Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(3):191-200. doi:10.7150/ijms.17718 This issue Cite
Review
Reviewing primary Sjögren's syndrome: beyond the dryness - From pathophysiology to diagnosis and treatment
Tim Both1, Virgil A.S.H. Dalm1,2, P. Martin van Hagen1,2, Paul L.A. van Daele1,2 ![]()
1. Department of Internal Medicine, Division of Clinical Immunology;
2. Department of Immunology, Erasmus MC, Rotterdam, The Netherlands.
Received 2016-9-26; Accepted 2016-12-21; Published 2017-2-23
Abstract
Primary Sjögren's syndrome (pSS) is a systemic autoimmune disease, characterized by lymphocytic infiltration of the secretory glands. This process leads to sicca syndrome, which is the combination of dryness of the eyes, oral cavity, pharynx, larynx and/or vagina. Extraglandular manifestations may also be prevalent in patients with pSS, including cutaneous, musculoskeletal, pulmonary, renal, hematological and neurological involvement. The pathogenesis of pSS is currently not well understood, but increased activation of B cells followed by immune complex formation and autoantibody production are thought to play important roles. pSS is diagnosed using the American-European consensus group (AECG) classification criteria which include subjective symptoms and objective tests such as histopathology and serology. The treatment of pSS warrants an organ based approach, for which local treatment (teardrops, moistures) and systemic therapy (including non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, disease-modifying antirheumatic drugs (DMARDS) and biologicals) can be considered. Biologicals used in the treatment of pSS mainly affect the total numbers of B cells (B cell depletion (Rituximab)) or target proteins required for B cell proliferation and/or activation (e.g. B cell activating factor (BAFF)) resulting in decreased B cell activity.
The aim of this review is to provide physicians a general overview concerning the pathogenesis, diagnosis and management of pSS patients.
Keywords: primary Sjögren syndrome, sicca, extraglandular syndrome, biologicals, epidemiology, pathogenesis, T-cell, B-cell.
Introduction
Sjögren's syndrome (SS) is a relatively common systemic autoimmune disease characterized by lymphocytic infiltration of the secretory glands. This process leads to sicca syndrome, which is the combination of dryness of the eyes, oral cavity, pharynx, larynx and/or vagina [1]. Sicca syndrome is often accompanied by symptoms resulting from systemic involvement. SS can be present as a primary disease without any other accompanying symptoms (primary Sjögren syndrome, pSS). When SS presents as a secondary disease with other autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and systemic sclerosis, it is then called secondary SS (sSS) [2, 3]. The prevalence of sSS is highest in RA patients and estimated to be around 20% [2, 4].
In this review we will focus on pSS. The incidence of pSS is estimated to be 4 per 1000 patients per year and overall prevalence of pSS in Europe is between 0.1-4.8% [5, 6]. However, as many symptoms are non-specific, the prevalence may be underestimated.
Pathogenesis
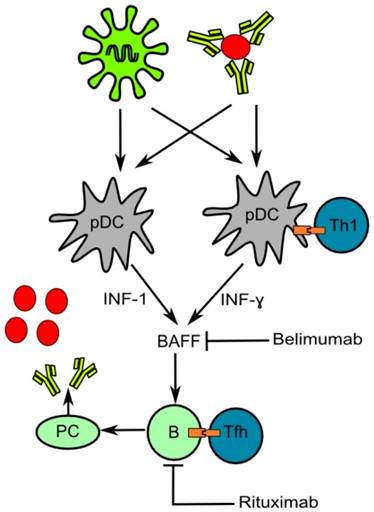
The pathogenesis of pSS is incompletely understood but appears to be multifactorial. Although T cells were originally considered to be the key players in the autoimmune process, there is now growing evidence that B cells play at least an equally important role in the pathophysiology of pSS (Figure 1). In the next section we will discuss in more detail the known and potential roles of the different immune cells in the pathogenesis of pSS.
Schematic showing a simplified overview of the pathogenesis of pSS with the targets for biologicals. An unknown cause (suggested to be a virus or immune complexes) may lead to activation of pDCs resulting in increased levels of interferons. Interferon-induced BAFF production leads to increased B cell proliferation and differentiation with autoantibody production as result. Abbreviations: pDC, plasmacytoid dendritic cell; Th1, T-helper 1 cell; IFN, interferon; B, B cell; PC, Plasma cell; Tfh, Follicular T-helper cell.
T cells in pSS
The presence, and sometimes predominance, of CD4+ T cells in salivary gland infiltrates underlines their potential contribution to the pathogenesis of pSS [7]. A meta-analysis showed the association between pSS and several major histocompatibility complex class 2 (MHC2) alleles suggesting that autoantigen presentation is important in the pathogeneses of pSS [8]. Th1 cells are hypothesized to be the main subtype contributing to pathogenesis, since they bind to the MHC2 molecules initiating an immune response. In addition, pro-inflammatory Th1 cell cytokines (e.g. IL-1b, IL-6, tumor necrosis factor-α and interferon-γ) are increased in saliva of patients with pSS [9]. Furthermore, a study in 2009 reported a pSS-like syndrome in mice with IL-12 overexpression, which is known to induce Th1 cell differentiation [10]. Besides Th1 cells, the numbers of T helper 17 (Th17) cells are also increased at sites of inflammation in salivary gland biopsies of pSS patients [11]. IL-17, produced by Th17 cells, is increased in both serum and salivary glands of patients with pSS as compared to healthy controls [12]. Co-expression of IL-17 and IL-18 has been associated with increased severity of pSS, probably due to maintaining the inflammatory process [11, 13]. Furthermore, regulatory T cells (Treg) have been identified in salivary glands of pSS patients and the increased presence of these cells has been associated with higher grade of inflammation in the local lesions [14, 15]. Tregs are known to have suppressive effects on the proliferation and function of effector T cells. It has been reported that the number of circulating Tregs are increased, while their function does not seem to be impaired in pSS, which suggests that Tregs do not play an important role in the pathogenesis of pSS [16].
