serum and 0.1% Triton X-100 for 15 min. The primary antisera and dilutions were as follows: mouse anti-pan-cadherin antibody at 1:200 (anti-CH19; abcan) for cell membrane, mouse anti-Golgin-97 at 1:200 (Invitrogen, Carisbad, CA) for Golgi apparatus. After incubation with primary antiserum at 4℃ overnight, the cells were rinsed in PBS three times before adding Alexa Fluor 488 and/or Alexa Fluor 594 conjugated secondary antibodies (Invitrogen, Carisbad, CA). Endoplasmic Reticulum (ER) was stained with ER-Tracker® Blue-white DPX Probes at 1:670 dilution (Invitrogen, Carisbad, CA) for 10 min at room temperature. Lysosomes were stained with LysoTracker® Probes (Invitrogen, Carisbad, CA) for 20 min at room temperature. The nuclei of cells were counterstained with DAPI (2 μg/ml) for 5 min and rinsed with PBS. Mounted slides were visualized and photographed using a fluorescence microscope (Zeiss Axioplam, Oberkochen, Germany).
Real-time Quantitative polymerase chain reaction (Q-PCR)
For quantitative real-time RT-PCR (q-PCR) analysis, total RNA was isolated from four positive stable cell lines using the Total RNA Extraction Miniprep System according to the manufacturer's directions (VIOGENE, Sunnyvale). Reverse transcription was performed using Improm-IITM Reverse Transcriptase (Promega, San Luis Obispo) in the presence of oligo-dT18 primer. Quantitative PCR for mRNA was performed using the SYBR Green I Master Mix (Applied Biosystems, Foster city, CA) and detected in a ABI7000 thermocycler (Applied Biosystems, Foster City, CA). Real-time PCR primers for mRNA were designed using PrimerExpress software [20]. The Primers, CX30.2/CX31.3 real-time F-5'CCTGGGATTCCGCCTTGT-3' and CX30.2/CX31.3 real-time R-5'-TGGGTGTGACACACGAATTCA-3' were using for CX30.2/CX31.3 detection. Each measurement was performed in triplicate and the results were normalized by the expression of the GAPDH reference gene.
DNA fragmentation analysis
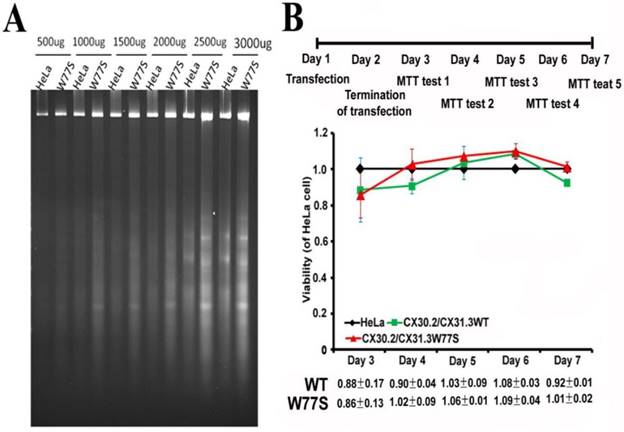
Both expressed CX30.2/CX31.3WT and CX30.2/CX31.3W77S HeLa cells (5x 106 cells) were cultured in DMEM medium for 4 days. After removing the nonadherent dead cells in the cultures by rinsing with PBS, the adherent cells were collected by centrifugation for 5 min (1000 rpm) at room temperature. DNAs were purified as previously describe [19]. Different DNA concentrations from 500μg to 3000μg were resolved in a 1 % (w/v) agarose gel in 1x TAE buffer. The DNA bands were stained with ethidium bromide (0.5 ug/ml) and photographed (Alpha Image 2200 analysis software).
Evaluation of cell viability
Cell viability was determined by MTT assay. Briefly, after MG63 cells were cultured on nanostructured alumina surface for 1, 2 and 4 days. 100 ml of MTT (5 mg/ml) (Wako, Japan) was added to each well and incubated at 37°C for another 4 h. Then, 0.5 ml dimethyl sulfoxide (DMSO) was added to each well to dissolve the formazan crystals. The absorbance of each solutionwas measured at the wavelength of 490 nm with a microplate reader (Bio-Rad 680, Bio-Rad, USA).
Statistical analysis
All data were calculated and presented as mean±standard error (mean±SE) in the present study. By GraphPad Prism Software, the statistical analyses between different groups at different time were performed via an unpaired t-test. Posterior comparisons were then followed by using Turkey's test HSD (honestly significant difference). A P-value of less than 0.05 was considered to be statistically significant. Star code for statistical significance is illustrated as follow: ***P<0.001, **P<0.01 and *P<0.05.