B cells in pSS
B cells are adaptive immune cells and are responsible for antibody secretion and antigen presentation. B cell development occurs in the bone marrow. One of the key factors in this process is B cell activating factor (BAFF). BAFF is a cytokine that promotes B cell proliferation, maturation and survival and is primarily induced by type I and type II interferons [17, 18]. These interferons are produced by plasmacytoid dendritic cells (pDCs) [13, 19]. It has been suggested that certain viruses (e.g. Epstein-Barr) and immune complex formation activate Toll-like receptors (TLRs) (e.g. TLR 3, 7 and 9), leading to activation of the innate immunity and interferon production. Although an increased activity of TLRs has been reported in pSS, a specific cause (virus or immune complex) has not yet been identified [20, 21]. In pSS patients, 55% have an increased IFN type I activity versus 4.5% in healthy controls [22]. The presence of this so-called 'IFN type I signature' in monocytes of patients with pSS was shown to be associated with higher EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI), the presence of biological markers of activity (increased levels of IgG and/or hypocomplementia) and increased levels of BAFF mRNA in monocytes [22]. Circulating and salivary gland tissue levels of BAFF are significantly elevated in patients with pSS, which is associated with increased disease activity but also with higher risk of development of B cell lymphoma [23-25]. These findings support the hypothesis that innate immune system activation leads to an autoimmune response by the adaptive immune system. Since BAFF is one of the links between innate and adaptive immune responses, it could be a potential target for therapy in pSS. The first results of studies on anti-BAFF therapy (belimumab) show a significant decrease in disease activity after twelve months of treatment as measured by the ESSDAI [26]. Upon antigen recognition in the germinal center, B cells proliferate and differentiate (e.g. class switching) into a specific B cell for this antigen [27]. In pSS, germinal centers are reported in the epithelium of non-lymphoid tissues such as the salivary glands [28]. The formation of germinal centers is probably important in the pathogenesis of pSS due to promotion of chronic stimulation and activation (by follicular T helper cells) of B cells. Patients with pSS often present with high levels of serum IgA and/or IgG [29]. Hyperglobulinemia may lead to the formation of immune complexes with the potential to precipitate in major organs leading to (irreversible) damage [30]. In addition, the presence of autoantibodies (anti-Ro52, anti-Ro60 and anti-La) is included in the diagnostic criteria for pSS [31, 32]. The presence of these autoantibodies is associated with early onset disease, parotid gland enlargement, extraglandular manifestations and lymphocytic glandular infiltration [13, 33].
In sum, it remains unclear how these changes in the adaptive immune system lead to the clinical manifestations of pSS. The traditional view that chronic inflammation results in tissue destruction of the exocrine glands will only partially contribute to the pathogenesis of pSS. There is a poor correlation between the amount of damage observed in tissue biopsies and the measured decrease in fluid production, as the reduction in salivary production is often larger than expected from both clinical and histological appearance [34].
Clinical presentation
There is not a standard clinical presentation for pSS, as many patients have various degrees of systemic involvement at the time of presentation. The symptoms of pSS can be divided into three groups, (1) sicca syndrome, (2) general symptoms and (3) systemic manifestations.
Sicca syndrome
Sicca syndrome is the combination of dryness of the eyes (xeropthalmia), oral cavity (xerostomia), pharynx and/or larynx, which are the classical symptoms of pSS. In woman, also vaginal dryness is a common feature of pSS [35]. These symptoms are part of the American-European classification criteria (AECG) of 2002 for the diagnosis pSS and occur in more than 95% of patients [31]. The positive predictive value of the AECG criteria is between 54-77% and the negative predictive value is between 94-98% as compared to the classification criteria of 1986 [31]. Xerostomia may lead to secondary problems like oral candidiasis (33%), dental carries (65%) and periodontal disease [36, 37]. Xerophtalmia may result in photosensitivity, chronic irritation and destruction of the corneal epithelium and ocular infections. Additionally, sicca syndrome also includes hoarseness, non-productive cough, skin dryness and, in woman, dyspareunia [38, 39]. Patients with pSS experience a significantly decreased quality of life compared to subjects with sicca syndrome without autoimmune features as measured by the SF-36 depression scale [40, 41].
General symptoms
The most prevalent general symptom is fatigue, occurring in up to 70-80% of pSS patients [42]. Fatigue in pSS has been well studied using the multidimensional fatigue inventory (MFI) on which pSS patients scores were two-fold worse on all dimensions as compared to healthy controls [43, 44]. In addition, chronic pain is often seen in pSS due to accompanying fibromyalgia and/or polyarthralgia [45]. Depression and anxiety are also more common in pSS patients compared to healthy controls [46]. A study showed that 47% of the working age pSS patients received disability compensation, because they were considered to be (partially) unfit for work [47]. The same study also reports that significantly more patients with the following demographic/disease characteristics receive disability compensation: male patients, patients with a high educational level, an increasing number of systemic manifestations and/or the use of artificial saliva and/or HCQ [40, 41, 47].
Systemic manifestations
Approximately 71% of the patients with pSS present with extraglandular manifestations [48] (Table 1). Of those, lymphoma has the highest mortality [49]. A large cohort study reported a nearly 5-fold higher relative risk in pSS patients with a life-time risk of approximately 10% [50, 51]. The most common subtype is the mucosa-associated lymphoid tissue (MALT) lymphoma often seen in the parotid glands, which is usually a low-grade indolent neoplasm [52, 53]. Clinical risk factors include persistent, unilateral salivary gland enlargement, lymphadenopathy, splenomegaly, skin vasculitis, cryoglobulinemia and the development of glomerulonephritis [54, 55]. Laboratory-assessed biological risk factors for lymphoma in pSS include cryoglobulinemia, lymphopenia (especially low total numbers of CD4+ T cells), hypocomplementia, increased serum BAFF and the presence of a monoclonal component in serum or urine [27, 54, 56]. Articular involvement in pSS predominantly consists of symmetric, intermittent, nonerosive arthropathy [57, 58]. Arthritis is less common and occurs in approximately 16% of pSS patients and mostly involves proximal interphalangeal joints (35%), metacarpal-phalangeal joints (35%) and wrists (30%) [58, 59]. Approximately 10-20% of pSS patients develop interstitial lung disease (ILD)[60]. In general, patients will have evidence of both airway disease and ILD by radiographs (plain X-ray and/or CT-scan) and pulmonary biopsy [60]. Another study reported that patients with pSS who do not have pulmonary symptoms already may have radiographic or computed tomography (CT) scan abnormalities (22%) or an impaired pulmonary function test [61]. The most frequently observed CT patterns consist of interstitial pneumonia, centrilobular abnormalities and lymphoproliferative disease [61]. This emphasizes that a frequent pulmonary function test or a high-resolution CT-scan should be performed in the follow-up of pSS patients with and without pulmonary complaints. The most common histopathological phenotype of ILD in pSS is nonspecific interstitial pneumonia (NSIP), which has been reported in approximately 45% of the pSS patients with ILD [62, 63]. ILD is difficult to treat and results in an increase of dry cough and dyspnea, leading to decreased quality of life. ILD is usually treated with glucocorticoids but other immunosuppressive drugs are also available, such as azathioprine, mycophenolate mofetil, cyclophosphamide and cyclosporine [64, 65]. Furthermore, renal involvement is common and includes a wide spectrum of manifestations, of which interstitial nephritis is the most prevalent [66, 67]. Consistent screening for renal function is important since renal failure (defined as a glomerular filtration rate < 60 ml/min) occurs in approximately 24% of the pSS patients [68]. There is no standardized treatment of renal involvement in pSS. Glucocorticoids are the treatment of first choice in tubulointerstitial nephritis, whereas other immunosuppressive drugs are only shown effective in a small study (mycophenolate mofetil) or not effective at all during the induction phase (cyclophosphamide) [69, 70]. Neurological involvement in pSS includes both the peripheral and central nervous systems. In many patients, neurologic symptoms precede the onset of other signs and symptoms of pSS [71, 72]. In general, intravenous corticosteroids are first-line therapy for patients with pSS associated neuropathy. Cyclophosphamide or intravenous immunoglobulins can be used in patients who do not improve with corticosteroids [72-74].
Systemic manifestations in primary Sjögren Syndrome.
| Domain | Prevalence (%) | Clinical manifestations | Investigations |
|---|---|---|---|
| Lymphadenopathy [51, 52] | 10 | persistent, unilateral salivary gland enlargement; lymphadenopathy; splenomegaly; skin vasculitis | Serology, biological markers, biopsy |
| Glandular [98] | 30-50 | firm, diffuse, nontender, swelling of mostly the parotid gland | - |
| Articular [57, 59] | 50 | Arthralgia; arthritis | Radiography |
| Skin [100-102] | 23-67 | Xerosis; Raynaud phenomenon; annular erythema, erythema nodosum; livedo reticularis; lichen planus; vitiligo; granuloma annulare; vasculitis | Biopsy (if required) |
| Lungs [60, 62, 63] | 10-20 | dry cough; nasal dryness; dyspnea,; interstitial lung disease | Radiography, CT, pulmonary function |
| Kidneys [66, 67, 103] | 30 | Distal renal tubular acidosis; nephrogenic diabetes insipidus; proximal tubular acidosis; hypokalemia | Systematic renal tests, acid loading test, biopsy |
| Muscles [104, 105] | 44 | Myalgia; muscle weakness; myositis | Biopsy |
| Peripheral nervous system [72, 106, 107] | 10 | painful, burning dysesthesias in the distal extremities; sensory ataxic neuropathy; axonal sensorimotor polyneuropathy; mononeuritis multiplex; cranial neuropathies; radiculoneuropathy; autonomic neuropathy | EMG |
| Central nervous system [71, 108] | 20-25 | motor or sensory deficits; seizures or cerebellar syndromes; psychiatric abnormalities; dementia and spinal cord involvement; subacute aseptic meningitis; chorea; optic neuritis; cognitive dysfunction | EMG, MRI, CSF investigation, psychiatric analysis |
| Haematological [29] | 20 | Normochromic, normocytic anemia; thrombocytopenia; mild leukopenia; lymphopenia | Biochemical tests, bone marrow |
| Biological [29, 109] | 36-62 | Hypergammaglobulinemia; hypogammaglobulinemia; hypocomplementia; cryoglobulinemia | Serology and biological tests, bone marrow |
Abbreviations: EMG, Electromyography; CSF, cerebrospinal fluid; CT, Computed tomography; MRI, Magnetic resonance imaging;
By performing the ESSDAI in pSS patients on a regular basis, all the above discussed systemic manifestations can be recognized. pSS is also associated with hepatitis C (12%), autoimmune thyroid disease (10%), autoimmune chronic active hepatitis (2%) and primary biliary cirrhosis (5%), but the ESSDAI does not include these diseases [75, 76].
pSS treatment requires a patient-specific approach that accounts for disease severity. In the Erasmus MC, we evaluate every pSS patient at least 1-2 times a year. In addition to recording the patient's self-reported symptoms and conducting a standard physical examination, we perform blood tests (including total blood count, liver and kidney tests, C3, C4 and IgG) and urinalysis to screen for organ involvement. In the case of mild disease activity (as measured by disease activity scores, ESSDAI), we do not perform additional invasive tests such as scans or functional tests (e.g. EMG, pulmonary function). In the case of self-reported symptoms or abnormal physical and/or laboratory examinations, additional testing for the presence (or change) of organ involvement is required. Also, patients with systemic immunosuppressive treatment or with increased organ involvement should be seen more frequently at the outpatient clinic (at least once every 3 months) to evaluate whether treatment is effective and potential side effects are tolerated.
Diagnosis
The diagnosis of pSS is based on the American-European consensus group (AECG) classification criteria for Sjögren syndrome [31] . These criteria include: 1) subjective presence of ocular dryness, 2) subjective presence of oral dryness, 3) objective measures of ocular dryness by Schirmer's test or corneal staining, 4) focus score > 2 in a salivary gland biopsy, 5) salivary scintigraphy showing reduced salivary flow (1.5 mL in 15 minutes) and/or diffuse sialectasias and 6) positive autoantibodies against SS-A and/or SS-B. SS is diagnosed when 4 out of 6 items are present; either salivary gland pathology or the presence of autoantibodies against SS-A/SS-B is mandatory.The specific questions (criteria 1 and 2) should reveal whether eye and mouth symptoms are characteristic for pSS and additional tests should be performed. If pSS is suspected, laboratory investigations should be performed (e.g. markers for inflammation, systemic biochemical tests, serology and haematology) and the patient should be referred to an ophthalmologist for evaluation of ocular dryness. Recently, the American Group of Rheumatology (ACR) has developed new diagnostic criteria for pSS since the increasing use of (expensive) biologic agents should be based on more objective rather than subjective criteria [77]. The newly proposed criteria by the ACR differ from the AECG criteria by focussing more on objective measurements. Therefore, ocular and oral dryness are no longer part of the classification criteria. It remains unclear whether the new criteria are more sensitive than the AECG criteria. Based on a comparison study in 646 subjects, the AECG criteria had an overall sensitivity in the general population of 88% compared to 83% of the ACR criteria. On all test characteristics (sensitivity, specificity etc.) the AECG criteria scores better compared to the ACR criteria, however, the results are not significantly different [78]. In conclusion, there is no clear evidence for increased value of the new ACR criteria over the old and familiar AECG criteria from the clinical or biological perspective [78]. Currently, the AECG criteria are still the most frequently used in clinical practice and research protocols. In Table 2, we summarize both sets of classification criteria [78].
Comparison of the Revised American-European Consensus Group (AECG) Classification criteria and the American College of Rheumatology (ACR) Classification criteria for Sjögren's syndrome.