Results
Schematic representation of the domain structure of the CX30.2/CX31.3 protein
The predictable protein structure of CX30.2/CX31.3 was acquired from UniProtKB/Swiss-Prot database entry P17302. We found that the topological model of CX30.2/CX31.3 protein is similarily other members of CX family. The structure of CX30.2/CX31.3 protein was described previously [19] and was displyed in supplemental Fig. 1. Our result indicated that p.W77S missense mutation is localized at the second membrane-spanning segments (TM2) and near border of the E1 domain of the CX30.2/CX31.3 protein (supplemental Fig. 1).
Multiple alignments of amino acid sequence in connexin proteins
Further, we examined and compared amino acid sequences of the CX30.2/CX31.3 domain among CX families in humans using Biology WorkBench Clustal W (1.81) Multiple Sequence Alignments (http://workbench.sdsc.edu/, San Diego Supercomputer Center). According to that comparison, the p.W77S amino acid of the CX30.2/CX31.3 protein was highly conserved among the human CX family members (supplemental Fig. 2A). Additionally, we compared amino acid sequences of CX30.2/CX31.3 in CX families of all species using a basic ConSeq analysis system (http://conseq.tau.ac.il/; 21). The contrast results revealed that p.W77 is also highly conserved (Conseq score = 8~9) in all species (supplemental Fig.2B). These results indicated that p.W77 may be play an important role in the function of CX30.2/CX31.3 protein.
Expression analysis of CX30.2/CX31.3WT and CX30.2/CX31.3W77S in HeLa cell
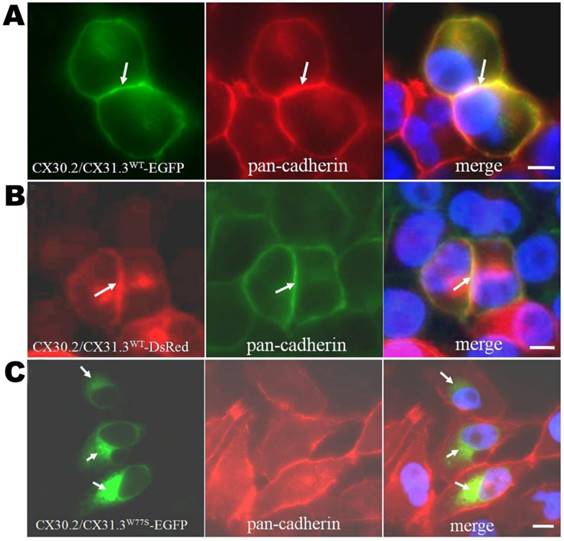
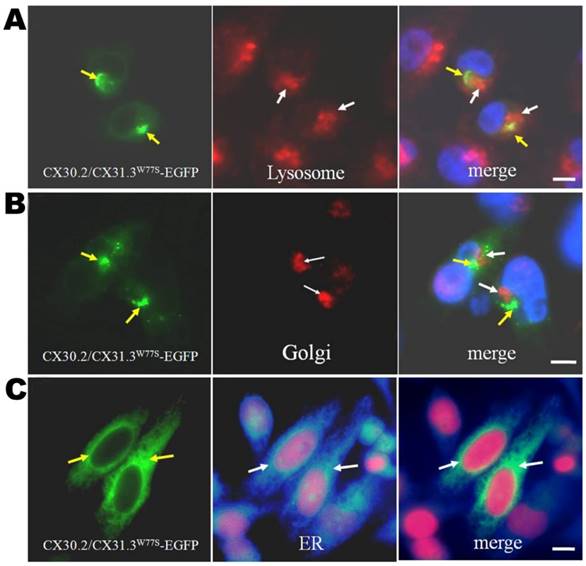
To confirm the contrast results and understand the effects of p.W77S missense mutation, we compared the functional properties and subcellular localization of the CX30.2/CX31.3 wild-type (WT) and mutant (p.W77S) protein in the gap junction-deficient HeLa cells. First, we transfected the cDNA constructs of WT (CX30.2/CX31.3WT-EGFP or CX30.2/CX31.3WT- DsRed) or mutant CX30.2/CX31.3 (CX30.2/CX31.3W77S-EGFP) into tet-on HeLa cells using lipofection. In the CX30.2/CX31.3WT-EGFP and CX30.2/CX31.3WT-DsRed expression cell line, the results indicated that the WT proteins were observed along apposed cell membranes between adjacent cell (Fig. 1A and 1B). Further, this membrane localization was confirmed by colocalization with pan-Cadherin (Fig. 1A and 1B). These results are consistent with our previous studies [18-19]. Contrary to CX30.2/CX31.3WT, as seen in the immunolabeling assay, CX30.2/CX31.3W77S mutant proteins were concentrated in the cytoplasm close to the nucleus (Fig. 1C). Following, we identify which organelles in the cytoplasm the mutant CX30.2/CX31.3 localized in HeLa cells. We analyzed the HeLa cell had been transfected with CX30.2/CX31.3W77S-EGFP cDNA by immunostaining with markers for lysosome, ER, and Golgi apparatus (Fig. 2). The results of the assay indicated that the most CX30.2/CX31.3W77S mutant protein was characteristically found in a reticular pattern co-localized with an ER marker (Fig. 2B). Base on above found, we consider that the p.W77S mutation interferes with normal CX30.2/CX31.3 trafficking.
Expression analysis of CX30.2/CX31.3WT and CX30.2/CX31.3W77S in transiently transfected HeLa cells by immunocytochemistry using pan-cadherin antibody. Fluorescence microscopy of HeLa cells expressing CX30.2/CX31.3WT-EGFP (A) and CX30.2/CX31.3WT- DsRed (B) shows expression of the CX30.2/CX31.3 fusion protein in the plasma membranes. However, CX30.2/CX31.3W77S -EGFP (C) transfected HeLa cells show impaired trafficking of the CX30.2/CX31.3 protein with localization near the nucleus. The cells were counterstained with 4'-6-Diamidino-2-phenylindole, DAPI, (blue) to highlight the nuclei. Scale bars: 10 μm.
Intercellular localization of mutant CX30.2/CX31.3 proteins. Photomicrographs of HeLa cells transfected with CX30.2/CX31.3W77S-EGFP cDNA after immunostaining for markers of the lysosome, Golgi apparatus, and ER (anti-PDI) (red in (A)-(C), respectively). Yellow signal in the image overlays (right column) indicates co-localization of CX30.2/CX31.3W77S-EGFP and the organelle of interest. Mutant CX30.2/CX31.3 shows moderate co-localization with the ER marker. The cells were counterstained with 4'-6-Diamidino-2-phenylindole, DAPI, (blue) to highlight the nuclei. Scale bars: 10 μm.
Expression of CX30.2/CX31.3WT and CX30.2/CX31.3W77S chimerae protein in tet-on HeLa cell
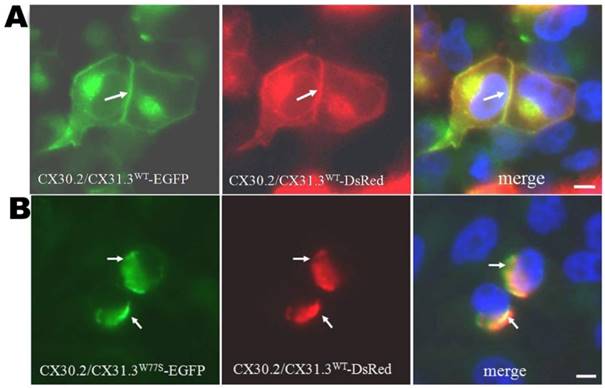
In our previous investigating, we showed that the p.W77S mutation in CX30.2/CX31.3 is a heterozygous mutation in patients with nonsyndromal hearing loss [12]. Therefore, co-expression studies were followed out to inspect the effects of the mutant protein on CX30.2/CX31.3WT using a bi-directional tet-on protein expression system with equal amounts of the two respective expression proteins. Our results showed that the CX30.2/CX31.3WT-DsRed and CX30.2/CX31.3W77S-EGFP co-expression pattern was similar to that in cells expressing only CX30.2/CX31.3W77S, which are concentrated in the cytoplasm close to the nucleus (Fig. 3). Based on this finding, we suggest that the p.W77S mutation seem to have a dominant negative effect on CX30.2/CX31.3WT.
Co-expression of mutant proteins with CX30.2/CX31.3WT revealed by tet-on protein expression system. (A) Tet-on HeLa cells co-expressing CX30.2/CX31.3WT-DsRed and CX30.2/CX31.3WT-EGFP. Co-localization of the two proteins is visible at the plasma membrane. (B) Tet-on HeLa cells co-expressing CX30.2/CX31.3WT-DsRed and CX30.2/CX31.3W77S-EGFP. Co-localization of the two proteins is visible near the nucleus regions. Arrows indicate co-expressed proteins. Cells were counterstained with DAPI to highlight the nuclei. Scale bars: 10 μm.