| # | AECG | ACR |
|---|---|---|
| 1 | Ocular symptoms: a positive response to at least one of the following questions: Have you had daily, persistent, troublesome dry eyes for more than 3 months? Do you have a recurrent sensation of sand or gravel in the eyes? Do you use tear substitutes more than 3 times a day? | |
| 2 | Oral symptoms: a positive response to at least one of the following questions: Have you had a daily feeling of dry mouth for more than 3 months? Have you had recurrently or persistently swollen salivary glands as an adult? Do you frequently drink liquids to aid in swallowing dry food? | |
| 3 | Objective ocular signs - a positive result for at least one of the following two tests: Schirmer's I test, performed without anesthesia (≤5 mm in 5 minutes) Rose Bengal score or other ocular dye score (≥4 according to van Bijsterveld's scoring system) | Keratoconjunctivitis sicca with ocular staining score ≥3 |
| 4 | Histopathology: in minor salivary glands (obtained through normal appearing mucosa) focal lymphocytic sialoadenitis, evaluated by an expert histopathologist, with a focus score ≥ 1, defined as number of lymphocytic foci (which are adjacent to normal-appearing mucous acini and contain more than 50 lymphocytes) per 4 mm2 of glandular tissue | Labial salivary gland biopsy exhibiting focal lymphocytic sialadenitis with a focus score ≥1 focus/4 mm2 |
| 5 | Salivary gland involvement: objective evidence of salivary gland involvement defined by a positive result for at least one of the following diagnostic tests: Unstimulated whole salivary flow (≤1.5 ml in 15 min) Parotid sialography showing the presence of diffuse sialectasias (punctate, cavitary or destructive pattern), without evidence of obstruction in major ducts Salivary scintigraphy showing delayed uptake, reduced concentration and/or delayed excretion of tracer | |
| 6 | Autoantibodies: presence in the serum of the following autoantibodies: Antibodies to Ro (SSA) and/or La (SSB) antigens | Autoantibodies: presence in the serum of the following autoantibodies: Antibodies to Ro (SSA) and/or La (SSB) antigens Positive rheumatoid factor and ANA titer ≥1:320 |
Classification rules | ||
| pSS may be diagnosed when: The presence of any 4 of the 6 items is indicative of primary SS, as long as either item 4 (Histopathology) or 6 (Serology) is positive | pSS may be diagnosed when: The classification of SS, which applies to individuals with signs/symptoms that may be suggestive of SS, will be met in patients who have at least 2 of the 3 objective features previously described | |
Treatment
Patients with pSS should be managed by a multidisciplinary team including at least a clinical immunologist/rheumatologist, ophthalmologist and dentist. Extensive clinical trials concerning the treatment of pSS are limited and thus, guidelines are lacking. Nowadays, multiple drugs are used in the treatment of pSS which can be divided in local and systemic therapy (Table 3).
Overview of treatment options in primary Sjögren Syndrome.
| Drug | Usual dose | Main indications | Main contra- indications | Main side-effects | Monitoring needed |
|---|---|---|---|---|---|
| Systemic | |||||
| Pilocarpin [82, 83] | 20-30mg/day orally | Dryness of oral cavity | Untreated cardiovascular condition, untreated asthma | Headache, transpiration, frequent miction | - |
| NSAID [110, 111] | 100-150 mg/day, orally | General symptoms (mainly arthralgia) | Peptic ulcer, GI-bleeding, IBD, CHF, CVA, liver- or renal failure. | GI effects, dizziness, rash, elevated liver enzyme test | Six monthly: Blood count, systemic liver and kidney test. Cardiovascular risk profile |
| Immunomodulatory | |||||
| Hydroxychloroquine [84, 85] | 200-400 mg/day orally | General symptoms (sicca, arthralgia and pain) | Retinopathy, breastfeeding | GI effects, rash, retinopathy, neuromyopathy | Six monthly: blood count and muscular strength Yearly: complete eye examination by ophthalmologist |
| Methotrexate [87] | 10-20mg/week orally or intramuscular Add folate to prevent GI toxic effects | Insufficient effect of HCQ on chronic complaints | Liver and severe renal failure, severe respiratory failure, alcohol abuse, pregnant or lactating women | GI effects, neutropenia, liver and renal toxicity, interstitial pneumonitis, alopecia | 3 monthly: blood count with differentiation, systemic liver and kidney test. Yearly: pulmonary function |
| Glucocorticoids [64, 112] | 20-40mg/day orally or intravenous 1g/day max. 3 days | Active systemic involvement (renal, pulmonary, neurological, muscular) | Active infections (viral, fungal), ulcus ventriculi / duodeni | Weight gain, hypertension, osteoporosis, diabetes, infection, neuropsychiatric reactions | Next outpatient visit: Weight, arterial blood pressure, glycaemia, bone density |
| Rituximab [24, 113] | 1000mg intravenous; repeat after 2 weeks. 30 minutes in prior: 100 mg methylprednisolone | Active systemic involvement not responsive to non-biologic immunosuppressive drugs | Pregnant or lactating women, active severe infection, severe CHF | Infections, allergic reaction | Next outpatient visit: Blood count with differentiation, systemic liver and kidney test. |
Abbreviations: GI, Gastrointestinal; IBD, inflammatory bowel disease; CHF, congestive heart failure; CVA, cerebrovascular accident.
Preventive and local therapy
Alcohol and smoking should be avoided and thorough oral hygiene is essential [79, 80]. Xeropthalmia can be managed with preservative-free teardrops and ocular lubricating ointments. Severe refractory dryness of the eyes can be treated with cyclosporin 0.05% [81]. Patients with xerostomia can manage the dry mouth by doing gustatory stimulation (chewing gum) and moisture replacement.
Systemic treatment
The majority of patients use pilocarpine, a muscarinic receptor agonist, which stimulates residual salivary gland function [82, 83]. Systemic treatment is indicated when: 1) general symptoms (e.g. arthralgia) cannot be managed with local treatment or adjustment of the patient's lifestyle and 2) in case of organ involvement. Non-steroidal anti-inflammatory drugs (NSAIDS) have beneficial effects on general symptoms, like arthralgia. When general symptoms become more chronic, hydroxychloroquine (HCQ) is indicated [84, 85]. It has been reported that patients with arthralgia benefit from HCQ [84]. A recent study shows, however, that fatigue does not improve by HCQ treatment [86]. In case of more severe organ involvement, other DMARDS or glucocorticoids should be added.
Since methothrexate (MTX) is effective in RA, MTX is also used in the treatment of arthritis in pSS patients [87]. Glucocorticoid treatment is predominantly indicated when (severe) cutaneous, pulmonary, renal, musculoskeletal and/or neurological involvement occurs [88]. In case of insufficient effect of glucocorticoid therapy, glucocorticoid intolerance due to side effects and/or to reduce glucocorticoid dose, adding or switching of a DMARD (mycophenolate mofetil, cyclosporine A, azathioprine) should be considered. Therapy resistant pSS with proven organ damage is an indication to start biologicals, with the B cell as the most promising target based on the aetiology of pSS.
Rituximab is a monoclonal antibody targeting the CD20 molecule (human B lymphocyte-restricted differentiation antigen) expressed on the surface of most B cells, including pre-B and mature B lymphocytes leading to B cell depletion [89]. Several studies have demonstrated a favourable effect of rituximab in pSS. Two studies combining 274 pSS patients reported that the severity of glandular, articular, renal, neurological, pulmonary and haematological involvement was significantly decreased in approximately 60% of the patients after six months [24, 90]. As a consequence of rituximab treatment, serum BAFF levels are increasing in order to stimulate B cell maturation, which can be countered by anti-BAFF treatment (belimumab) to achieve a longer B cell depletion and associated longer treatment effect [91]. Based on these findings and the pathophysiology of pSS, combination therapy with belimumab and rituximab would be an expensive but promising option [26]. The combination of rituximab and belimumab may be effective since this combination leads to an effective depletion of both the tissue and circulating B cells as well as a depletion of one of the stimulators (BAFF) required for B cell differentiation. Currently, new potential anti B cell therapies are being evaluated in (pre)clinical trials including anti-CD40 (decreases antigen presentation by B cells), anti-BAFF receptor (inhibits the effects of BAFF), anti-inducible costimulatory ligand (ICOSL, decreases activation of T-cells) and phosphoinositide 3-kinase delta inhibitor (PI3Kδ, inhibition of B cell development and activation) [92-95].