Effect of CX30.2/CX31.3W77S mutation protein in the HeLa cell
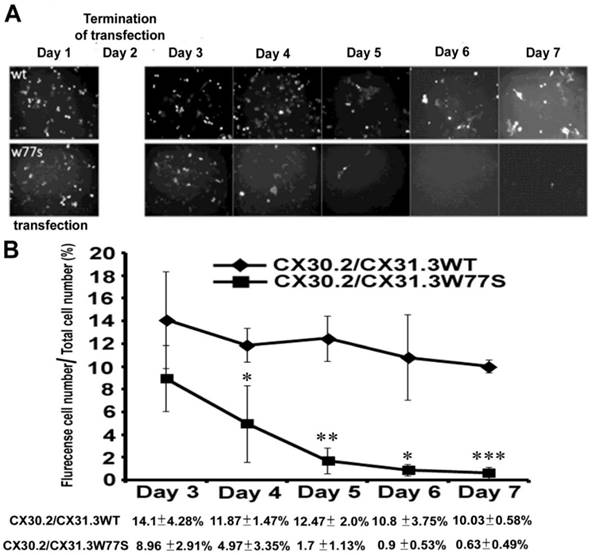
Under a fluorescence microscope, we found that the positive cells displaying green fluorescence had decreased noticeably from 8.96±0.91% to 0.63±0.49% in the days following the culture in post-transfect mutant CX30.2/CX31.3W77S plasmid (Fig. 4). In contrast, the post-transfect CX30.2/CX31.3WT cell with green fluorescence had decreased only slightly, from 14.1±4.28% to 10.03±0.58% in the days following the culture (Fig. 4). Therefore, one possibility reason is the accumulation of a great quantity of mutant proteins in the ER switches on unfolded protein response (UPR) within the ER that leads to the programmed cell death (apoptosis).
Quantitative analysis of positive cells displaying after transiently transfected CX30.3/CX31.3WT and CX30.2/CX31.3W77s into HeLa cells. Fluorescence microscopy of HeLa cells expressing CX30.2/CX31.3WT and CX30.2/CX31.3W77S shows expression of the CX30.2/CX31.3 fusion protein in the transiently transfected HeLa cell. (B) Data represent average ± SD of the percent, after transiently transfected, cells expressing flurencense (EGFP) versus the total cells. Total cell numbers are 1500. Results are representative of three separate experiments. Star code for statistical significance is illustrated as follow: ***P<0.001, **P<0.01 and *P<0.05 (unpaired t-test).
Analysis of DNA fragmentation and MTT
To explanation the possibility, we further analyzed cell death using two methods, DNA fragmentation and MTT assay. Both expressed CX30.2/CX31.3WT and CX30.2/CX31.3W77S HeLa cells were incubated for 4 days before being subjected to cell viability assays by DNA fragmentation. DNAs were purified from expressed HeLa cells, which are post-transfect CX30.2/CX31.3WT and CX30.2/CX31.3W77S plasmids and were then resolved by conventional agarose gel electrophoresis to assay the potential apoptotic DNA fragmentation. The results obviously proved absence of the characteristic DNA laddering of those cells expressing CX30.2/CX31.3W77S (Fig. 5A). Simultaneously, the characteristics of cell viability were also determined by the MTT analysis in the days following the culture in post-transfect WT CX30.2/CX31.3 and mutant CX30.2/CX31.3W77S plasmids (Fig. 5B). This result of cell viability is consistent with DNA fragmentation analysis, which is not different between expressed WT and mutant HeLa cell. Thus, we suggest that the accumulation of CX30.2/CX31.3W77S mutant protein in the ER did not trigger cell death.
Cell viability analysis on expressed CX30.2/CX31.3WT and stably CX30.2/CX31.3W77S HeLa cells. Both cells were incubated in DMEM medium for 1-7 day and were then harvested for DNA fragmentation assay (A) and analysis of MTT (B). (A) DNA was prepared for agarose gel electrophoresis as described in the Materials and Methods. (B) The results showed that when the HeLa cells expressed p.W77S mutant proteins, the performance showed unobvious downward trend compared to the number of cells in WT CX30.2/CX31.3. Results are representative of three separate experiments.
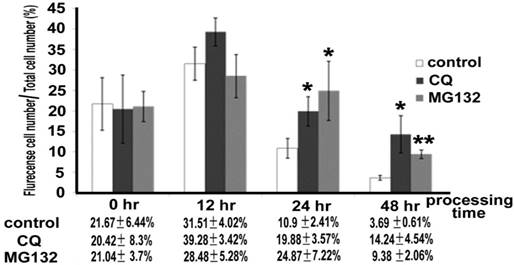
Quantitative analysis of positive cells displaying in the HeLa with transiently expressed CX30.2/CX31.3W77S after chloroquine (CQ) and MG132 treatment. HeLa cells that expressed mutant CX30.2/CX31.3W77S were cultured in the presence of 20μg/ml CQ or 3μg/ml MG132 medium, respectively. After 0, 12, 24, 48 hour, cells were washed and the culture medium was replaced using normal culture medium. Then, these cells were visualized and photographed using a fluorescence microscope (Zeiss Axioplam, Oberkochen, Germany). Data represent average ± SD of the percent, after transiently transfected, cells expressing flurencense (EGFP) versus the total cells. Total cell numbers are 1500. Results are representative of three separate experiments. Star code for statistical significance is illustrated as follow: ***P<0.001, **P<0.01 and *P<0.05 (unpaired t-test).
Analysis of real-time Quantitative-PCR
Further, we tried to find the factors contributing to green fluorescence decrease in the positive cells. To understand p.W77S of GJC3 mutant mRNA expression, real-time Quantitative-PCR was performed to assess the expression of transgenes in post-transfect HeLa cells (supplemental Fig. 3). Our results indicate that p.W77S of GJC3 mRNA expression is consistent with normal GJC3 mRNA expression in the post-transfect HeLa cells. Therefore, we suggest that the p.W77S mutant does not interfere with mRNA expression in the transcription process.
Protein degradation in cultured cells
As well, we used the lysosome inhibitor chloroquine (CQ) 20μg/ml and the proteasome inhibitor MG132 3μg/ml to understand whether the CX30.2/CX31.3W77S mutant proteins underwent degradation. These concentrations were decided according to cell viability using MTT assay (data not show). After treatment with both chloroquine (CQ) and MG132, we found that the positive cells were higher in number than in the untreated group 12, 24, and 48 hours after treatment (Figure. 6). The rate of positive cells was 20.42±8.3%, 39.28±3.42%, 19.88±3.57%, and 14.24±4.54% for CQ treatment at 0, 12, 24, and 48 hours respectively. At 0, 12, 24, and 48 hours after MG132 treatment, the rate of positive cells was 21.04±3.7%, 28.48±5.28%, 24.87±7.22%, and 9.38±2.06%. After unpaired t-test analysis, both CQ (p=0.0224 and p=0.0162, respectively) and MG132 treatment (p=0.0335 and p=0.0013, respectively) were highly significant than no-treatment (control) at the treatment 24 and 48 hours group. Based on these results, we suggest that a missense mutation of the CX30.2/CX31.3 p.W77S protein was degraded in the HeLa cell.
Discussion
At least seven heterozygous mutations and two heterozygous polymorphisms of the GJC3 gene have been detected in Taiwanese patients with nonsyndromic deafness [10-12]. Of the seven mutation, four heterozygous missense mutations [c.43C>G (p.R15G), c.68T>A(p.L23H), c.230C>G (p.W77S) and c.807A>T (p.E269D)] of the GJC3 gene were identified. The phenotype/genotype correlation of connexin (CX) gene family variants have described in our previous study [12]. Previously, we had indicated that p.E269D missense mutation in the GJC3 gene resulted in accumulation of the CX30.3/CX31.2 mutant protein in the endoplasmic reticulum (ER), and had a dominant negative effect on the formation and function of the gap junction [18]. In the p.R15G and p.L23H mutants, we have indicated that p.R15G and p.L23H mutants exhibited continuous staining along apposed cell membranes in the fluorescent localization assay, which is the same as the wild type. However, two mutations in GJC3 genes resulted in a loss of ATP release (hemichannel function) function of the CX30.2/CX31.3 protein [19]. In this study, we found that the p.W77S mutation proteins in the intracellular distribution were different from those in CX30.2/CX31.3WT, which showed continuous staining along apposed cell membranes, and an accumulation of the mutant protein in the endoplasmic reticulum (ER). Simultaneously, we also found that the p.W77S heterozygous mutation has a dominant negative effect on the formation and function of the gap junction. These results of p.W77S mutation are consistent with p.E269D mutation, but there is no consistency with p.R15G and p.L23H mutations.