Prognosis
Patients with pSS should be closely monitored to evaluate the development of systemic manifestations and the effects of treatment. Compared to the general population, pSS patients have an increased mortality risk. The standardized mortality ratio (ratio of observed deaths in the study group to expected deaths in the general population, SMR) of pSS patients is on average 2.86, showing that pSS has an impact on patients' survival [96, 97]. The leading cause of mortality in pSS is lymphoma with a lymphoma-specific SMR of 7.89, associating lymphoproliferative disorders directly with death in pSS [49]. However, once lymphoma is diagnosed, the prognosis is relatively favourable with a 15-year survival of almost 80% [98]. Other causes of death in pSS include vasculitis, renal failure due to glomerulonephritis and infections after the administration of immunosuppressive medication [69, 99]. Morbidity in pSS is mainly due to extreme fatigue and the presence of systemic manifestations and should be evaluated for each patient individually. Patients with systemic complications and lymphoma development have an increased mortality risk. Therefore, risk factors (clinical and biological) for lymphoma and other organ involvement (e.g. pulmonary function, renal function, neurological evaluation) should be assessed frequently.
Conclusion
This review summarizes the clinical aspects of pSS with special emphasis on the systemic manifestations. Physicians should be aware of pSS in patients presenting with sicca or general symptoms since the systemic manifestations are severe and are associated with increased morbidity and mortality. The treatment of pSS is effective and includes both local and systemic therapy. In recent years, changes in treatment of pSS have developed into a more organ-based approach, mainly due to the introduction of biologicals. Further research should focus on revealing new aetiological targets for therapy and to evaluate the effectiveness of current treatment protocols.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Asmussen K, Andersen V, Bendixen G, Schiodt M, Oxholm P. A new model for classification of disease manifestations in primary Sjogren's syndrome: evaluation in a retrospective long-term study. J Intern Med. 1996;239:475-82
2. Carmona L, Gonzalez-Alvaro I, Balsa A, Angel Belmonte M, Tena X, Sanmarti R. Rheumatoid arthritis in Spain: occurrence of extra-articular manifestations and estimates of disease severity. Ann Rheum Dis. 2003;62:897-900
3. Manoussakis MN, Georgopoulou C, Zintzaras E, Spyropoulou M, Stavropoulou A, Skopouli FN. et al. Sjogren's syndrome associated with systemic lupus erythematosus: clinical and laboratory profiles and comparison with primary Sjogren's syndrome. Arthritis Rheum. 2004;50:882-91
4. Skoumal M, Wottawa A. Long-term observation study of Austrian patients with rheumatoid arthritis. Acta Med Austriaca. 2002;29:52-6
5. Bowman SJ, Ibrahim GH, Holmes G, Hamburger J, Ainsworth JR. Estimating the prevalence among Caucasian women of primary Sjogren's syndrome in two general practices in Birmingham, UK. Scand J Rheumatol. 2004;33:39-43
6. Drosos AA, Andonopoulos AP, Costopoulos JS, Papadimitriou CS, Moutsopoulos HM. Prevalence of primary Sjogren's syndrome in an elderly population. Br J Rheumatol. 1988;27:123-7
7. Singh N, Cohen PL. The T cell in Sjogren's syndrome: force majeure, not spectateur. J Autoimmun. 2012;39:229-33
8. Cruz-Tapias P, Rojas-Villarraga A, Maier-Moore S, Anaya JM. HLA and Sjogren's syndrome susceptibility. A meta-analysis of worldwide studies. Autoimmun Rev. 2012;11:281-7
9. Fox RI, Kang HI. Pathogenesis of Sjogren's syndrome. Rheum Dis Clin North Am. 1992;18:517-38
10. Vosters JL, Landek-Salgado MA, Yin H, Swaim WD, Kimura H, Tak PP. et al. Interleukin-12 induces salivary gland dysfunction in transgenic mice, providing a new model of Sjogren's syndrome. Arthritis Rheum. 2009;60:3633-41
11. Sakai A, Sugawara Y, Kuroishi T, Sasano T, Sugawara S. Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren's syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J Immunol. 2008;181:2898-906
12. Katsifis GE, Rekka S, Moutsopoulos NM, Pillemer S, Wahl SM. Systemic and local interleukin-17 and linked cytokines associated with Sjogren's syndrome immunopathogenesis. The American journal of pathology. 2009;175:1167-77
13. Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary Sjogren's syndrome. Nat Rev Rheumatol. 2013;9:544-56
14. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos NM, Moutsopoulos HM. Foxp3+ T-regulatory cells in Sjogren's syndrome: correlation with the grade of the autoimmune lesion and certain adverse prognostic factors. The American journal of pathology. 2008;173:1389-96
15. Sarigul M, Yazisiz V, Bassorgun CI, Ulker M, Avci AB, Erbasan F. et al. The numbers of Foxp3 + Treg cells are positively correlated with higher grade of infiltration at the salivary glands in primary Sjogren's syndrome. Lupus. 2010;19:138-45
16. Gottenberg JE, Lavie F, Abbed K, Gasnault J, Le Nevot E, Delfraissy JF. et al. CD4 CD25high regulatory T cells are not impaired in patients with primary Sjogren's syndrome. J Autoimmun. 2005;24:235-42
17. Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann Rheum Dis. 2007;66:700-3
18. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annual review of immunology. 2003;21:231-64
19. Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. B-cell activating factor of the tumour necrosis factor family expression in blood monocytes and T cells from patients with primary Sjogren's syndrome. Scand J Immunol. 2008;67:185-92
20. Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T. et al. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J Exp Med. 2009;206:2091-9
21. Zheng L, Zhang Z, Yu C, Yang C. Expression of Toll-like receptors 7, 8, and 9 in primary Sjogren's syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:844-50
22. Brkic Z, Maria NI, van Helden-Meeuwsen CG, van de Merwe JP, van Daele PL, Dalm VA. et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren's syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis. 2013;72:728-35