Similarly, the different site mutations in the GJC gene caused different effect mechanisms that were also found in the GJB2 (CX26) gene, which is a major CX gene linked to hearing loss either alone or as part of a syndrome. For example, p.W44S and p.W44C of CX26 result in a protein that is trafficked to the plasma membrane. In contrast, the p.G59A and p.D66H mutations resulted in protein with impaired trafficking and were concentrated close to the nucleus [22-23]. Despite p.R127H mutant proteins of the Cx26 were mainly localized in the cell membrane and prominent in the region of cell-cell contact, but this mutant proteins was a formation of defective junctional channels [24]. In the CX30.2/CX31.3 protein, the p.E269D mutation occurred in the putative C-terminal cytoplasmic domain, and the p.W77S mutation was localized in the second membrane-spanning segments (TM2) and near border of the E1 domain protein. The p.L23H mutation was at the border of the N-terminal (NT) domain, and the first membrane-spanning segments (M1) and p.R15G occurred in the putative NT cytoplasmic domain (Supplemental Fig. 1). Based on these results, we believe that mutant sites within the protein are important in determining the functional effects of protein.
In a previous study, we found a novel p.W77S mutation in the GJC3 gene from patients with nonsyndromic hearing loss [12]. Tryptophan [W; Ph-NH-CH=C-CH2-CH (NH2)-COOH] and serine [S; HO-CH2-CH(NH2)-COOH] are similar, each having polar and unchanged side-chain amino acids [25]. There are, however, some differences between tryptophan and serine. Tryptophan has heterocyclic aromatic amino side chains, weak basic, and is the largest of the amino acids [26]. Tryptophan is also a very hydrophobic amino acid and prefers to be buried in protein-hydrophobic cores. Tryptophan also can be involved in interactions with non-protein ligands that themselves contain aromatic groups via stacking interactions and in binding to polyproline-containing peptides, for example, in SH3 or WW domains. Serine is generally considered a slightly polar, weakly acidic, and small amino acid. Serine can reside both within the interior of a protein and on the protein surface. It is quite common in protein functional centers, and it is possible for the serine side-chain hydroxyl oxygen to form a hydrogen bond with the protein's backbone or with a variety of polar substrates. In addition, a common role for serine within intracellular proteins is phosphorylation [25-27].
Position 77 of the CX30.2/CX31.3 protein is located at the second membrane-spanning segments (M2) and near the border of the first extracellular loop (E1), which is highly conserved among CX/Cx family members and throughout evolution by Multiple Sequence Alignments and ConSeq analysis (Fig 1). The E1 domain of CX is involved in the interactions between the two adjoining connexons of the gap junction channel [4, 28]. Previous X-ray structure of the CX26 monomer study found that CX26 comprises a typical four-helical bundle in which any pair of neighboring helices is antiparallel. Moreover, this study also indicated that M1, E1, and M2 face the pore; meanwhile, TM3, E2, and TM4 are on the border of the hemichannel facing the lipid or extracellular environments. Additionally, the results disclosed that the cytoplasmic half of TM1 and TM2 are sheltered by the facing the lipid or extracellular environments [29]. In the CX26, the prominent intra-protomer interactions are in the extracellular part of the transmembrane region. Two hydrophobic cores around p.W44 (E1) and p.W77 (TM2) stabilize the protomer structure of CX26 [28]. In this study, our data confirmed that the p.W77S mutant protein of CX30.2/CX31.3 was retained in the ER of HeLa cells. Moreover, we observed significant inhibition of the functional activity of CX30.2/CX31.3-WT in HeLa cells when expressed in a manner mimicking a heterozygous genotype. Thus, p.W77S mutation has dominant negative effect on the function of WT CX30.2/CX31.3. Based on these findings, we suggested that the p.W77 amino acid likely plays a critical role in CX30.2/CX31.3, and as a result a mutation in this residue (W changed to S in position 77) will lead to loss of function of the protein. However, these are predictions, and these cell experiment results are restricted to this study. Therefore, we suggest that the X-ray and 3D structure of the CX30.2/CX31.3 protein needs to be studied to understand further the influence this mutation has at the protein level.
In eukaryotic cells, most secreted and transmembrane proteins fold and mature in the lumen of the endoplasmic reticulum (ER). Previously studies have indicated that CXs can assemble into functional hexameric connexons in the ER membrane [30]. That CXs pass through the Golgi apparatus to reach the plasma membrane has been demonstrated by subcellular fractionation studies and immunocolocalization analyses [31-33]. In this study, we found that the mutant p.W77S proteins accumulate in the ER. The result is similar to the p.E269D mutation. Our previous study described that great quantities of mutant proteins accumulating in the ER might cause unfolded protein responses (UPR), which is a cellular stress response (ER stress) [34]. The ER responds to the accumulation of unfolded proteins in its lumen by activating intracellular signal transduction pathways, cumulatively called UPR [35]. UPR increases the biosynthetic capacity of the secretory pathway through upregulation of ER chaperone and foldase expression. In addition, the UPR decreases the biosynthetic burden of the secretory pathway by downregulating the expression of genes encoding secreted proteins [36]. At least three such mechanisms were found in an imbalance (called ER stress) between the load of unfolded proteins that enter the ER and the capacity of the cellular machinery, the first two of which are rectifying. The first mechanism is a transient adaptation, which reduces the protein load that enters the ER by lowering protein synthesis and translocation into the ER. The second mechanism is a longer-term adaptation that increases the capacity of the ER to handle unfolded proteins by transcriptional activation of UPR target genes, including those that function as part of the ER protein-folding mechanism. If homeostasis cannot be re-established, then a third mechanism, cell apoptosis, is triggered, presumably to protect the organism from rogue cells that display misfolded proteins [35]. In our study, we discovered via cell-viability analysis that the accumulation of p.W77S mutant protein in the ER did not cause cell apoptosis. In conclusion, we found that CX30.2/CX31.3W77S missense mutant proteins were degraded by lysosomes and proteosomes in the transfected-HeLa cell. We have suggested that the accumulation of p.W77S mutant proteins in the ER triggered their degradation, which was insufficient to cause cell apoptosis.
Conclusions
In the article, our result indicated that the p.W77S missense mutation proteins in the intracellular distribution are different CX30.2/CX31.3WT, which showed continuous staining along apposed cell membranes, and an accumulation of the mutant protein in the endoplasmic reticulum (ER) of the HeLa cell. In addition, our results indicated that p.W77S of the CX30.2/CX31.3 mutation has a dominant negative effect on CX30.2/CX31.3WT using tet-on protein expression system. Our study also demonstrated CX30.2/CX31.3W77S missense mutant proteins were degraded by lysosomes and proteosomes in the transfected HeLa cell. Our study highlights the importance of p.W77 amino acid in the CX30.2/CX31.3, and as a result a mutation in this residue will lead to loss of function of the protein.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by Ministry of Science and Technology (MOST 102-2320-B-040-022 and MOST 103-2320-B-040-021-MY3) and Chung Shan Medical University (CSMU-INT-101-04 and CSMU-INT-102-04).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Oghalai JS. The cochlear amplifier: augmentation of the traveling wave within the inner ear. Curr Opin Otolaryngol Head Neck Surg. 2004;12:431-438
2. KiKuchi T, Adams JC, Miyabe Y, So E, Kobayashi T. Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness. Med Electron Microsc. 2000;33:51-56
3. Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Sohl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2000;383:725-737
4. Krutovskikh V, Yamasaki H. Connexin gene mutations in human genetic diseases. Mutat Res. 2000;462:197-207
5. Dinh EH, Ahmad S, Chang Q, Tang W, Stong B, Lin X. Diverse deafness mechanisms of connexin mutations revealed by studies using in vitro approaches and mouse models. Brain Res. 2009;1277:52-69
6. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary nonsyndromic sensorineural deafness. Nature. 1997;387:80-83
7. Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet. 1998;20:370-373
8. Grifa A, Wagner CA, D'Ambrosio L, Melchionda S, Bernardi F, López-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet. 1999;23:16-18
9. López-Bigas N, Melchionda S, Gasparini P, Borragán A, Arbonés ML, Estivill X. A Common Frameshift Mutation and Other Variants in GJB4 (Connexin 30.3): Analysis of Hearing Impairment Families. Hum Muta. 2002;19:458