23. Daridon C, Devauchelle V, Hutin P, Le Berre R, Martins-Carvalho C, Bendaoud B. et al. Aberrant expression of BAFF by B lymphocytes infiltrating the salivary glands of patients with primary Sjogren's syndrome. Arthritis Rheum. 2007;56:1134-44
24. Gottenberg JE, Cinquetti G, Larroche C, Combe B, Hachulla E, Meyer O. et al. Efficacy of rituximab in systemic manifestations of primary Sjogren's syndrome: results in 78 patients of the AutoImmune and Rituximab registry. Ann Rheum Dis. 2013;72:1026-31
25. Mariette X, Roux S, Zhang J, Bengoufa D, Lavie F, Zhou T. et al. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjogren's syndrome. Ann Rheum Dis. 2003;62:168-71
26. Quartuccio L, Salvin S, Corazza L, Gandolfo S, Fabris M, De Vita S. Efficacy of belimumab and targeting of rheumatoid factor-positive B-cell expansion in Sjogren's syndrome: follow-up after the end of the phase II open-label BELISS study. Clin Exp Rheumatol. 2016
27. Zhang Y, Garcia-Ibanez L, Toellner KM. Regulation of germinal center B-cell differentiation. Immunological reviews. 2016;270:8-19
28. Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J. et al. Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjogren's syndrome. Arthritis Rheum. 2001;44:2633-41
29. Ramos-Casals M, Font J, Garcia-Carrasco M, Brito MP, Rosas J, Calvo-Alen J. et al. Primary Sjogren syndrome: hematologic patterns of disease expression. Medicine. 2002;81:281-92
30. Bendaoud B, Pennec YL, Lelong A, Le Noac'h JF, Magadur G, Jouquan J. et al. IgA-containing immune complexes in the circulation of patients with primary Sjogren's syndrome. J Autoimmun. 1991;4:177-84
31. Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE. et al. Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002;61:554-8
32. Scofield RH, Farris AD, Horsfall AC, Harley JB. Fine specificity of the autoimmune response to the Ro/SSA and La/SSB ribonucleoproteins. Arthritis Rheum. 1999;42:199-209
33. Fox RI, Howell FV, Bone RC, Michelson P. Primary Sjogren syndrome: clinical and immunopathologic features. Semin Arthritis Rheum. 1984;14:77-105
34. Vitali C, Gravili C, Scamardella M. et al. Do the epithelial cells of salivary glands in Sjögren's syndrome express the La antigen? Preliminary results of an immunohistochemical study (abstract). Arthritis Rheum. 1995;38(suppl):S403
35. Haga HJ, Gjesdal CG, Irgens LM, Ostensen M. Reproduction and gynaecological manifestations in women with primary Sjogren's syndrome: a case-control study. Scand J Rheumatol. 2005;34:45-8
36. Daniels TE, Silverman S Jr, Michalski JP, Greenspan JS, Sylvester RA, Talal N. The oral component of Sjogren's syndrome. Oral Surg Oral Med Oral Pathol. 1975;39:875-85
37. Tapper-Jones L, Aldred M, Walker DM. Prevalence and intraoral distribution of Candida albicans in Sjogren's syndrome. J Clin Pathol. 1980;33:282-7
38. Lehrer S, Bogursky E, Yemini M, Kase NG, Birkenfeld A. Gynecologic manifestations of Sjogren's syndrome. Am J Obstet Gynecol. 1994;170:835-7
39. Provost TT, Watson R. Cutaneous manifestations of Sjogren's syndrome. Rheum Dis Clin North Am. 1992;18:609-16
40. Champey J, Corruble E, Gottenberg JE, Buhl C, Meyer T, Caudmont C. et al. Quality of life and psychological status in patients with primary Sjogren's syndrome and sicca symptoms without autoimmune features. Arthritis Rheum. 2006;55:451-7
41. Milin M, Cornec D, Chastaing M, Griner V, Berrouiguet S, Nowak E. et al. Sicca symptoms are associated with similar fatigue, anxiety, depression, and quality-of-life impairments in patients with and without primary Sjogren's syndrome. Joint Bone Spine. 2016
42. Jung H, Bobba R, Su J, Shariati-Sarabi Z, Gladman DD, Urowitz M. et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum. 2010;62:863-8
43. Godaert GL, Hartkamp A, Geenen R, Garssen A, Kruize AA, Bijlsma JW. et al. Fatigue in daily life in patients with primary Sjogren's syndrome and systemic lupus erythematosus. Ann N Y Acad Sci. 2002;966:320-6
44. Segal B, Thomas W, Rogers T, Leon JM, Hughes P, Patel D. et al. Prevalence, severity, and predictors of fatigue in subjects with primary Sjogren's syndrome. Arthritis Rheum. 2008;59:1780-7
45. Jousse-Joulin S, Morvan J, Devauchelle-Pensec V, Saraux A. Ultrasound assessment of the entheses in primary Sjogren syndrome. Ultrasound Med Biol. 2013;39:2485-7
46. Inal V, Kitapcioglu G, Karabulut G, Keser G, Kabasakal Y. Evaluation of quality of life in relation to anxiety and depression in primary Sjogren's syndrome. Mod Rheumatol. 2010;20:588-97
47. Meijer JM, Meiners PM, Huddleston Slater JJ, Spijkervet FK, Kallenberg CG, Vissink A. et al. Health-related quality of life, employment and disability in patients with Sjogren's syndrome. Rheumatology (Oxford). 2009;48:1077-82
48. Ramos-Casals M, Solans R, Rosas J, Camps MT, Gil A, Del Pino-Montes J. et al. Primary Sjogren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine. 2008;87:210-9
49. Theander E, Manthorpe R, Jacobsson LT. Mortality and causes of death in primary Sjogren's syndrome: a prospective cohort study. Arthritis Rheum. 2004;50:1262-9
50. Fallah M, Liu X, Ji J, Forsti A, Sundquist K, Hemminki K. Autoimmune diseases associated with non-Hodgkin lymphoma: a nationwide cohort study. Ann Oncol. 2014;25:2025-30
51. Zufferey P, Meyer OC, Grossin M, Kahn MF. Primary Sjogren's syndrome (SS) and malignant lymphoma. A retrospective cohort study of 55 patients with SS. Scand J Rheumatol. 1995;24:342-5
52. Ekstrom Smedby K, Vajdic CM, Falster M, Engels EA, Martinez-Maza O, Turner J. et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood. 2008;111:4029-38
53. Papageorgiou A, Ziogas DC, Mavragani CP, Zintzaras E, Tzioufas AG, Moutsopoulos HM. et al. Predicting the outcome of Sjogren's syndrome-associated non-hodgkin's lymphoma patients. PLoS One. 2015;10:e0116189
54. Solans-Laque R, Lopez-Hernandez A, Bosch-Gil JA, Palacios A, Campillo M, Vilardell-Tarres M. Risk, predictors, and clinical characteristics of lymphoma development in primary Sjogren's syndrome. Semin Arthritis Rheum. 2011;41:415-23
55. Voulgarelis M, Tzioufas AG. Pathogenetic mechanisms in the initiation and perpetuation of Sjogren's syndrome. Nat Rev Rheumatol. 2010;6:529-37
56. Gottenberg JE, Seror R, Miceli-Richard C, Benessiano J, Devauchelle-Pensec V, Dieude P. et al. Serum levels of beta2-microglobulin and free light chains of immunoglobulins are associated with systemic disease activity in primary Sjogren's syndrome. Data at enrollment in the prospective ASSESS cohort. PLoS One. 2013;8:e59868
57. Pease CT, Shattles W, Barrett NK, Maini RN. The arthropathy of Sjogren's syndrome. Br J Rheumatol. 1993;32:609-13
58. Ryu YS, Park SH, Lee J, Kwok SK, Ju JH, Kim HY. et al. Follow-up of primary Sjogren's syndrome patients presenting positive anti-cyclic citrullinated peptides antibody. Rheumatology international. 2013;33:1443-6
59. Baldini C, Pepe P, Quartuccio L, Priori R, Bartoloni E, Alunno A. et al. Primary Sjogren's syndrome as a multi-organ disease: impact of the serological profile on the clinical presentation of the disease in a large cohort of Italian patients. Rheumatology (Oxford). 2014;53:839-44
60. Kreider M, Highland K. Pulmonary involvement in Sjogren syndrome. Semin Respir Crit Care Med. 2014;35:255-64
61. Matsuyama N, Ashizawa K, Okimoto T, Kadota J, Amano H, Hayashi K. Pulmonary lesions associated with Sjogren's syndrome: radiographic and CT findings. Br J Radiol. 2003;76:880-4
62. Ito I, Nagai S, Kitaichi M, Nicholson AG, Johkoh T, Noma S. et al. Pulmonary manifestations of primary Sjogren's syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171:632-8
63. Parambil JG, Myers JL, Lindell RM, Matteson EL, Ryu JH. Interstitial lung disease in primary Sjogren syndrome. Chest. 2006;130:1489-95
64. Kondoh Y, Taniguchi H, Yokoi T, Nishiyama O, Ohishi T, Kato T. et al. Cyclophosphamide and low-dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. The European respiratory journal. 2005;25:528-33
65. Nanki N, Fujita J, Yamaji Y, Maeda H, Kurose T, Kaji M. et al. Nonspecific interstitial pneumonia/fibrosis completely recovered by adding cyclophosphamide to corticosteroids. Internal medicine. 2002;41:867-70
66. Both T, Hoorn EJ, Zietse R, van Laar JA, Dalm VA, Brkic Z. et al. Prevalence of distal renal tubular acidosis in primary Sjogren's syndrome. Rheumatology (Oxford). 2015;54:933-9
67. Maripuri S, Grande JP, Osborn TG, Fervenza FC, Matteson EL, Donadio JV. et al. Renal involvement in primary Sjogren's syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4:1423-31
68. Ramos-Casals M, Brito-Zeron P, Seror R, Bootsma H, Bowman SJ, Dorner T. et al. Characterization of systemic disease in primary Sjogren's syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology (Oxford). 2015;54:2230-8
69. Goules AV, Tatouli IP, Moutsopoulos HM, Tzioufas AG. Clinically significant renal involvement in primary Sjogren's syndrome: clinical presentation and outcome. Arthritis Rheum. 2013;65:2945-53
70. Evans RD, Laing CM, Ciurtin C, Walsh SB. Tubulointerstitial nephritis in primary Sjogren syndrome: clinical manifestations and response to treatment. BMC Musculoskelet Disord. 2016;17:2
71. Delalande S, de Seze J, Fauchais AL, Hachulla E, Stojkovic T, Ferriby D. et al. Neurologic manifestations in primary Sjogren syndrome: a study of 82 patients. Medicine. 2004;83:280-91
72. Mori K, Iijima M, Koike H, Hattori N, Tanaka F, Watanabe H. et al. The wide spectrum of clinical manifestations in Sjogren's syndrome-associated neuropathy. Brain: a journal of neurology. 2005;128:2518-34
73. Rist S, Sellam J, Hachulla E, Sordet C, Puechal X, Hatron PY. et al. Experience of intravenous immunoglobulin therapy in neuropathy associated with primary Sjogren's syndrome: a national multicentric retrospective study. Arthritis Care Res (Hoboken). 2011;63:1339-44
74. Rogers SJ, Williams CS, Roman GC. Myelopathy in Sjogren's syndrome: role of nonsteroidal immunosuppressants. Drugs. 2004;64:123-32
75. Lazarus MN, Isenberg DA. Development of additional autoimmune diseases in a population of patients with primary Sjogren's syndrome. Ann Rheum Dis. 2005;64:1062-4
76. Montano-Loza AJ, Crispin-Acuna JC, Remes-Troche JM, Uribe M. Abnormal hepatic biochemistries and clinical liver disease in patients with primary Sjogren's syndrome. Ann Hepatol. 2007;6:150-5
77. Shiboski SC, Shiboski CH, Criswell L, Baer A, Challacombe S, Lanfranchi H. et al. American College of Rheumatology classification criteria for Sjogren's syndrome: a data-driven, expert consensus approach in the Sjogren's International Collaborative Clinical Alliance cohort. Arthritis Care Res (Hoboken). 2012;64:475-87
78. Rasmussen A, Ice JA, Li H, Grundahl K, Kelly JA, Radfar L. et al. Comparison of the American-European Consensus Group Sjogren's syndrome classification criteria to newly proposed American College of Rheumatology criteria in a large, carefully characterised sicca cohort. Ann Rheum Dis. 2014;73:31-8
79. Kassan SS, Moutsopoulos HM. Clinical manifestations and early diagnosis of Sjogren syndrome. Arch Intern Med. 2004;164:1275-84
80. Ramos-Casals M, Tzioufas AG, Font J. Primary Sjogren's syndrome: new clinical and therapeutic concepts. Ann Rheum Dis. 2005;64:347-54
81. Sall K, Stevenson OD, Mundorf TK, Reis BL. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology. 2000;107:631-9
82. Papas AS, Sherrer YS, Charney M, Golden HE, Medsger TA Jr, Walsh BT. et al. Successful Treatment of Dry Mouth and Dry Eye Symptoms in Sjogren's Syndrome Patients With Oral Pilocarpine: A Randomized, Placebo-Controlled, Dose-Adjustment Study. J Clin Rheumatol. 2004;10:169-77
83. Chitapanarux I, Kamnerdsupaphon P, Tharavichitkul E, Sumitsawan Y, Sittitrai P, Pattarasakulchai T. et al. Effect of oral pilocarpine on post-irradiation xerostomia in head and neck cancer patients: a single-center, single-blind clinical trial. J Med Assoc Thai. 2008;91:1410-5
84. Fox RI, Dixon R, Guarrasi V, Krubel S. Treatment of primary Sjogren's syndrome with hydroxychloroquine: a retrospective, open-label study. Lupus. 1996;5(Suppl 1):S31-6