10. Yang JJ, Huang SH, Chou KH, Liao PJ, Su CC, Li SY. Identification of mutations in members of connexin gene family as a cause of nonsyndromic deafness in Taiwan. Audio Neurootol. 2007;12:198-208
11. Wang WH, Yang JJ, Lin YC, Yang JT, Chan CH, Li SY. Identification of novel variants in the Cx29 gene of nonsyndromic hearing loss patients using buccal cells and RFLP method. Audio Neurootol. 2010;15:81-87
12. Yang JJ, Wang WH, Lin YC, Weng HH, Yang JT, Hwang CF, Wu CM, Li SY. Prospective variants screening of connexin genes in children with hearing impairment: genotype/phenotype correlation. Hum Genet. 2010;128:303-313
13. Sohl G, Eiberger J, Jung YT, Kozak CA, Willecke K. The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. J Biol Chem. 2001;382:973-978
14. Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci. 2002;22:6458-6470
15. Ahmad S, Chen S, Sun J, Lin X. Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mouse. Biochem Biophys Res Comm. 2003;307:362-368
16. Yang JJ, Liao PJ, Su CC, Li SY. Expression patterns of connexin 29 (GJE1) in mouse and rat cochlea. Biochem Biophys Res Comm. 2005;338:723-728
17. Tang W, Zhang Y, Chang Q, Ahmad S, Dahlke I, Yi H, Chen P, Paul DL, Lin X. Connexin29 is highly expressed in cochlear Schwann cells, and it is required for the normal development and function of the auditory nerve of mice. J Neurosci. 2006;26:1991-1999
18. Hong HM, Yang JJ, Su CC, Chang JY, Li TC, Li SY. A novel mutation in the connexin 29 gene may contribute to nonsyndromic hearing loss. Hum Genet. 2010;127:191-199
19. Su CC, Li SY, Yen YC, Nian JH, Liang WG, Yang JJ. Mechanism of two novel human GJC3 missense mutations in causing nonsyndromic hearing loss. Cell Biochemistry and Biophysics. 2013;66:277-286
20. Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Method. 2007;3:12
21. Berezin C, Glaser F, Rosenberg J, Paz I, Pupko T, Fariselli P, Casadio R, Ben-Tal N. ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20:1322-1324
22. Martin PE, Coleman SL, Casalotti SO, Forge A, Evans WH. Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal heriditary deafness. Hum Mol Genet. 1999;8:2369-2376
23. Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet. 2003;12:805-812
24. Wang HL, Chang WT, Li AH, Yeh TH, Wu CY, Chen MS, Hung PC. Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. Journal of Neurochemistry. 2003;84:735-742
25. Baldwin Y, Lapointe M. The Chemistry of Amino Acids. The Biology Project [Online], 2003. http://www.biology.arizona.edu/biochemistry
26. Nelson DL, Cox MM. Lehninger Principles of Biochemistry (3rd ed.) Worth Publishers. ISBN 1-57259-153-6. 2000
27. Betts MJ, Russell RB. Amino acid properties and consequences of subsitutions. In Bioinformatics for GENETICISTS, m.r. Barnes, I.C. Gray eds, Wiley. 2003
28. Maeda S1, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597-602
29. Nakagawa S, Maedal S, Tsukihara T. Structural and functional studies of gap junction channels. Current Opinion in Structural Biology. 2010;20:423-430
30. Falk MM, Buehler LK, Kumar NM, Gilula NB. Cell-free synthesis of connexins into functional gap junction membrane channels. EMBO J. 1997;10:2703-2716
31. Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junction plaques. J Cell Biol. 1991;115:1357-1374
32. Falk MM, Kumar NM, Gilula NB. Membrane insertion of gap junction connexins: polytopic channel forming membrane proteins. J. Cell Biol. 1994;127:343-355
33. Laird DW, Castillo M, Kasprzak L. Gap junction turnover, intracellular trafficking, and phosphorylation of connexin43 in brefeldin A-treated rat mammary tumor cells. J Cell Biol. 1995;131:1193-1203
34. Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Bio Chem. 2004;279:25935-25938
35. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Bio. 2007;8:519-529
36. Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res/Funda and Mol Mechan of Mutagen. 2005;569:29-63
Author contact
![]() Corresponding authors: Dr. J-J Yang, Department of BioMedical Sciences, Chung Shan Medical University, Taichung, Taiwan, Republic of China. Tel: 886-4- 24730022, ext. 11804; Fax: 886-4-24757412; E-mail: jiannjouedu.tw or Dr. S-Y Li, Genetics Laboratory, Department of BioMedical Sciences, Chung Shan Medical University, Taichung, Taiwan, Republic of China. Tel: 886-4- 24730022, ext. 11800; Fax: 886-4-24757412; E-mail: syledu.tw.
Corresponding authors: Dr. J-J Yang, Department of BioMedical Sciences, Chung Shan Medical University, Taichung, Taiwan, Republic of China. Tel: 886-4- 24730022, ext. 11804; Fax: 886-4-24757412; E-mail: jiannjouedu.tw or Dr. S-Y Li, Genetics Laboratory, Department of BioMedical Sciences, Chung Shan Medical University, Taichung, Taiwan, Republic of China. Tel: 886-4- 24730022, ext. 11800; Fax: 886-4-24757412; E-mail: syledu.tw.