85. Rihl M, Ulbricht K, Schmidt RE, Witte T. Treatment of sicca symptoms with hydroxychloroquine in patients with Sjogren's syndrome. Rheumatology (Oxford). 2009;48:796-9
86. Gottenberg JE, Ravaud P, Puechal X, Le Guern V, Sibilia J, Goeb V. et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjogren syndrome: the JOQUER randomized clinical trial. Jama. 2014;312:249-58
87. Skopouli FN, Jagiello P, Tsifetaki N, Moutsopoulos HM. Methotrexate in primary Sjogren's syndrome. Clin Exp Rheumatol. 1996;14:555-8
88. Saraux A, Pers JO, Devauchelle-Pensec V. Treatment of primary Sjogren syndrome. Nat Rev Rheumatol. 2016;12:456-71
89. Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant. 2006;6:859-66
90. Ramos-Casals M, Garcia-Hernandez FJ, de Ramon E, Callejas JL, Martinez-Berriotxoa A, Pallares L. et al. Off-label use of rituximab in 196 patients with severe, refractory systemic autoimmune diseases. Clin Exp Rheumatol. 2010;28:468-76
91. Pollard RP, Abdulahad WH, Vissink A, Hamza N, Burgerhof JG, Meijer JM. et al. Serum levels of BAFF, but not APRIL, are increased after rituximab treatment in patients with primary Sjogren's syndrome: data from a placebo-controlled clinical trial. Ann Rheum Dis. 2013;72:146-8
92. Sharma A, Kiripolsky J, Klimatcheva E, Howell A, Fereidouni F, Levenson R. et al. Early BAFF receptor blockade mitigates murine Sjogren's syndrome: Concomitant targeting of CXCL13 and the BAFF receptor prevents salivary hypofunction. Clin Immunol. 2016;164:85-94
93. Belkhir R, Gestermann N, Koutero M, Seror R, Tost J, Mariette X. et al. Upregulation of membrane-bound CD40L on CD4+ T cells in women with primary Sjogren's syndrome. Scand J Immunol. 2014;79:37-42
94. Nakamura H, Horai Y, Suzuki T, Okada A, Ichinose K, Yamasaki S. et al. TLR3-mediated apoptosis and activation of phosphorylated Akt in the salivary gland epithelial cells of primary Sjogren's syndrome patients. Rheumatology international. 2013;33:441-50
95. Le KS, Thibult ML, Just-Landi S, Pastor S, Gondois-Rey F, Granjeaud S. et al. Follicular B lymphomas generate regulatory T cells via the ICOS/ICOSL pathway and are susceptible to treatment by anti-ICOS/ICOSL therapy. Cancer Res. 2016
96. Brito-Zeron P, Kostov B, Solans R, Fraile G, Suarez-Cuervo C, Casanovas A. et al. Systemic activity and mortality in primary Sjogren syndrome: predicting survival using the EULAR-SS Disease Activity Index (ESSDAI) in 1045 patients. Ann Rheum Dis. 2016;75:348-55
97. Skopouli FN, Dafni U, Ioannidis JP, Moutsopoulos HM. Clinical evolution, and morbidity and mortality of primary Sjogren's syndrome. Semin Arthritis Rheum. 2000;29:296-304
98. Vazquez A, Khan MN, Sanghvi S, Patel NR, Caputo JL, Baredes S. et al. Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue of the salivary glands: a population-based study from 1994 to 2009. Head Neck. 2015;37:18-22
99. Tsokos M, Lazarou SA, Moutsopoulos HM. Vasculitis in primary Sjogren's syndrome. Histologic classification and clinical presentation. American journal of clinical pathology. 1987;88:26-31
100. Fox RI, Liu AY. Sjogren's syndrome in dermatology. Clin Dermatol. 2006;24:393-413
101. Garcia-Carrasco M, Siso A, Ramos-Casals M, Rosas J, de la Red G, Gil V. et al. Raynaud's phenomenon in primary Sjogren's syndrome. Prevalence and clinical characteristics in a series of 320 patients. J Rheumatol. 2002;29:726-30
102. Roguedas AM, Misery L, Sassolas B, Le Masson G, Pennec YL, Youinou P. Cutaneous manifestations of primary Sjogren's syndrome are underestimated. Clin Exp Rheumatol. 2004;22:632-6
103. Bossini N, Savoldi S, Franceschini F, Mombelloni S, Baronio M, Cavazzana I. et al. Clinical and morphological features of kidney involvement in primary Sjogren's syndrome. Nephrol Dial Transplant. 2001;16:2328-36
104. Colafrancesco S, Priori R, Gattamelata A, Picarelli G, Minniti A, Brancatisano F. et al. Myositis in primary Sjogren's syndrome: data from a multicentre cohort. Clin Exp Rheumatol. 2015;33:457-64
105. Lindvall B, Bengtsson A, Ernerudh J, Eriksson P. Subclinical myositis is common in primary Sjogren's syndrome and is not related to muscle pain. J Rheumatol. 2002;29:717-25
106. Brito-Zeron P, Akasbi M, Bosch X, Bove A, Perez-De-Lis M, Diaz-Lagares C. et al. Classification and characterisation of peripheral neuropathies in 102 patients with primary Sjogren's syndrome. Clin Exp Rheumatol. 2013;31:103-10
107. Pavlakis PP, Alexopoulos H, Kosmidis ML, Mamali I, Moutsopoulos HM, Tzioufas AG. et al. Peripheral neuropathies in Sjogren's syndrome: a critical update on clinical features and pathogenetic mechanisms. J Autoimmun. 2012;39:27-33
108. Binder A, Snaith ML, Isenberg D. Sjogren's syndrome: a study of its neurological complications. Br J Rheumatol. 1988;27:275-80
109. Ramos-Casals M, Nardi N, Brito-Zeron P, Aguilo S, Gil V, Delgado G. et al. Atypical autoantibodies in patients with primary Sjogren syndrome: clinical characteristics and follow-up of 82 cases. Semin Arthritis Rheum. 2006;35:312-21
110. Bickham K, Kivitz AJ, Mehta A, Frontera N, Shah S, Stryszak P. et al. Evaluation of two doses of etoricoxib, a COX-2 selective non-steroidal anti-inflammatory drug (NSAID), in the treatment of Rheumatoid Arthritis in a double-blind, randomized controlled trial. BMC Musculoskelet Disord. 2016;17:331
111. Hochberg MC, Altman RD, April KT, Benkhalti M, Guyatt G, McGowan J. et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64:465-74
112. Fox PC, Datiles M, Atkinson JC, Macynski AA, Scott J, Fletcher D. et al. Prednisone and piroxicam for treatment of primary Sjogren's syndrome. Clin Exp Rheumatol. 1993;11:149-56
113. Devauchelle-Pensec V, Mariette X, Jousse-Joulin S, Berthelot JM, Perdriger A, Puechal X. et al. Treatment of primary Sjogren syndrome with rituximab: a randomized trial. Ann Intern Med. 2014;160:233-42
Author contact
![]() Corresponding author: Dr. P.L.A. van Daele, PO Box 2040 - Room D-419, 3000 CA Rotterdam, The Netherlands. Phone: +31-10-7035954 Fax: +31-10-7040704 E-mail: p.l.a.vandaelenl.
Corresponding author: Dr. P.L.A. van Daele, PO Box 2040 - Room D-419, 3000 CA Rotterdam, The Netherlands. Phone: +31-10-7035954 Fax: +31-10-7040704 E-mail: p.l.a.vandaelenl.